

Abstract
Leukemia stem cells (LSC) are resistant to conventional chemotherapy and persistent LSC after chemotherapy are supposed to be a major cause of relapse. However, information on genetic or epigenetic regulation of stem cell properties is still limited and LSC-targeted drugs have scarcely been identified. Epigenetic regulators are associated with many cellular processes including maintenance of stem cells. Of note are polycomb group proteins, because they potentially control stemness, and can be pharmacologically targeted by a selective inhibitor (DZNep). Therefore, we investigated the therapeutic potential of EZH2 inhibition in mixed lineage leukemia (MLL) fusion leukemia. Intriguingly, EZH2 inhibition by DZNep or shRNA not only suppressed MLL fusion leukemia proliferation but also reduced leukemia initiating cells (LIC) frequency. Expression analysis suggested that p16 upregulation was responsible for LICs reduction. Knockdown of p16 canceled the survival advantage of mice treated with DZNep. Chromatin immunoprecipitation assays demonstrated that EZH2 was highly enriched around the transcription-start-site of p16, together with H3K27 methylation marks in MLL/ENL and Hoxa9/Meis1 transduced cells but not in E2A/HLF transduced cells. Although high expression of Hoxa9 in MLL fusion leukemia is supposed to be responsible for the recruitment of EZH2, our data also suggest that there may be some other mechanisms independent of Hoxa9 activation to suppress p16 expression, because expression levels of Hoxa9 and p16 were not inversely related between MLL/ENL and Hoxa9/Meis1 transduced cells. In summary, our findings show that EZH2 is a potential therapeutic target of MLL fusion leukemia stem cells.
Keywords: 3-Deazaneplanocin, histones, homeobox proteins, mixed-lineage acute leukemias, polycomb repressive complex 2
It has been reported that epigenetic changes including histone modulation are broadly linked to leukemogenesis.1–3 Many drugs modulating histone acetylation have been available for patients with hematological malignancy and they have shown a certain level of anti-tumor effect.4–6 Although aberrant histone methylation or mutations of genes concerning histone methylation have been reported in patients with various types of leukemia,7 drugs targeting histone methylation have not been applied in clinical settings so far. As for aberrant histone methylation, mixed lineage leukemia (MLL) rearranged leukemia is associated with poor prognosis and has been closely investigated over the decades.8,9 For instance, it has been demonstrated that Disruptor of Telomere Silencing 1-like (Dot1L)-mediated di-methylation of Histone H3 lysine 79 (H3K79) is aberrantly methylated in MLL fusion leukemia.10,11 Bernt et al. have shown that Dot1L itself is indispensable for leukemia initiation and maintenance,11 and Dot1L appears to be an ideal target for MLL fusion leukemia. However, pharmacological inhibition of Dot1L resulted in the limited effect against MLL-AF9 leukemia mouse models.12 Furthermore, depletion of Dot1L is considered to suppress normal hematopoiesis.11,13 Thus identification of other therapeutic targets for MLL fusion leukemia is highly anticipated.
Enhancer of Zeste Homolog 2 (EZH2) is the main component of polycomb repressive complex 2 (PRC2) which catalyzes tri-methylation of histone H3 lysine 27 (H3K27) and recruits polycomb repressive complex 1 (PRC1) to the promoter region of target genes.14,15 A growing body of evidence demonstrates that EZH2 and other polycomb group proteins are frequently mutated in patients with hematological malignancy including MLL fusion leukemia.10,16–25 Moreover, inhibitor of EZH2, 3-deazaneplanocin A (DZNep), was shown to be effective to kill leukemia cells.26,27 However, the effect of EZH2 inhibitor in vivo is not fully investigated.
Here we show that EZH2 plays a crucial role in maintenance of MLL fusion leukemia and that inhibition of EZH2 can specifically target leukemia initiating cells (LIC) of MLL fusion leukemia.
Materials and Methods
Leukemia cell lines
Human leukemia cell lines K562, HEL, Kasumi-1, ME-1, Mv4-11 and MOLM13 were cultured in Roswell Park Memorial Institute 1640 (RPMI1640) medium (Wako 189-02025) with 20% fetal calf serum (FCS) and 1% penicillin/streptomycin (PS).
Plasmid construction
The plasmids pMSCV-neo-FLAG-MLL/ENL, pMSCV-IRES-GFP-MLL/AF9, pMXs-neo-E2A/HLF and pMYs-Hoxa9-IRES-Meis1 have been described previously.28 pMSCV-TEL/PDGFβR-IRES-AML1/ETO (TPAE) is a gift from Dr. Michael H. Tomasson (Washington University School of Medicine, St. Louis). Mouse p16 DNA was synthesized by PCR using primers (Forward, 5′-GCGAATTCACCATGGGTCGCAGGTTCTTGG-3′; Reverse, 5′-GCCTCGAGCAGCTACTTGTCGTCATCGTCTTTGTAGTCTTTTGCCCGTCGGTCTGG-3′) and cDNA extracted from mouse total bone marrow cells as a template. The product was inserted into pMYs-IRES-GFP at EcoR1 and Xho1 site.
Short hairpin RNA (shRNA)
Specific siRNA oligos targeting murine EZH2 and p16 mRNAs were designed as indicated by Takara Bio (Shiga, Japan) and cloned into pSIREN-RetroQ (harboring puromycin resistant gene) and pSIREN-ZsGreen vectors. Control shRNA is a nonfunctional construct provided from Takara Bio. The target sequences are as follows; EZH2: 5′-ggtggaagacgaaactgtt-3′, p16: 5′-caggaaaggaatggcatga-3′.
Retrovirus transduction
Retrovirus transduction was performed to produce immortalized cells, to transplant pre-leukemic cells to mice, and to transduce shRNA into cells. To produce retrovirus, Plat-E packaging cells29 were transiently transfected with retroviral constructs as described previously.30 To generate immortalized cells, at least three times of passages were performed in methocult M3434 semisolid medium (Stemcell technologies, Tokyo, Japan).
Transplantation assay
All transplantation assays were performed using secondary transplantation of leukemic cells. To obtain primary leukemic cells, MLL/ENL, MLL/AF9 or TPAE oncogene was transduced into c-Kit positive bone marrow (BM) cells which were isolated from 8 to 10 week-old C57BL/6 mice (Sankyo Laboratory Service, Tokyo, Japan) with anti-CD117 magnetic beads using the autoMACS apparatus (Miltenyi Biotec, Tokyo, Japan) according to the manufacturer's instructions. Recipient mice were sublethally irradiated (7.5 Gy) and injected with these pre-leukemic cells. After several months, primary leukemic cells were collected from BM and utilized for transplantation assays.
Flow cytometry
Cell sorting and flow cytometry analysis were performed on FACS AriaII (BD, Tokyo, Japan). Leukemic cells flushed from the tibia, femur, ilium and vertebra were isolated by density centrifugation over Histopaque-1083 (Sigma-Aldrich Japan, Tokyo, Japan) and prepared for GFP positive cell sorting or leukemic granulocyte macrophage progenitor (L-GMP) analysis. For L-GMP analysis, cells were stained with CD34-Alexa647, Fcγreceptor II/III-PE, c-Kit-PE-Cy7, Sca-1-PerCP-Cy5.5, and lineage-biotin (Lin; CD3e, CD4, CD8a, CD127, Gr-1, Ter119 and B220), followed by visualization with streptavidin-APC-Cy7. Stained cells were analyzed as described previously.31
Quantitative real-time polymerase chain reaction
Real-time PCR was performed using the LightCycler 480 (Roche Diagnostics, Tokyo, Japan) following the manufacturers' instructions. Results were normalized to GAPDH levels. PCR primers used for quantitative PCR were shown in Table S1.
Western blotting
For protein detection, cells were lysed with lysis buffer (10 mM Tris-HCl, 0.15 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% Aprotinnin, 1 mM Na3NO4, 50 mM β-glycerophosphate, 2.5 mM phenylmethylsulfonylsluoride, and complete protease inhibitor cocktail [Roche Diagnostics]). Lysates were boiled with sample buffer (0.1% Tris-HCl, 4% SDS, 20% Glycerol, 7.5% bromophenol blue) at 100 degrees Celsius for 5 min. For histone detection, 1 × 106 cells were lysed with 100 μl of lysis buffer (0.1 M Tris-HCl, 0.15 M NaCl, 0.15 M MgCl2, 0.65% NP-40, and complete protease inhibitor cocktail) and centrifuged. The deposit were treated with 100 μl of 0.2 M sulfuric acid and centrifuged for 10 min. The supernatant were soluble in 25 μl of 100% trichloroacetic acid and centrifuged for 10 min. The deposit were washed with acetone and boiled with sample buffer at 100 degrees Celsius for 5 min. The samples were subjected to sodium dodecylsulfate-polyacrylamide gel (10% for protein detection and 15% for histone detection) electrophoresis (SDS-PAGE) and analyzed by western blotting. Antibodies used for Western blotting are shown in Table S2. ECL detection (GE Healthcare Japan, Tokyo, Japan) was carried out according to the manufacturer's recommendations. Protein and histone levels were quantified with ImageJ Version 1.41o software (National Institutes of Health, Bethesda, MD, USA).
Chromatin immunoprecipitation (ChIP)
Cells were crosslinked, lysed, sonicated and immunoprecipitated as previously described.32 Antibodies used for ChIP are shown in Table S2. Bound DNA fragments were eluted and quantified by subsequent quantitative real-time polymerase chain reaction (qRT-PCR). The sequences and locations for mouse p16 gene of primers used for PCR are shown in Table S3.
Results
EZH2 inhibitor is therapeutically active for mixed lineage leukemia fusion leukemia mouse models
To clarify whether in vivo administration of EZH2 inhibitor has therapeutically effective against MLL fusion leukemic mice, we employed DZNep, an inhibitor for H3K27 methyltransferase which is feasible for in vivo administration.26 First, we administered DZNep intraperitoneally to MLL fusion leukemia mouse models and analyzed epigenetic and transcriptional changes in the BM cells. In vivo administration of DZNep prolonged survival of MLL/ENL and MLL/AF9 leukemic mice (Fig.1a). The BM cells of mice treated with DZNep were globally hypomethylated at H3K27 (Fig.1b). Next, we investigated the expression of specific genes concerning apoptosis and cellular senescence that are known to be targets of EZH2.26,27,33 As previously reported,34 DZNep did not suppress EZH2 transcript level (Fig. S1a) but decreased protein level (Fig. S1b). Most of the genes tested showed a tendency to upregulation by DZNep treatment in vitro (Fig. S1a), among which only p16 was significantly upregulated by in vivo administration of DZNep (Fig.1c,d). p16 is known to be a tumor suppressor which is typified by association with oncogene-induced senescence (OIS) and is epigenetically regulated by PRC2 and PRC1.35–37 Loss of epigenetic suppression of p16 is reported to induce depletion of stem cells.38 Thus, we investigated whether LIC ratio in MLL/AF9 leukemic mice is influenced by administration of DZNep. The LICs in MLL/AF9 leukemic mice are known to be enriched in L-GMP fraction.31 In FACS analysis, the ratios of L-GMP in mice treated with DZNep were significantly decreased compared with control mice, while administration of typical cytotoxic agent did not reduce them (Fig.1e, Fig. S2). Moreover, the BM cells from MLL/AF9 leukemic mice treated with DZNep had impaired capacity to reconstitute leukemia in vivo and attenuated colony forming capacity in vitro (Fig.1f,g, Fig. S3). These data suggest that LIC are decreased by DZNep administration in MLL/AF9 leukemic mice.
Figure 1.
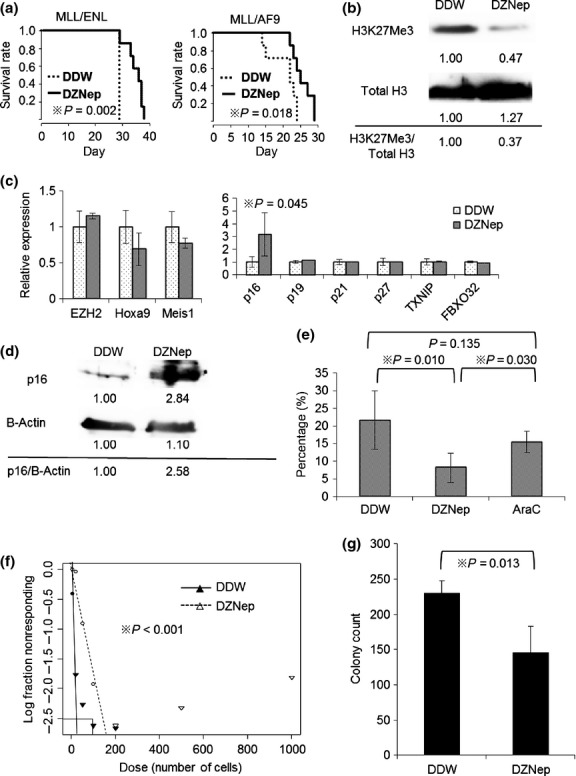
Administration of EZH2 inhibitor is therapeutically effective in MLL fusion leukemic mouse models. (a) Mice were injected intravenously with 1 × 104 of leukemia cells. From 7 days after injection, mice were treated intraperitoneally with DZNep, 2 mg/kg 3 days per week, until death. Survival of the mice in both groups (DDW as vehicle control and DZNep) is represented by Kaplan–Meier plot. P-values were calculated by log-rank test. N = 7 for each group. (b) Total BM cells were collected and lysed at 21 days after transplantation. Immunoblot analysis was performed for H3K27Me3. The levels of total H3 served as the loading control. Values indicate relative density scale. (c) GFP positive BM cells were sorted and analyzed by qRT-PCR at 21 days after transplantation. Relative expression of EZH2 and its target genes are shown. P-values were calculated by unpaired T test. N = 6 for DDW group and N = 5 for DZNep group. (d) Immunoblot analysis was performed for p16. The levels of β-actin served as the loading control. (e) The BM cells of MLL/AF9 leukemic mice treated with DDW, DZNep or AraC (100 mg/kg intraperitoneally from day 14–18) were collected and analyzed by flow cytometry at 21 days after transplantation. The percentages of L-GMP in GFP positive cells are shown (N = 6, 4 and 5). P-values were calculated by unpaired T test. (f) GFP positive BM cells were sorted and limiting dilution assay was performed. Poisson distribution of LIC frequency is shown. P-value was calculated by chi-square test. (g) 1 × 104 of sorted GFP positive BM cells were placed in 1 ml of methocult M3434 and cultured for 5 days. Colony counts for each group are shown. P-value was calculated by unpaired T test. N = 4 for each.
Knockdown of EZH2 reveals similar phenotype to DZNep administration in MLL fusion leukemic mice
To confirm whether the effect of DZNep against MLL related leukemia is due to EZH2 inhibition, we transduced shRNAs for EZH2 (shEZH2) into MLL/ENL leukemic cells (Fig.2a). The transduced cells were FACS sorted and transplanted to sublethally irradiated mice. Knockdown of EZH2 prolonged the survival of MLL/ENL leukemic mice (Fig.2b). The BM cells transduced with shEZH2 were globally hypomethylated (Fig.2c), and had diminished colony forming capacity with elevated p16 expression (Fig.2d,e). The ratios of L-GMP in mice transplanted with shEZH2 transduced cells were markedly reduced (Fig. S4). These data indicate that therapeutic efficacy of DZNep is due to EZH2 inhibition.
Figure 2.
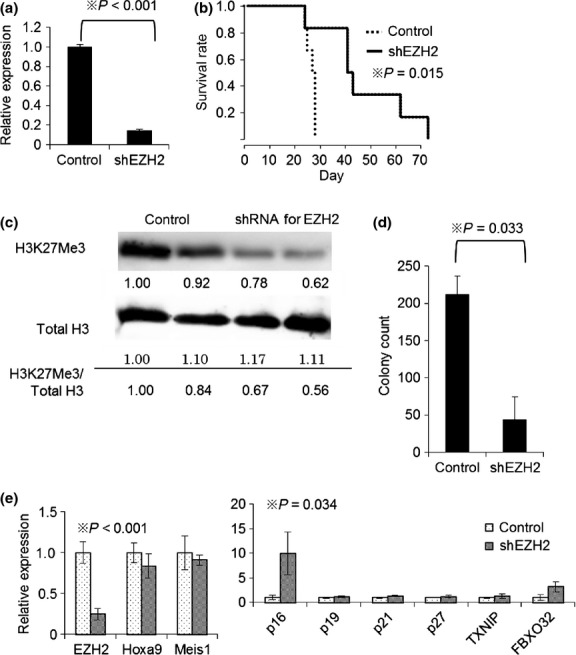
Knockdown of EZH2 reproduces the effect of EZH2 inhibitor for MLL fusion leukemic mice. (a) MLL/ENL leukemia cells were transduced with pSIREN-ZsGreen control vector or pSIREN-ZsGreen-shEZH2 (shEZH2) and cultured for 3 days. The total RNAs were isolated and analyzed for EZH2 by qRT-PCR. N = 2 for each. (b) Mice were injected intravenously with 1 × 104 ZsGreen (shRNA targeting EZH2 or control vector) positive leukemia cells. Survival of the mice in both groups is represented by Kaplan–Meier plot. P-values were calculated by log-rank test. N = 6 for each group. (c) Immunoblot analysis of H3K27Me3 and total H3. Values indicate relative density scale. (d) 1 × 104 of sorted ZsGreen positive BM cells were placed in 1 ml of methocult M3434 and cultured for 5 days. Colony counts for each group are shown. P-value was calculated by unpaired T test. N = 3 for each. (e) ZsGreen positive cells were sorted and analyzed by qRT-PCR. Relative expression of EZH2 and its target genes are shown. P-values were calculated by T test. N = 3 for each group.
Upregulation of p16 as a candidate anti-leukemic effect by EZH2 inhibition
To validate whether upregulation of p16 is responsible for anti-leukemic effect of EZH2 inhibition, we tested shRNA for p16 (shp16) (Fig.3a). Colony forming capacity of MLL/ENL leukemia cells attenuated by shEZH2 was markedly restored by p16 knockdown (Fig.3b). Furthermore, similar results were obtained using MLL/ENL leukemia mice treated with shp16 and DZNep (Fig.3c). In contrast, overexpression of p16 diminished colony number of MLL/ENL leukemia cells (Fig.3d) and showed decreased leukemia initiating capacity in transplantation assay (Fig.3e). These data support the idea that p16 is one of the main targets of EZH2 inhibition.
Figure 3.
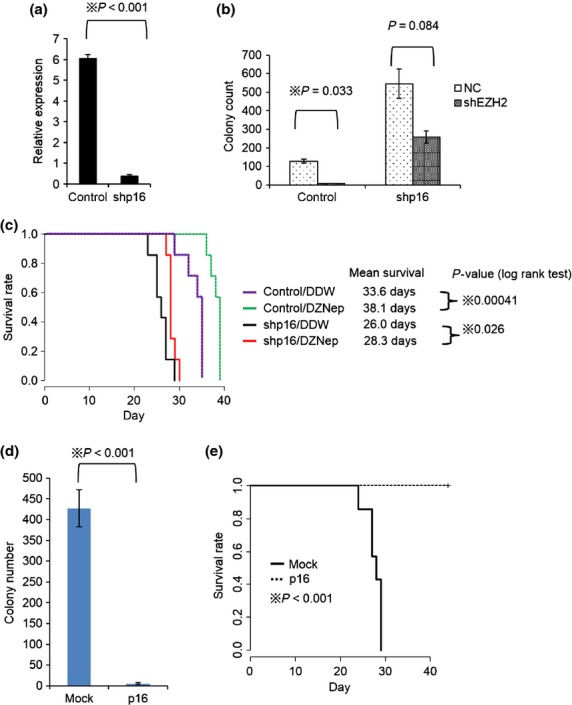
Suppression of p16 restores anti-leukemic effect of EZH2 inhibition. (a) MLL/ENL leukemia cells were transduced with pSIREN-RetroQ-shp16 (shp16) or control vector and cultured in 2.5 μg/ml of puromycin containing medium for 5 days. The total RNAs were isolated and analyzed for p16 by qRT-PCR. N = 2 for each. (b) shp16 transduced leukemia cells were transduced with pSIREN-ZsGreen control or shEZH2. 1 × 104 of sorted ZsGreen positive cells were placed in 1 ml of methocult M3434 (2.5 μg/ml of puromycin containing) and cultured for 5 days. Colony counts for each group are shown. P-value was calculated by unpaired T test. N = 2 for each. (c) Mice were injected intravenously with 1 × 104 of control or shp16 transduced MLL/ENL leukemic cells. Mice were treated with DZNep or DDW as previously shown in Figure1a. N = 7 for each group. (d) Retrovirus encoding p16-IRES-GFP was infected into MLL/ENL leukemia cells. GFP positive cells were sorted and placed in 1 ml of methocult M3434 and cultured for 5 days. Colony counts for each group are shown. P-value was calculated by unpaired T test. N = 5 for each. (e) Mice were intravenously injected with 1 × 104 of p16-IRES-GFP or mock transduced MLL/ENL leukemic cells. N = 7 for each group.
Anti-leukemic effect of EZH2 inhibition may be specific for MLL fusion leukemia
Next, we studied whether anti-leukemic effect for MLL fusion leukemia by EZH2 inhibition can be applied to various kinds of leukemia. We exposed several human leukemia cell lines to DZNep. K562, HEL, Kasumi-1 and ME-1 cells, all of which do not harbor MLL fusion genes were not sensitive to low concentration of DZNep while both of MLL leukemia cell lines Mv4-11 and MOLM13 were sensitive to 50 nM of DZNep (Fig.4a). Knockdown of EZH2 led to minimal effect on E2A/HLF transduced BM cells or c-Kit positive BM cells as we previously reported,32 while colony forming capacity of MLL/ENL and Hoxa9/Meis1 transduced cells were strongly suppressed (Fig.4b). Knockdown of p16 rescued colony forming capacity of MLL/ENL or Hoxa9/Meis1 transduced cells by EZH2 inhibition (Fig.4c). Expression levels of p16 were not influenced by knockdown of EZH2 in the BM cells transduced with leukemia fusion genes other than MLL/ENL (Fig. S5). In addition, survival of mice transplanted with leukemia cells induced by TPAE is not prolonged by DZNep administration (Fig.4d), although H3K27 were globally hypomethylated (Fig.4e). These data imply that anti-leukemic effect of EZH2 inhibition through p16 upregulation is specific for MLL fusion leukemia.
Figure 4.
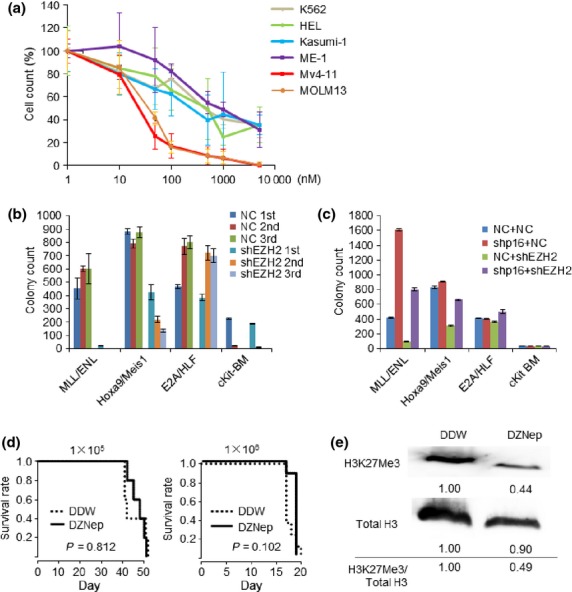
Anti-leukemic effect of EZH2 inhibition is specific for MLL fusion leukemia. (a) Cell proliferation analysis of several types of leukemia cell lines. Leukemia cell lines are cultured for 3 days in medium containing indicated concentration of DZNep. Cell counts are plotted as percentage treating counts of DZNep non-containing cultures as 100%. (b) Leukemic fusion gene transduced cells or c-Kit positive BM cells (cKit-BM) were transduced with pSIREN-RetroQ-shEZH2 or control vector. 1 × 104 of cells were placed in 1 ml of methocult M3434 (2.5 μg/ml of puromycin containing) and cultured for 5 days. Replating was performed for three times. Colony counts for each round are shown. N = 2 for each. (c) Leukemic fusion gene transduced cells or cKit-BM cells were transduced with pSIREN-RetroQ control or shp16, and cultured in 2.5 μg/ml of puromycin containing medium for 5 days. The cells were transduced with pSIREN-ZsGreen-shEZH2 or control vector. 1 × 104 of sorted ZsGreen positive cells were placed in 1 ml of methocult M3434 (2.5 μg/ml of puromycin containing) and cultured for 5 days. Colony counts for each group are shown. N = 2 for each. (d) Mice were injected intravenously with 1 × 105 or 1 × 106 of TPAE leukemic cells. From 7 days after injection, mice were treated intraperitoneally with DZNep, 2 mg/kg 3 days per week, until death. Survival of the mice in both groups is represented by Kaplan–Meier plot. P-values were calculated by log-rank test. N = 5 for 1 × 105 and N = 8 for 1 × 106 cohort. (e) Total BM cells of TPAE leukemic mice (injected with 1 × 106 cells) treated with DDW or DZNep were collected and lysed at 15 days after transplantation. Immunoblot analysis was performed for H3K27Me3.
EZH2 is enriched at the p16 transcription-start-site in MLL fusion leukemia
To clarify the molecular mechanism of p16 regulation by EZH2 in MLL fusion leukemia, we investigated p16 transcription levels and epigenetic status of p16 promoter in MLL/ENL, Hoxa9/Meis1 and E2A/HLF transduced cells. Among several types of immortalized cells and leukemia cells, the transcript level of p16 is remarkably low in MLL/ENL immortalized or leukemia cells (Fig.5a,b). This suggests existence of specific mechanism of p16 suppression in MLL fusion leukemia and we speculated that difference in epigenetic status between MLL fusion leukemia and other types of leukemia is a possible cause. The promoter of p16 is suggested to locate around the transcription-start-site (TSS), and a previous study using mouse embryonic fibroblasts revealed that H3K27 methylation status around this region is altered by OIS.37 As expected, our ChIP assay revealed that EZH2 is enriched at the p16 TSS together with Bmi1 and H3K27Me3 in MLL/ENL or Hoxa9/Meis1 transduced cells, while they are not enriched at the p16 TSS of E2A/HLF transduced cells (Fig.5c, Fig. S6). In contrast, H3K4Me3 was enriched at the TSS of p16, not only in MLL/ENL and Hoxa9/Meis1 cells, but also in E2A/HLF cells (Fig. S6), which is consistent with the report that H3K4Me3 in the p16 TSS is an initial event after transduction of oncogene leading to p16 upregulation.37,39 Our data suggest that enriched EZH2 at the p16 TSS maintains H3K27Me3 methylation status against p16 upregulation, leading to suppressed expression of p16 compared with other types of leukemia. Recently, Hoxa9 is reported to be important to recruit EZH2 to the TSS of p16.40 In fact, Hoxa9/Meis1 transduced cells which mimic downstream signaling of MLL fusion leukemia displayed similar behavior of EZH2 and H3K27Me3 on the p16 gene (Fig.5c). At this point, our data are compatible to the previous report showing a role of Hoxa9 to recruit EZH2 to p16 gene. However, despite the higher expression of Hoxa9 in Hoxa9/Meis1 transduced cells compared with MLL/ENL transduced cells, the expression of p16 in Hoxa9/Meis1 transduced cells is higher than that of MLL/ENL transduced cells (Fig.5d). These data suggest that there may be some specific mechanisms other than Hoxa9-mediated recruitment of EZH2 to suppress p16 in MLL/ENL transduced cells.
Figure 5.
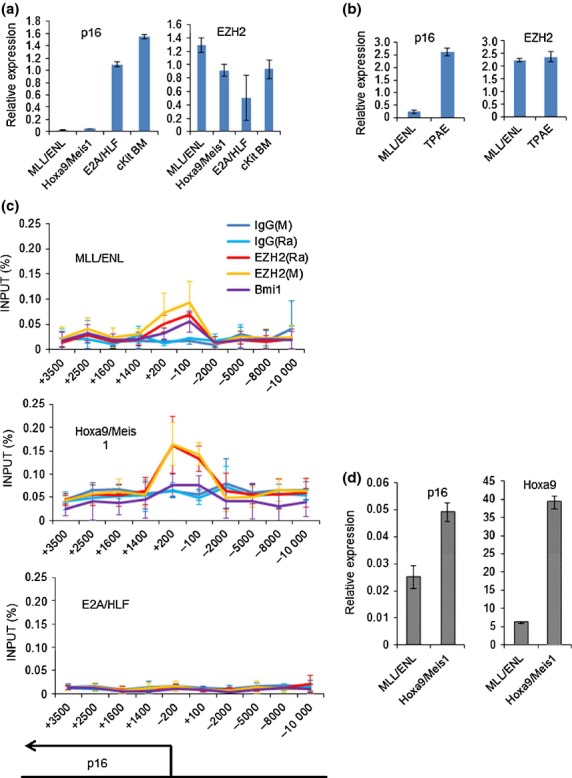
EZH2 is enriched at the TSS of p16 in MLL fusion leukemia. (a) Relative expression of EZH2 and p16 in immortalized cells harboring several types of leukemic fusion genes. N = 4 for all fusion genes. (b) Relative expression of EZH2 and p16 in BM cells of wild type mice (Wt BM) or mice harboring several kinds of leukemic fusion genes. N = 4 for Wt BM, N = 3 for MLL/ENL and N = 2 for TPAE. (c) 5 × 105 of MLL/ENL, Hoxa9/Meis1 or E2A/HLF transduced cells were harvested from methocult M3434 for ChIP assays using indicated antibodies. N = 3 for each. (d) Relative expression of p16 and Hoxa9 in MLL/ENL and Hoxa9/Meis1 immortalized cells. N = 4 for each.
Discussion
We investigated the impact of EZH2 inhibition in leukemia models. Our data suggest that inhibition of EZH2 in MLL fusion leukemia is therapeutically effective by diminishing LIC through upregulation of p16. Recently, genetic deletion of EZH2 is shown to decrease susceptibility to MLL/AF9-induced leukemic transformation.33,41 Although these reports suggested the efficacy of EZH2 inhibitors for the treatment of MLL fusion leukemia, in vivo administration of EZH2 inhibitors for MLL rearranged leukemia mouse models had never been verified. We confirmed that treatment with DZNep or knockdown of EZH2 prolonged the survival of MLL fusion leukemic mice in vivo. Previous reports33,41 revealed that conditional knock out of EZH2 after transplantation of MLL/AF9 pre-leukemic cells does not prolong survival in the primary transplantation. However, they observed prolonged survival of MLL fusion leukemic mice in the secondary transplantation of EZH2 depleted leukemia cells. In our study, we transplanted the primary MLL fusion leukemia cells after knockdown of EZH2 and found that EZH2 suppression before transplantation reduces LIC and extends survival period. Similarly, we observed that administration of DZNep from 7 days after transplantation also prolonged survival. We presume that the engraftment of MLL fusion leukemia cells is at day 14–18 because white blood cell counts of mice are almost zero within 2 weeks after transplantation. Therefore, transplanted cells are supposed to be exposed by DZNep before engraftment. Tanaka et al. discussed41 that EZH2 is required for the maintenance of LICs against the stress imposed by transplantation but is dispensable for initiating leukemia. We suppose that their idea also explain our findings, and that depleting EZH2 before engraftment of MLL fusion leukemia cells is important for eradication of LICs. On the other hand, Bmi1, a component of PRC1, was not shown to be a therapeutic target whereas it is required for initiating MLL/AF9 leukemia,42 which highlights the importance of EZH2 as a therapeutic target among polycomb repressive complexes. At the same time, EZH2 inhibition spares normal hematopoiesis, which suggests that EZH2 is one of the rational therapeutic targets in MLL rearranged leukemia and endorses the future development of EZH2 inhibitors in a clinical setting.
The expression profile of genes targeted by EZH2 is different between in vivo and in vitro models of EZH2 inhibition. Only p16 was significantly upregulated by in vivo administration of DZNep (Fig.1c), while many genes were upregulated by in vitro exposure of DZNep (Fig. S1a). We suppose that this difference is due to drug concentration and exposure pattern. The BM cells of mice are virtually exposed to DZNep intermittently, because its concentration in mice plasma is lowered to less than 10% within 30 minutes after administration.43 This fact supports our idea that the effects of DZNep in vitro and in vivo are not the same, and that p16 is a bona fide target of EZH2 in MLL-related leukemia.
Our data also imply that EZH2 inhibition exerts the unique anti-leukemic effect against MLL fusion leukemia. The expression of p16 is remarkably low in MLL/ENL transduced cells. Surprisingly, although basal expression of p16 is suppressed, additional knockdown of p16 increased colony formation in MLL/ENL immortalized cells (Fig.3b and 4c). Knockdown of p16 also shortened the survival of MLL/ENL leukemic mice (Fig.3c). Although these results indicate that knockdown of p16 itself exacerbates leukemia, we suppose that down-regulation of p16 also prevent the anti-leukemic effect of EZH2 inhibitor, because the difference of mean survival time between Control/DZNep and shp16/DZNep (9.8 days) is longer than that between Control/DDW and shp16/DDW (7.6 days). Furthermore, overexpression of p16 diminished leukemia initiating capacity of MLL/ENL leukemia cells (Fig.3d,e). These data support the idea that suppression of p16 is one of the important processes to maintain MLL fusion leukemia, and it is more sensitive to restoration of p16 expression compared with other leukemias. Clinically, chromosomal deletion around p16 is associated with lower survival in MLL rearranged leukemia.44 The upregulation of p16, which is negatively controlled by EZH2 and Bmi1, is known to be the initial event after transduction of oncogene.35,36,45 We suppose that removal of EZH2 at the p16 promoter restores its expression and induces OIS. Our data suggest that MLL fusion leukemia depends on EZH2 to prevent p16 upregulation while other leukemias do not (Fig.4, Fig. S5), and it would explain, at least partially, the specificity of EZH2 inhibition for MLL rearranged leukemia. Moreover, a previous report showed that EZH2 is essential for development of fetal hematopoiesis but dispensable for maintenance of adult hematopoiesis.46 They also revealed that EZH2 is recruited to the TSS of p16 in fetal hematopoiesis, but not in normal adult hematopoiesis. This point is one of the crucial rationales for the clinical use of EZH2 inhibitors because their toxicity potentially spares normal hematopoietic stem cells. It is also important to unveil the factor that recruits EZH2 to the p16 TSS, as it may lead to clarification of other novel targets for the treatment of MLL fusion leukemia.
The recruitment of EZH2 to the p16 TSS is supposed to depend on high expression of Hoxa9 which is upregulated by MLL fusion protein, because the transduction of Hoxa9/Meis1 also recruited EZH2 to the TSS of p16 (Fig.5c). These data are well consistent with a recent report.40 Our data also suggest some other mechanisms independent of Hoxa9 activation to suppress p16 in MLL/ENL transduced cells because the expression levels of Hoxa9 and p16 are not inversely related (Fig.5d).
Collectively, our data indicate that EZH2 plays a crucial role to maintain MLL fusion leukemia through inhibition of p16. This mechanism is specific for MLL fusion leukemia because high expression of Hoxa9 and other possible factors act jointly to recruit EZH2 to the p16 TSS. Removal of EZH2 from the TSS of p16 activates its transcription, and it supposed to be the reason why LIC of MLL fusion leukemia is diminished by EZH2 inhibition. Our findings encourage the development of EZH2 inhibitors that are tolerated in human.
Acknowledgments
The authors thank F. Komine, T. Nosaka, J. Hess, M. Tomasson, T. Nakamura and T. Inaba for providing essential plasmids; P. Liu for ME-1 cell line; Kyowa Hakko Kirin Co., Ltd. for cytokines; Y. Izawa and Y. Hokama for expert technical assistance. This study was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology (KAKENHI 24890045 and KAKENHI 24249055), and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Disclosure
Satoshi Nishikawa is a member of Kyowa Hakko Kirin Co., Ltd., and Mineo Kurokawa received lecture fee and research funding from Kyowa Hakko Kirin Co.
Funding information
Ministry of Education, Culture, Sports, Science and Technology (KAKENHI 24890045 and 24249055). NIH, National Cancer Institute, Center for Cancer Research.
Supporting Information
Additional supporting information may be found in the online version of this article:
In vitro effect of DZNep.
Fig. S2 Representative presentation of FACS analysis of MLL/AF9 leukemic mice treated with DDW, DZNep, or AraC.
Fig. S3 Result of limiting dilution transplantation assay of MLL/AF9 secondary leukemia cells.
Fig. S4 Representative presentation of FACS analysis of MLL/AF9 leukemic mice treated with shRNA.
Fig. S5 Relative expression of EZH2 and p16 in immortalized cells.
Fig. S6 Results of ChIP analysis.
Table S1 Primer sequence for expression analysis.
Table S2 Antibodies for Immunoprecipitation, Immunoblotting and ChIP.
Table S3 Primer sequence for ChIP analysis.
References
- 1.Bonifer C, Bowen DT. Epigenetic mechanisms regulating normal and malignant haematopoiesis: new therapeutic targets for clinical medicine. Expert Rev Mol Med. 2010;12:e6. doi: 10.1017/S1462399410001377. [DOI] [PubMed] [Google Scholar]
- 2.Yoshimi A, Kurokawa M. Key roles of histone methyltransferase and demethylase in leukemogenesis. J Cell Biochem. 2011;112:415–24. doi: 10.1002/jcb.22972. [DOI] [PubMed] [Google Scholar]
- 3.Uribesalgo I, Di Croce L. Dynamics of epigenetic modifications in leukemia. Brief Funct Genomics. 2011;10:18–29. doi: 10.1093/bfgp/elr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prebet T, Vey N. Vorinostat in acute myeloid leukemia and myelodysplastic syndromes. Expert Opin Investig Drugs. 2011;20:287–95. doi: 10.1517/13543784.2011.542750. [DOI] [PubMed] [Google Scholar]
- 5.Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011;25:226–35. doi: 10.1038/leu.2010.276. [DOI] [PubMed] [Google Scholar]
- 6.Masetti R, Serravalle S, Biagi C, Pession A. The role of HDACs inhibitors in childhood and adolescence acute leukemias. J Biomed Biotechnol. 2011;2011:148046. doi: 10.1155/2011/148046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012;2:405–13. doi: 10.1158/2159-8290.CD-12-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 9.Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94:984–93. doi: 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–78. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 11.Bernt KM, Zhu N, Sinha AU, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117:6912–22. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–96. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 15.Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 16.Chowdhury M, Mihara K, Yasunaga S, Ohtaki M, Takihara Y, Kimura A. Expression of Polycomb-group (PcG) protein BMI-1 predicts prognosis in patients with acute myeloid leukemia. Leukemia. 2007;21:1116–22. doi: 10.1038/sj.leu.2404623. [DOI] [PubMed] [Google Scholar]
- 17.Mohty M, Yong AS, Szydlo RM, Apperley JF, Melo JV. The polycomb group BMI1 gene is a molecular marker for predicting prognosis of chronic myeloid leukemia. Blood. 2007;110:380–3. doi: 10.1182/blood-2006-12-065599. [DOI] [PubMed] [Google Scholar]
- 18.Mihara K, Chowdhury M, Nakaju N, et al. Bmi-1 is useful as a novel molecular marker for predicting progression of myelodysplastic syndrome and patient prognosis. Blood. 2006;107:305–8. doi: 10.1182/blood-2005-06-2393. [DOI] [PubMed] [Google Scholar]
- 19.Pizzatti L, Binato R, Cofre J, et al. SUZ12 is a candidate target of the non-canonical WNT pathway in the progression of chronic myeloid leukemia. Genes Chromosom Cancer. 2010;49:107–18. doi: 10.1002/gcc.20722. [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharyya J, Mihara K, Yasunaga S, et al. BMI-1 expression is enhanced through transcriptional and posttranscriptional regulation during the progression of chronic myeloid leukemia. Ann Hematol. 2009;88:333–40. doi: 10.1007/s00277-008-0603-8. [DOI] [PubMed] [Google Scholar]
- 21.Grubach L, Juhl-Christensen C, Rethmeier A, et al. Gene expression profiling of Polycomb, Hox and Meis genes in patients with acute myeloid leukaemia. Eur J Haematol. 2008;81:112–22. doi: 10.1111/j.1600-0609.2008.01083.x. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Cuellar MP, Zilles O, Schreiner SA, Birke M, Winkler TH, Slany RK. The ENL moiety of the childhood leukemia-associated MLL-ENL oncoprotein recruits human Polycomb 3. Oncogene. 2001;20:411–9. doi: 10.1038/sj.onc.1204108. [DOI] [PubMed] [Google Scholar]
- 23.Hemenway CS, de Erkenez AC, Gould GC. The polycomb protein MPc3 interacts with AF9, an MLL fusion partner in t(9;11)(p22;q23) acute leukemias. Oncogene. 2001;20:3798–805. doi: 10.1038/sj.onc.1204478. [DOI] [PubMed] [Google Scholar]
- 24.Mueller D, Bach C, Zeisig D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–54. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci USA. 2003;100:8342–7. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiskus W, Wang Y, Sreekumar A, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–43. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, Bi C, Cheong LL, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118:2830–9. doi: 10.1182/blood-2010-07-294827. [DOI] [PubMed] [Google Scholar]
- 28.Arai S, Yoshimi A, Shimabe M, et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117:6304–14. doi: 10.1182/blood-2009-07-234310. [DOI] [PubMed] [Google Scholar]
- 29.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–6. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 30.Nakagawa M, Shimabe M, Watanabe-Okochi N, et al. AML1/RUNX1 functions as a cytoplasmic attenuator of NF-kappaB signaling in the repression of myeloid tumors. Blood. 2011;118:6626–37. doi: 10.1182/blood-2010-12-326710. [DOI] [PubMed] [Google Scholar]
- 31.Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 32.Yoshimi A, Goyama S, Watanabe-Okochi N, et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood. 2011;117:3617–28. doi: 10.1182/blood-2009-12-261602. [DOI] [PubMed] [Google Scholar]
- 33.Neff T, Sinha AU, Kluk MJ, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci USA. 2012;109:5028–33. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–30. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaneda A, Fujita T, Anai M, et al. Activation of Bmp2-Smad1 signal and its regulation by coordinated alteration of H3K27 trimethylation in Ras-induced senescence. PLoS Genet. 2011;7:e1002359. doi: 10.1371/journal.pgen.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med. 2006;203:2247–53. doi: 10.1084/jem.20052477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kotake Y, Zeng Y, Xiong Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res. 2009;69:1809–14. doi: 10.1158/0008-5472.CAN-08-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin N, Popov N, Aguilo F, et al. Interplay between Homeobox proteins and Polycomb repressive complexes in p16INK(4)a regulation. EMBO J. 2013;32:982–95. doi: 10.1038/emboj.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107–17. doi: 10.1182/blood-2011-11-394932. [DOI] [PubMed] [Google Scholar]
- 42.Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia. 2011;25:1335–43. doi: 10.1038/leu.2011.85. [DOI] [PubMed] [Google Scholar]
- 43.Coulombe RA, Jr, Sharma RP, Huggins JW. Pharmacokinetics of the antiviral agent 3-deazaneplanocin A. Eur J Drug Metab Pharmacokinet. 1995;20:197–202. doi: 10.1007/BF03189670. [DOI] [PubMed] [Google Scholar]
- 44.Ohnishi H, Guo SX, Ida K, et al. Alterations of p16 and p15 genes in acute leukemia with MLL gene rearrangements and their correlation with clinical features. Leukemia. 1997;11:2120–4. doi: 10.1038/sj.leu.2400872. [DOI] [PubMed] [Google Scholar]
- 45.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 46.Mochizuki-Kashio M, Mishima Y, Miyagi S, et al. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood. 2011;118:6553–61. doi: 10.1182/blood-2011-03-340554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vitro effect of DZNep.
Fig. S2 Representative presentation of FACS analysis of MLL/AF9 leukemic mice treated with DDW, DZNep, or AraC.
Fig. S3 Result of limiting dilution transplantation assay of MLL/AF9 secondary leukemia cells.
Fig. S4 Representative presentation of FACS analysis of MLL/AF9 leukemic mice treated with shRNA.
Fig. S5 Relative expression of EZH2 and p16 in immortalized cells.
Fig. S6 Results of ChIP analysis.
Table S1 Primer sequence for expression analysis.
Table S2 Antibodies for Immunoprecipitation, Immunoblotting and ChIP.
Table S3 Primer sequence for ChIP analysis.