

Abstract
Mutations in Kirsten rat-sarcoma (KRAS) are well appreciated to be major drivers of human cancers through dysregulation of multiple growth and survival pathways. Similar to many other non-kinase oncogenes and tumor suppressors, efforts to directly target KRAS pharmaceutically have not yet materialized. As a result, there is broad interest in an alternative approach to develop therapies that induce synthetic lethality in cancers with mutant KRAS, therefore exposing the particular vulnerabilities of these cancers. Fueling these efforts is our increased understanding into the biology driving KRAS mutant cancers, in particular the important pathways that mutant KRAS governs to promote survival. In this mini-review, we summarize the latest approaches to treat KRAS mutant cancers and the rationale behind them.
Keywords: Apoptosis, Kirsten rat-sarcoma, MEK, phosphatidylinositol 3-kinase, synthetic lethality
Onocogenic mutations in Kirsten rat-sarcoma (KRAS) occur in up to 25% of human cancers, positioning them as the most common gain-of-function mutations in human cancer.1–3 Despite the development of small-molecule inhibitors that interfere with the localization of KRAS or inhibit the activity of mutant KRAS,4,5 oncogenic KRAS remains a largely elusive target of drug development. Thus, blocking mutant KRAS may require a strategy more akin to one designed to counter the loss of a tumor suppressor – via targeting of vital downstream effector pathways. Along these lines, a number of studies in KRAS mutant cancers have led to strategies to target these pathways. Below, we will discuss the main effector pathways of KRAS and current approaches to develop combination therapies targeting these KRAS-effector pathways. Also, other approaches targeting KRAS, including synthetic lethal screening, will be summarized.
Downstream Effectors of KRAS
Kirsten rat-sarcoma protein cycles between an inactive GDP-bound state and an active GTP-bound state. A number of stimuli, including ligands that activate growth factor receptors and G-protein coupled receptors on the cell membrane, lead to the activation of RAS guanine exchange factors (GEFs).6 This, in turn, results in the formation of active GTP-bound KRAS. In wild-type KRAS cells, KRAS is subsequently inactivated by Ras-GTPase activating proteins (RasGAPs). However, oncogenic KRAS mutations, which occur most frequently at amino acids 12, 13, and 61, render KRAS proteins resistant to RasGAP-mediated GTP-hydrolysis. This leads to constitutive activation of KRAS protein. Mutant KRAS activates multiple downstream effector pathways, resulting in the uncontrolled growth, proliferation, and survival of cancer cells (Fig.1). Amongst these, three major effector pathways have emerged as being critical to mutant KRAS-mediated transformation and will be discussed in greater detail: the RAF-MEK-ERK pathway, the phosphatidylinositol 3-kinase (PI3K) pathway, and the Ral-NF-kB pathway.
Figure 1.
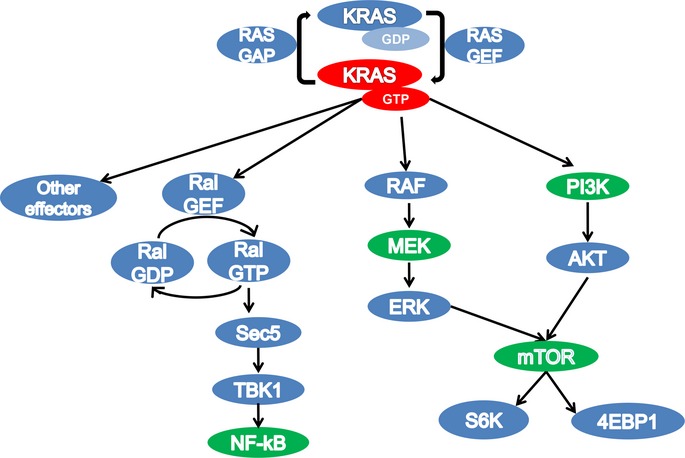
Effector pathways of Kirsten rat-sarcoma (KRAS). Proteins highlighted green are pharmacologically targetable.
RAF-MEK-ERK pathway
The RAF serine/threonine kinases bind KRAS via their RAS Binding Domain (RBD). RAF activation in turn activates the serine/threonine kinases MEK1 and MEK2, which in turn activate ERK. The requirement for the RAF-MEK-ERK (MAPK) pathway in KRAS-mediated transformation and tumorigenesis has been well established.7 However, inhibition of the MAPK pathway alone is not sufficient to eradicate KRAS mutant tumors. MEK inhibitors exhibit cytostatic rather than cytotoxic activity, inhibiting proliferation but not inducing significant apoptosis.8,9 In accordance with these preclinical studies, the MEK inhibitor selumetinib (AstraZeneca, Macclesfield, UK) failed to show clinical activity in an unselected pretreated patient population with a high-rate of KRAS mutations.10–12
PI3K pathway
The precise role of KRAS in regulating PI3K has been difficult to elucidate because PI3K can be activated by multiple upstream signals, not all of which integrate KRAS to promote downstream signaling. Several lines of evidence suggest PI3K associates with, and is activated by KRAS, thus serving as a principal mechanism of PI3K regulation. The binding of KRAS to p110α induces a conformational change in p110α, which opens and orients the active site of KRAS toward its substrate. Although RBD mutants of p110α fail to bind KRAS, they still maintain enzymatic activity. Interestingly, mice engineered to express RBD-mutant p110α cannot develop mutant Kras-driven lung tumors.13 Furthermore, by using an inducible mouse model of mutant Kras-driven lung cancer, Downward and colleagues showed that loss of Kras-p110α binding leads to long-term tumor stasis and partial regression.14 These elegant studies showed that the interaction between mutant KRAS and p110α is not only required for tumorigenesis but also for tumor maintenance.
In addition to direct activation by KRAS, PI3K can also be activated by receptor tyrosine kinases (RTKs) in KRAS mutant cancers. We have reported in colorectal cancers that insulin-like growth factor 1 receptor (IGF-IR) exerts dominant control over PI3K signaling through binding to insulin receptor substrate (IRS) adaptor proteins even in the presence of mutant KRAS.15 PI3K activity is also dependent on basal IGF-IR activity in KRAS mutant lung cancer, although in this context mutant KRAS is still thought to be involved in PI3K activation. It has been shown that IGF-IR activation causes IRS-1:p85 complex formation, which in turn relieves an inhibitory effect of p85 on PI3K signaling.16 Additionally, a recent study showed the KRAS mutant NCI-H358 non-small cell lung cancer (NSCLC) cell line still remains dependent on ERBB3 for PI3K signaling.17 Altogether, these studies suggest numerous contributors, including mutant KRAS and RTKs, activate PI3K signaling in KRAS mutant cancers. Another confounding issue is that the role of mutant KRAS may further differ depending on other mutations that may be more or less prevalent among the different tissue types of origin. For example, oncogenic mutations in KRAS and PIK3CA often coexist in colorectal cancer but less often in pancreatic cancer.18 The coexistence of KRAS and PIK3CA mutations in colorectal cancers suggests that mutant KRAS is not sufficient for robust PI3K activity. Similar to MEK inhibitors, single agent PI3K inhibitors are also ineffective for treatment of KRAS mutant cancers; murine lung cancers driven by oncogenic Kras do not respond to the PI3K/mammalian target of rapamycin (mTOR) inhibitor, NVP-BEZ235.19 Furthermore, KRAS mutations predict resistance to PI3K inhibitors in cell culture experiments.20,21
Ral-NF-κB pathway
While the RAF-MEK-ERK and PI3K pathways have been established as key KRAS-effector pathways, KRAS has a number of additional effectors. Among them, the guanine exchange factors of the Ras-like (Ral) GTPases (RalGEFs) have emerged as important effectors of KRAS. Ras-like GTPases directly interact with RAS, and subsequently activates Ral small GTPases.22,23 Two Ral small GTPases, RalA and RalB, appear to have distinct biological roles in KRAS mutant cancers. For instance, inhibition of RalA alone is enough to inhibit tumor initiation, while RalB is vital for tumor invasion and metastasis.24–26 Similar to KRAS, activated Ral-GTP interacts with multiple downstream effector proteins including RalBP1, which promotes membrane ruffling and filopodia formation through Rac1 and CDC42, as well as receptor trafficking via endocytic regulation.27 Additional effectors of Ral are the octometric exocyst subunits Sec5 and Exo84, important for secretory vesicle delivery to different membrane compartments.28 Lastly, active RalB signaling causes the association of Sec5 complex with the atypical IkB-related protein kinase TBK1 to promote cell survival through activation of the oncogenic transcription factor NF-κB.30
Targeting PI3K-AKT and MEK-ERK Signaling by Combinatorial Approaches
The lack of efficacy seen following suppression of single effector pathway (e.g. use of MEK inhibitors or PI3K inhibitors) in KRAS mutant cancers suggests that a combinatorial approach targeting multiple effector pathways is needed. When cancer cells exhibit dependency on a single oncogene (“oncogene addiction”), inhibition of the oncogene leads to downregulation of both PI3K/AKT and MEK/ERK signaling in most instances. Importantly, combination of both a PI3K inhibitor and a MEK inhibitor is sufficient to recapitulate much of the apoptosis and suppression of tumor growth induced by EGFR inhibitors in EGFR mutant NSCLC.31 Moreover, HER2 amplified and/or PIK3CA mutant breast cancers are particularly sensitive to single agent PI3K inhibitors, which surprisingly downregulate both PI3K and MEK/ERK signaling in these cancers, resulting in apoptosis.32 These results suggest that concomitant disruption of PI3K/AKT and MEK/ERK signaling may underlie much of the antitumor effects observed with targeted therapies in oncogene-addicted models. Consistent with this concept, pharmaceutical inhibition of both the MEK and PI3K pathways has shown durable responses in KRAS mutant cancers in vivo.8,19
Currently, a large number of clinical trials to assess the combination of PI3K inhibitors and MEK inhibitors are ongoing (Table1). A recent dose-escalation trial tested the combination of the dual PI3K/mTOR inhibitor SAR245409 (Sanofi, Paris, France) with the MEK1/2 inhibitor pimasertib (Merck KGAA, Darmstadt, Germany) in 46 cancer patients. Among the patients, two partial responses were observed: one in a patient with KRAS mutant colorectal cancer whose tumor exhibited neuroendocrine features, and a low-grade ovarian cancer patient with simultaneous KRAS and PI3KCA mutations. Grade 3 and 4 toxicities were infrequent, with the most common grade 3 event being skin rash in 14% of patients.33 In a separate trial combining the PI3K inhibitor BKM120 (Novartis, Basel, Switzerland) and the MEK inhibitor trametinib (GlaxoSmithKline, Brentford, UK), three patients with KRAS mutant ovarian cancer achieved partial responses among 66 patients in an unselected population.34 Based on these three responses, this trial is expanding cohorts to specifically include patients with KRAS or BRAF mutant tumors. These results suggest that the combination of PI3K and MEK inhibitors has activity, but the activity appears relatively limited. This lack of robust activity seems to be attributed to the difficulty of sufficiently suppressing both pathways without toxicities in a given patient. For example, a trial combining MK-2206 (Merck), an AKT inhibitor, and selumetinib, four of eight patients demonstrated biologically significant inhibition in one marker; however, at the maximum tolerated dose no patient had ≥70% inhibition of both targets.35
Table 1.
Currently ongoing trials combining phosphatidylinositol 3-kinase (PI3K) inhibitor and MEK inhibitor
| NCT no. | Phase | Company | PI3K inhibitor | MEK inhibitor | Patient selection |
|---|---|---|---|---|---|
| 01347866 | I | Pfizer (New York, NY, USA) | PF-05212384 (PI3K/mTOR inhibitor) | PD-0325901 | At the MTD dose, further assessment of these combinations will be done in patients with KRAS mutated colorectal cancer |
| 01363232 | Ib | Novartis | BKM120 (pan PI3K inhibitor) | MEK162 | At the MTD dose, this combination is explored in patients with EGFR mutant NSCLC, whom have progressed on EGFR inhibitors and triple negative breast cancer, as well as other advanced solid tumors with KRAS, NRAS, and/or BRAF mutations |
| 01390818 | I | EMD Serono (Rockland, MA, USA) | SAR245409 (PI3K/mTOR inhibitor) | Pimasertib | Locally advanced or metastatic solid tumors |
| 01155453 | Ib | Novartis | BKM120 (pan PI3K inhibitor) | Trametinib | At the MTD dose, further assessment will be done in patients with KRAS or BRAF mutated NSCLC, ovarian, and pancreatic cancer |
| 01859351 | I | Wilex (München, Germany) | WX-037 (pan PI3K inhibitor) | WX-554 | Solid tumor |
| 01337765 | Ib | Novartis | BEZ235 (PI3K/mTOR inhibitor) | MEK162 | At the MTD dose, this combination was assessed in patients with EGFR mutant NSCLC, whom have progressed on EGFR inhibitors and triple negative breast cancer, as well as other advanced solid tumors with KRAS, NRAS, and/or BRAF mutations |
| 01392521 | Ib | Bayer (Leverkusen, Germany) | BAY80-6946 (pan class I PI3K inhibitor) | BAY86-9766 | Advanced cancer |
| 00996892 | Ib | Genentech (San Francisco, CA, USA) | GDC-0941 (Pan PI3K inhibitor) | GDC-0973 | Locally advanced or metastatic solid tumors |
| 01449058 | Ib | Novartis | BYL719 (PI3K alpha-specific inhibitor) | MEK162 | Advanced solid tumors or AML or high risk and very high risk MDS, with documented RAS or BRAF mutations |
| 01248858 | I | GlaxoSmithKline | GSK2126458 (pan PI3K/mTOR inhibitor) | Trametinib | Advanced solid tumors |
AML, acute myeloid leukemia; EGFR, epidermal growth factor receptor; MDS, myelodysplastic syndromes; MEK, mitogen-activated protein kinase kinase; MTD, Maximum Tolerated Dose; mTOR, mammalian target of rapamycin; NCT, national clinical trial that is given to each registered clinical trial; NSCLC, non–small-cell lung cancer; PI3K, phosphatidylinositol 3-kinase.
Alternative therapeutic strategies targeting RTKs that indirectly suppress the PI3K pathway in combination with MEK inhibition may be more tolerable, and as a consequence more effective. As mentioned, the IGF-IR is largely responsible for PI3K activation in KRAS mutant colorectal and lung cancer cell lines, and the combination of IGF-IR and MEK inhibitors results in tumor regressions in these xenografts.15,16 This approach is currently being evaluated in a phase I/II trial of IGF-IR antibody ganitumab (Amgen, Thousand Oaks, CA, USA) combined with the MEK inhibitor MEK162 (Novartis) in KRAS mutant colorectal and pancreatic cancer and BRAF mutant melanoma (ClinicalTrilas.gov registry number, NCT01562899).
Targeting the Apoptotic Machinery
As mentioned above, in cancers addicted to a single oncogene, effective target inhibition generally results in apoptosis. This process involves the downstream BCL-2 family of proteins, which act as guardians of mitochondria-mediated apoptosis. For example, in EGFR mutant NSCLCs, treatment with an EGFR inhibitor shifts the balance of pro- and anti-apoptotic BCL-2 family members, reducing the expression of anti-apoptotic MCL-1 as a result of PI3K/mTORC1 inhibition,31 and increasing the expression of pro-apoptotic BIM as a result of MEK/ERK suppression, leading to apoptosis.31,36 In addition, a recent study using engineered mice deficient for the pro-apoptotic BCL-2 family members BIM or PUMA provided evidence that BIM and PUMA are both key apoptotic effectors of tyrosine kinase inhibitors in EGFR mutant NSCLC and HER2 amplified breast cancer.37
The TBK1/BCL-XL pathway
In addition to the PI3K and MEK/ERK pathway, mutant KRAS maintains proliferation and evades apoptosis through other pathways. For instance, shRNA screening using KRAS mutant cancer cell lines identified TBK1 as a synthetic lethal partner of oncogenic KRAS. Interestingly, BCL-XL, a known NF-κB target, was identified as a TBK1-regulated gene. Overexpression of BCL-XL rescued apoptosis induced by KRAS or TBK1 knockdown in the NCI-H23 KRAS mutant cell line.38
Combination of MEK inhibitor with BCL-XL inhibitor
Pharmacological inhibition of the MEK/ERK pathway is relatively more achievable compared with the PI3K pathway.38,39 Therefore, MEK inhibitor therapy could be a backbone for combinatorial approaches for KRAS mutant cancers. To this point, shRNA screening was performed to identify genes that, when inhibited, cooperate with MEK inhibitors to reduce cell survival in KRAS mutant cell lines.41 BCL-XL emerged as a top hit through this approach. That is, BIM induction following MEK inhibition is not enough to cause apoptosis, but BCL-XL knockdown disrupts an inhibitory complex between BIM and BCL-XL, leading to apoptosis in the presence of MEK inhibitor. Induction of apoptosis is recapitulated by combining the BCL-2/BCL-XL inhibitor navitoclax (ABT-263) with a MEK inhibitor. Two additional studies have also shown the efficacy of this combination.41,42
Combination of mTORC1/2 inhibitor and BCL-2/BCL-XL inhibitor
We have recently showed KRAS mutant colorectal cancers are particularly vulnerable to simultaneous inhibition of the BCL-2 anti-apoptotic proteins BCL-2, BCL-XL and MCL-1.44 Pure mTORC catalytic site inhibitors downregulated MCL-1 in KRAS mutant colorectal cancers, and targeting KRAS with shRNA similarly reduced mTORC1 signaling and MCL-1 levels, suggesting MCL-1 to be a vital KRAS-effector molecule in these cancers. When combined with the BCL-2/BCL-XL inhibitor navitoclax, the mTORC1/2 inhibitor AZD8055 induced tumor regressions in KRAS mutant human colorectal cancer xenografts and Kras mutant genetically engineered mouse models of colorectal cancers. In all, this study provides the rationale to use mTORC inhibitors in combination with BCL-2/BCL-XL inhibitors in KRAS mutant colorectal cancers. Altogether, these data mark the apoptotic machinery as an attractive target to treat KRAS mutant cancers (Fig.2).
Figure 2.
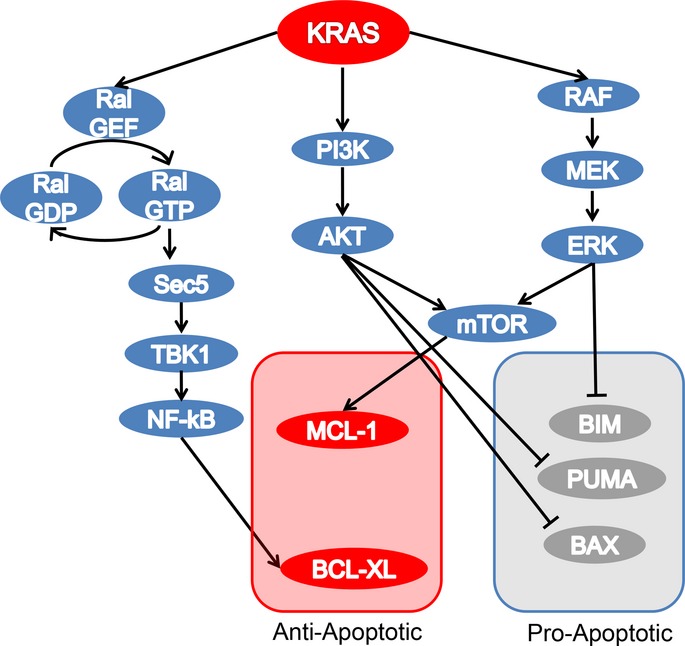
Effector proteins of Kirsten rat-sarcoma (KRAS) and apoptosis. The BCL-2 family of proteins regulates mitochondrial-driven apoptosis in KRAS mutant cancers. The BCL-2 family consists of three subfamilies: the pro-survival members such as BCL-2 or MCL1, the pro-apoptotic BCL-2 homology domain 3 (BH3)-only proteins such as BIM and PUMA, and the pro-apoptotic BAX and BCL-2 antagonist/killer (BAK; not shown in this figure). The anti-apoptotic function of oncogenic KRAS is mediated by several effector pathways that converge on the BCL-2 family of proteins. The PI3K effector pathway suppresses pro-apoptotic protein PUMA and BAX, the RAS–RAF pathway downregulates the pro-apoptotic protein BIM, and the mTORC1 pathway regulates MCL-1. In addition, the Ral-NF-κB pathway has been implicated in the regulation of BCL-XL. Thus, KRAS suppresses cell death responses through regulation of both pro-apoptotic and anti-apoptotic BCL-2 family proteins.
Combination of MEK inhibitor and docetaxel
Several studies have demonstrated that cytotoxic agents, including microtubule stabilizing drugs, stimulate MAPK signaling upon administration. Combining inhibitors of MAPK signaling with one such drug, docetaxel, results in an enhanced anti-tumorigenic phenotype.45 One of the key mechanisms of this synergy is induction of pro-apoptotic proteins by inhibiting MAPK signaling, which reduces the threshold for apoptosis induction by cytotoxic agents. In fact, prolonged exposure to the MEK inhibitor selumetinib induced BIM expression in the KRAS mutant HCT-116 xenograft model. A prospective randomized phase II study assessing the impact of adding selumetinib to docetaxel in previously treated patients with advanced KRAS mutant NSCLC was conducted based on these pre-clinical results. Despite no differences in median overall survival, there was significant improvements in both progression-free survival and objective response rate in patients administered selumetinib.46
Concurrently with the clinical trials in human subjects, a Kras mutant transgenic mouse model was used to optimize treatment modalities, a so-called “co-clinical” trial.47 This mouse study revealed that adding selumetinib was beneficial for mice with Kras or Kras / p53 mutant lung cancer, but not with Kras and Lkb1 mutations. Interestingly, Kras/Lkb1 tumors show substantially less phosphorylation of ERK, suggesting that the ERK pathway is less active in these cancers. Furthermore, integrated genomic and proteomic profiles revealed SRC is activated in Kras/Lkb1 tumors,48 suggesting that Kras/Lkb1 mutant tumors are a distinct subset of KRAS mutant cancers that may be less dependent on ERK signaling and more dependent on other pathways. Intriguingly, another recent report suggests that NSCLCs harboring mutations both in KRAS and LKB1 are addicted to coatomer complex I (COPI)-dependent lysosome acidification, which participates in retrograde transport, is required for endosome maturation and is a CDC42 effector required for CDC42 transformation.49
Identifying Synthetic Lethal Interaction with KRAS
Recent high-throughput screening has provided an expanded list of targets for KRAS mutant tumors (Table2). For example, siRNA screening in KRAS mutant NSCLC cell lines identified the transcription factor GATA2 as necessary for the survival of these cancers.50 GATA2 maintains cell survival via the proteasome machinery, the IL-1/NF-κB signaling pathway, and the Rho-signaling cascade. Combined inhibition of the proteasome and Rho signaling recapitulates the effect of GATA2 loss on KRAS-driven tumorigenesis. CDC6, a critical regulator of DNA replication, has also been identified as a synthetic lethal protein with mutant KRAS.51 Bioinformatic analysis suggests proteasome components functionally interact with CDC6, and knockdown of CDC6 showed additional synthetic lethal effects with proteasome inhibitor treatment. Other targets identified by synthetic lethal approaches include, as discussed above, TBK1,38 as well as COPI,48 STK33,52 TAK1,53 APC/C,54 CDK4,55 Polo-like kinase (PLK) 1,54 and reactive oxygen species (ROS).56 It should be cautioned that a major caveat associated with RNAi screening is potential off-target effects and the potential disconnect between reduction of total expression and inhibition of kinase function. For example, while STK33 knockdown was synthetic lethal for KRAS mutant cancers, inhibition of STK33 kinase activity does not appear to be effective therapy for KRAS mutant cancers.57
Table 2.
Candidate genes showing synthetic lethal interaction with Kirsten rat-sarcoma (KRAS)
| Synthetic lethal genes or pathways | Methodology | Pharmacological inhibition | References |
|---|---|---|---|
| TBK1 | shRNA screening | Not assessed | 38 |
| Coatomer complex I (COPI) | Parallel screening of chemical and genetic perturbations | Saliphenylhalamide A | 49 |
| GATA2 | siRNA screening | Bortezomib with Fasudil | 50 |
| CDC6 | siRNA screening | Bortezomib and topotecan | 51 |
| STK33 | shRNA screening | Specific inhibitor was subsequently developed, but failed to suppress growth of cells | 52,57 |
| TAK1 | Expression data based bioinfomatic analysis | 5Z-7-oxozeaenol | 53 |
| Polo-like kinase (PLK) 1 and 2 | shRNA screening and outlier kinase analysis | BI-2536 | 54,58 |
| CDK4 | Mouse genetic studies | PD0332991 | 55 |
| Reactive oxygen species | Chemical screening | Lanperisone | 56 |
Fasudil is a Rho signaling inhibitor, approved for the treatment of cerebrovascular spasm in Japan.
Other Means to Target KRAS
“Outlier kinase” approach
Using an innovative approach of identifying “outlier kinase” expression through analysis of transcriptome sequencing data from a large number of cancers, polo-like kinases (PLKs) were noted to be overexpressed in a subset of KRAS mutant pancreatic cancers, and these cancers had specific sensitivity to the PLK-pan inhibitor, BI-6727.58
HSP90 inhibitor combinations
Pharmaceutically targeting HSP90 has attracted significant interest. HSP90 inhibitors target HSP90 client proteins resulting in their rapid degradation. Although KRAS is not a client protein of HSP90, KRAS mutant NSCLCs are exquisitely sensitive to HSP90 inhibition,59 most likely through the HSP90-inhibitor-mediated degradation of downstream signaling proteins such as C-RAF60 as well as the production of ROS.61 Interestingly, HSP90 inhibitors may have particular activity in combination with the mTOR inhibitor rapamycin in KRAS/p53 mutant NSCLCs through rapamycin-mediated suppression of glutathione in the presence of HSP90-inhibitor induced ROS.61
Targeting posttranslational modification of KRAS
Lastly, targeting mutant KRAS by interfering with important KRAS post-translational modifications has recently been explored. The phosphorylation of KRAS on Serine 181, which is mediated by PKC,62 is indispensable for full KRAS oncogenic activity.63,64 As such, treatment of KRAS mutant cancers with PKC inhibitors has anti-proliferative and pro-apoptotic activity,63,64 marking PKC as an intriguing therapeutic target.
Conclusion
Targeted therapies that directly disrupt oncogene function have changed the way cancers are treated. While one of the most obvious targets is oncogenic KRAS, mutated in roughly one-fourth of all cancers, direct targeting of KRAS has remained largely elusive. Instead, co-targeting pathways downstream of mutant KRAS has emerged in pre-clinical studies as a promising therapeutic strategy. However, validation of these pre-clinical studies has been hindered by unanticipated challenges, such as dose-limiting toxicity of combinatorial inhibition of PI3K and MEK/ERK signaling. Alternatively, blocking upstream activators of PI3K, such as IGF-IR, in combination with MEK inhibition, may be a less toxic and thus more successful strategy. More recently, targeting the apoptotic machinery in KRAS mutant cancers has garnered attention. For instance, mTORC inhibitors in combination with BCL-2/BCL-XL inhibitors showed dramatic pre-clinical efficacy in KRAS mutant colorectal cancers in vivo. Moreover, the identification of novel targets that offer synthetic lethality with mutant KRAS has paved the way toward new therapeutic strategies. However, whether effective drugs can be designed to disrupt these targets, and whether these drugs can be administered at doses high enough to inhibit their targets, remains to be seen. Lastly, the identification of already clinically available drugs that show efficacy in subsets of KRAS mutant cancers, such as the combination of docetaxel and selumetinib in KRAS mutant NSCLC with wild type LKB1, may speed up the implementation of much needed novel therapies.
Acknowledgments
We thank Drs Matt Niederst and Erin Coffee for critical reading of the manuscript.
Disclosure Statement
The authors have no conflict of interest.
Funding information
None declared.
References
- 1.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–42. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 6.Buday L, Downward J. Many faces of Ras activation. Biochim Biophys Acta. 2008;1786:178–87. doi: 10.1016/j.bbcan.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Khosravi-Far R, White MA, Westwick JK, et al. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923–33. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sos ML, Fischer S, Ullrich R, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci USA. 2009;106:18351–6. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brognard J, Dennis PA. Variable apoptotic response of NSCLC cells to inhibition of the MEK/ERK pathway by small molecules or dominant negative mutants. Cell Death Differ. 2002;9:893–904. doi: 10.1038/sj.cdd.4401054. [DOI] [PubMed] [Google Scholar]
- 10.Hainsworth JD, Cebotaru CL, Kanarev V, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5:1630–6. doi: 10.1097/JTO.0b013e3181e8b3a3. [DOI] [PubMed] [Google Scholar]
- 11.Bennouna J, Lang I, Valladares-Ayerbes M, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs. 2011;29:1021–8. doi: 10.1007/s10637-010-9392-8. [DOI] [PubMed] [Google Scholar]
- 12.Bodoky G, Timcheva C, Spigel DR, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30:1216–23. doi: 10.1007/s10637-011-9687-4. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Ramjaun AR, Haiko P, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 14.Castellano E, Sheridan C, Thin MZ, et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell. 2013;24:617–30. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebi H, Corcoran RB, Singh A, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311–21. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013;3:548–63. doi: 10.1158/2159-8290.CD-12-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salt MB, Bandyopadhyay S, McCormick F. Epithelial to mesenchymal transition rewires the molecular path to PI3-Kinase-dependent proliferation. Cancer Discov. 2014;4:186–99. doi: 10.1158/2159-8290.CD-13-0520. [DOI] [PubMed] [Google Scholar]
- 18. COSMIC (Catalog of Somatic Mutations in Cancer) database Wellcome Trust Sanger Institute. [Cited 14 Feb 2014.] Available from URL: http://www.sanger.ac.uk/cosmic.
- 19.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torbett NE, Luna-Moran A, Knight ZA, et al. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008;415:97–110. doi: 10.1042/BJ20080639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ihle NT, Lemos R, Jr, Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–50. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neel NF, Martin TD, Stratford JK, Zand TP, Reiner DJ, Der CJ. The RalGEF-Ral effector signaling network: The road less traveled for anti-ras drug discovery. Genes Cancer. 2011;2:275–87. doi: 10.1177/1947601911407329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133–40. doi: 10.1038/nrc2296. [DOI] [PubMed] [Google Scholar]
- 24.Chien Y, White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 2003;4:800–6. doi: 10.1038/sj.embor.embor899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–45. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 26.Lim KH, O'Hayer K, Adam SJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385–94. doi: 10.1016/j.cub.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 27.Jullien-Flores V, Dorseuil O, Romero F, et al. Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J Biol Chem. 1995;270:22473–7. doi: 10.1074/jbc.270.38.22473. [DOI] [PubMed] [Google Scholar]
- 28.Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA. The exocyst is a Ral effector complex. Nat Cell Biol. 2002;4:66–72. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- 29.Kashatus DF. Ral GTPases in tumorigenesis: emerging from the shadows. Exp Cell Res. 2013;319:2337–42. doi: 10.1016/j.yexcr.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chien Y, Kim S, Bumeister R, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–70. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 31.Faber AC, Li D, Song Y, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009;106:19503–8. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebi H, Costa C, Faber AC, et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc Natl Acad Sci USA. 2013;110:21124–9. doi: 10.1073/pnas.1314124110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Infante JR, Gandi L, Shapiro G, et al. Combination of the MEK inhibitor, pimasertib (MSC1936369B), and the PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: results of a phase Ib dose-escalation trial. Cancer Res. 2013;73:LB–147. [Google Scholar]
- 34.Bedard P, Tabernero J, Kurzrock R, et al. 2012. ASCO Meeting Abstracts A phase lb, open-label, multicenter, dose-escalation study of the oral pan-PI3K inhibitor BKM120 in combination with the oral MEK1/2 inhibitor GSK1120212 in patients (pts) with selected advanced solid tumors; Abst 3003.
- 35.Speranza G, Kinders RJ, Khin S, et al. 2012. ASCO Meeting Abstracts Pharmacodynamic biomarker-driven trial of MK-2206, an AKT inhibitor, with AZD6244 (selumetinib), a MEK inhibitor, in patients with advanced colorectal carcinoma (CRC); Abst 3529.
- 36.Gong Y, Somwar R, Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bean GR, Ganesan YT, Dong Y, et al. PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci Signal. 2013;6:ra20. doi: 10.1126/scisignal.2003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–53. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 41.Corcoran RB, Cheng KA, Hata AN, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan N, Wong M, Nannini MA, et al. Bcl-2/Bcl-xL inhibition increases the efficacy of MEK inhibition alone and in combination with PI3 kinase inhibition in lung and pancreatic tumor models. Mol Cancer Ther. 2013;12:853–64. doi: 10.1158/1535-7163.MCT-12-0949. [DOI] [PubMed] [Google Scholar]
- 43.Sale MJ, Cook SJ. The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem J. 2013;450:285–94. doi: 10.1042/BJ20121212. [DOI] [PubMed] [Google Scholar]
- 44.Faber AC, Coffee EM, Costa C, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014;4:42–52. doi: 10.1158/2159-8290.CD-13-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holt SV, Logie A, Odedra R, et al. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106:858–66. doi: 10.1038/bjc.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janne PA, Shaw AT, Pereira JR, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z, Cheng K, Walton Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–7. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carretero J, Shimamura T, Rikova K, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–59. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim HS, Mendiratta S, Kim J, et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell. 2013;155:552–66. doi: 10.1016/j.cell.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar MS, Hancock DC, Molina-Arcas M, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642–55. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 51.Steckel M, Molina-Arcas M, Weigelt B, et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012;22:1227–45. doi: 10.1038/cr.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scholl C, Frohling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 53.Singh A, Sweeney MF, Yu M, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148:639–50. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puyol M, Martin A, Dubus P, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 56.Shaw AT, Winslow MM, Magendantz M, et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci USA. 2011;108:8773–8. doi: 10.1073/pnas.1105941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Babij C, Zhang Y, Kurzeja RJ, et al. STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res. 2011;71:5818–26. doi: 10.1158/0008-5472.CAN-11-0778. [DOI] [PubMed] [Google Scholar]
- 58.Kothari V, Wei I, Shankar S, et al. Outlier kinase expression by RNA sequencing as targets for precision therapy. Cancer Discov. 2013;3:280–93. doi: 10.1158/2159-8290.CD-12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sos ML, Michel K, Zander T, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–40. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Acquaviva J, Smith DL, Sang J, et al. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol Cancer Ther. 2012;11:2633–43. doi: 10.1158/1535-7163.MCT-12-0615. [DOI] [PubMed] [Google Scholar]
- 61.De Raedt T, Walton Z, Yecies JL, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–13. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ballester R, Furth ME, Rosen OM. Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem. 1987;262:2688–95. [PubMed] [Google Scholar]
- 63.Alvarez-Moya B, Lopez-Alcala C, Drosten M, Bachs O, Agell N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene. 2010;29:5911–22. doi: 10.1038/onc.2010.298. [DOI] [PubMed] [Google Scholar]
- 64.Barcelo C, Paco N, Morell M, et al. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014;74:1190–9. doi: 10.1158/0008-5472.CAN-13-1750. [DOI] [PubMed] [Google Scholar]