

Abstract
p62/SQSTM1 (p62) is a multifunctional protein implicated in several signal transduction pathways and selectively degraded by autophagy, a process for lysosomal degradation of both protein and organelle. p62 was also recently reported to be overexpressed in various malignancies and its inhibition to suppress carcinoma cell proliferation. However, its correlation with autophagy in carcinoma cells has remained largely unknown. Therefore, in this study, we examined the effects of p62 inhibition on the regulation of autophagy and cell survival in p62-positive carcinoma cells. p62-silencing dramatically suppressed cell proliferation and induced autophagy in p62 expressing PC9 and A549 cells. Electron microscopical analysis revealed the formation of autophagosomes with multilayer membranes caused by p62-silencing. p62 silencing-mediated reduced cell viability was restored by both genomic and pharmacological inhibition of autophagy but not that of apoptosis. These findings were also detected in several types of carcinoma cell lines including adenocarcinomas and squamous cell carcinomas. Results of our present study revealed that an inhibition of p62 resulted in the formation of mis-regulated autophagosomes with multilayer membranes and an autophagic cell death, and p62 can therefore be an attractive target for the development of anti-neoplastic agents.
Keywords: Autophagy, cell death, electron microscopy, p62, SQSTM1
Autophagy is a catabolic pathway that maintains cellular homeostasis and cell growth.1,2 When autophagy is activated in the cells, double-membrane autophagic vesicles (autophagosomes) were formed and engulf cytoplasm and/or cytoplasmic organelles and proteins. Autophagosomes then fuse to lysosomal vesicles to degrade the contents of the autophagic vesicle and provide essential building blocks such as amino acids back to the cell.3 Therefore, autophagy contributes to cell survival via reproduction of nutrients and restoration of metabolic stress but its excess also results in the type 2 cell death (autophagic cell death).4 However, it is not clearly identified how the equilibrium between its pro-survival and pro-death activity of autophagy is regulated in the cells.
p62/SQSTM1 (p62) is one of the polyubiquitin-binding proteins and selective autophagic substrate, and can act as a cargo receptor for degradation of damaged or long-lived proteins.5–7 In particular, p62 was also recently reported to be overexpressed in several human malignancies,8 partly because an inhibition of autophagy resulted in tumorigenesis and p62 accumulation.9–11 In addition, p62-overexpression was also reported to be significantly correlated with aggressive biological behavior of both breast and prostate carcinoma patients.12,13 Several reports revealed that accumulated p62 activates several intracellular signal transduction pathways, which promotes cell proliferation and carcinoma progression.14–16 Results of these studies all indicated that genomic p62 inhibition could suppress carcinoma cell proliferation via an inactivation of p62-associated intracellular signal pathways, and p62 can be a novel therapeutic target for carcinoma treatment.14–16
However, autophagy was also recently reported to be activated in carcinoma cells, conversely to protect carcinoma cells from hypoxia, metabolic stress and anti-neoplastic agents.17,18 Thus, overexpressed p62 could contribute to the development of autophagy but the significance of p62 as an autophagy regulator in carcinoma cells has remained largely unknown. Therefore, in this study, we hypothesized that autophagy could be strictly regulated by p62 in carcinoma cells, and an inhibition of p62 could result mis-regulated autophagy and autophagic carcinoma cell death. Toward resolving this particular hypothesis above, we first examined the possible association of p62 expression with autophagic activity. We then evaluated the effects of autophagy upon carcinoma progression when p62 was silenced by p62-specific small interference RNAs (siRNAs). Results of our present study clearly shed light on the novel roles of p62 inhibition, which activates the formation of mis-regulated autophagosomes with multilayer membranes and induces autophagic cell death.
Materials and Methods
Reagents and antibodies
The following materials were obtained as indicated: 20 mM ammonium chloride, 100 μM leupeptin, chloroquine and 3-methyladenine (Sigma-Aldrich, St Louis, MO, USA). Antibodies were obtained from the following source: p62 (MBL, Nagoya, Japan); β-actin (Sigma-Aldrich); LC3B, p-Akt, Akt, p-mTOR, mTOR, Atg5, Atg7, PARP and Caspase-3 (Cell Signaling Technology, Beverly, MA, USA).
Cell culture
We used NCI-H23 (H23), NCI-H1975 (H1975), A549, LCSC#1, LK87, PC9, PC3, PANC-1, ECGI-10, HSC-4 and HSC-1 cells. The original tissue and source were summarized in Suppl. Table S1. The whole cells were grown in RPMI-1640 (Sigma-Aldrich) containing 10% FBS (Nichirei, Tokyo, Japan). Cell viability evaluated by WST-8 colorimetric assay (Cell Counting Kit-8; Dojindo Laboratories, Kumamoto, Japan) was used as an indicator of cell viability in these cell lines. In order to monitor the cell death rate, cells were suspended with regular medium and mixed with trypan blue for a minute. Cell count was conducted using TC20 Automated Cell Counter (BioRad Laboratories, Richmond, CA, USA). For starvation conditions, cells were placed in HBSS (Invitrogen, Carlsbad, CA, USA).
RNA interference
Silencing of p62, Atg5 and Atg7 was performed using specific siRNAs purchased from Sigma-Aldrich (sip62-1, sip62-2, siAtg5 and siAtg7). p62-targeting siRNAs purchased from Ambion was also used in this study (sip62-3 and sip62-4). Silencer Select Negative Control #1 and #2 siRNA (Ambion, Austin, TX, USA) was served as negative control (Control siRNA/Control siRNA-1 and Control siRNA-2, respectively) because MISSION siRNA Universal Negative Control (Sigma-Aldrich) demonstrated marked cytotoxicity in PC9 cells (Suppl. Fig. S1). siRNA was transfected in cells using Lipofectamine RNAi MAX reagent (Invitrogen). Silencing efficiency was assessed by immunoblotting.
Immunoblotting
Cells were dissolved in NP-40 buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% NP-40) with Halt Protease Inhibitor Cocktail and Phosphatase Inhibitor (Pierce Biotechnology, Rockford, IL, USA). Proteins were resolved by SDS-PAGE and transferred onto PVDF membrane. After blocking with TBS containing 0.05% Tween 20 and 5% non-fat dry skim milk, membranes were incubated overnight at 4°C with primary antibody. Membranes were then washed and incubated for 1 h with horseradish peroxidase-conjugated goat anti-mouse/rabbit IgGs (GE Healthcare, Buckinghamshire, UK) at room temperature. Immunoreactivity was subsequently visualized by chemiluminescence (ECL Prime western blotting detection reagents: GE Healthcare).
Immunofluorescence
The cells were fixed in 10% trichloroacetic acid for 15 min, permeabilized with 0.2% Triton X-100 for 10 min. After blocking with 1% BSA, the cells were stained with primary antibody for 1 h at room temperature. Immunoreactivity was visualized using Alexa Fluor488/594 goat anti-rabbit/mouse IgGs (Invitrogen). Images were obtained using BZ-9000 fluorescence microscope (Keyence, Osaka, Japan) with ×60 objective.
Transmission electron microscopy
The cells were fixed in 2.5% glutaraldehyde and paraformaldehyde in 100 mM sodium cacodylate, and post-fixed in 1% osmium tetroxide in sodium cacodylate. After ethanol dehydration and embedding in Epon 812 (TAAB Laboratories Equipment, Berkshire, UK), ultrathin sections were stained with uranyl acetate and lead citrate. Images were obtained using JEM-1400 (JEOL, Tokyo, Japan) or HT7700 (Hitachi, Tokyo, Japan) transmission electron microscope at an accelerating voltage of 80 kV. Autophagic vacuoles were identified based upon previously established criteria.19,20
cDNA constructs and transfection
The expression vector encoding human p62 was constructed by inserting the full-length cDNA fragment prepared by RT-PCR into pcDNA 3.1 vector (Invitrogen). The insertion was subsequently confirmed by sequencing using ABI 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Constructs were transfected into cells using Lipofectamine 2000 reagent (Invitrogen).
Statistical analysis
Results were expressed as mean ± SD. Statistical analysis was performed using the statview 5.0 J software (SAS Institute, Cary, NC, USA). The statistical difference of the two groups was determined by Bonferroni/Dunnet, and statistical significance was defined as P < 0.05 in this study.
Results
p62-silencing inhibited carcinoma cell proliferation
We first examined p62 expression in seven lung carcinoma cells by immunoblotting, because p62-overexpression was reported in lung carcinoma cells compared with normal lung epithelial cells.15,21 p62 expression was detected in all carcinoma cells examined in this study but PC9 and A549 cells exhibited higher p62 expression than other cells (Fig.1a). Figure1(b) demonstrated immunoblotting of p62, and two siRNAs targeting p62 (sip62-1 and sip62-2) successfully decreased p62 expression. Cell viability was significantly decreased by two siRNAs transfection in PC9 and A549 cells at either 3 or 6 days after transfection (Fig.1c,d). These results all indicated that p62 was expressed in some carcinoma cells and played pivotal roles on carcinoma cell survival.
Figure 1.

Effects of p62-silencing on cell proliferation. (a) Protein homogenates were isolated and immunoblotting was used to determine the levels of p62 and β-actin expression. (b–d) PC9 and A549 cells were transfected with indicated siRNAs for 3 days. After transfection, protein homogenates were isolated and immunoblotting was used to measure the levels of p62 and β-actin expression (b), and cell viability was measured after indicated times (c), and light microscopical images were obtained (d). Scale bars, 500 μm.
p62-silencing induced autophagy activation
The conversion of LC3B-I into LC3B-II was determined by immunoblotting analysis in order to evaluate the changes of autophagic activities in the cells being treated with two different sip62s. In PC9 and A549 cells, both sip62s inhibited an activation of mTOR (mammalian target of rapamycin; prominent autophagy suppressor) but an activation of Akt, located at the upstream of mTOR signaling pathway, was not changed. In addition, both of these sip62s notably induced the switch of LC3B-I to LC3B-II, indicating that autophagy was activated by both sip62s (Fig.2a). Significant increment of LC3B-II in p62-silenced cells was also detected when treated with lysosomal or autophagic inhibitors (ammonium chloride and Leupeptin or chloroquine, respectively) which indicated that LC3B-II was not simply accumulated but rather induced or synthesized at least partly (Fig.2b). We further confirmed this change using double immunofluorescence evaluations. p62 expression was not detected in p62-silenced cells, and LC3B dots were more frequently detected in p62-silenced cells (Fig.2c). These results all demonstrated that p62 inhibition certainly activated autophagy in PC9 and A549 cells.
Figure 2.

Effects of p62-silencing on autophagic activity. (a) PC9 and A549 cells were transfected with indicated siRNAs. After 3 days, protein homogenates were isolated and immunoblotting was used to measure p62, p-Akt, Akt, p-mTOR, mTOR, LC3B and β-actin. (b) PC9 and A549 cells were treated with ammonium chloride and leupeptin (N/L) or chloroquine (CQ.) and transfected with indicated siRNAs for 3 days. Protein homogenates were isolated and immunoblotting was used to measure p62, LC3B and β-actin. (c) Double immunofluorescent analysis of p62 and LC3B after indicated siRNAs transfection in PC9 cells. At least 100 cells were counted, and frequency of cells with LC3B dots was indicated. Scale bars, 10 μm.
p62-silencing induced conformation of multilayer vesicles
We further elucidated the mechanisms of p62 silencing-induced autophagy by an ultrastructural examination. Figure3(a) showed representative electron microscopical images of siRNA-transfected PC9 cells. Autophagic vesicles were not detected in PC9 cells transfected with Control siRNA but when transfected with two sip62s, morphologically identified autophagic vesicles were clearly detected in cytoplasm (Fig.3a). The great majority of autophagic vesicles detected in p62-silenced PC9 cells formed multilayer structures but its average diameter was 0.5–1 μm, the same as representative autophagic vesicles. We also found the small number of the cells with large multilayer vesicles (5–15 μm) containing other multilayer vesicles (Fig.3b). Autophagy inhibition as a result of the transfection of siRNA targeting autophagy-related gene Atg5 reduced the number of multilayer autophagosomes (Fig.3c). These ultrastructural studies indicated that an inhibition of p62 induced the formation of autophagosomes with multilayer membranes.
Figure 3.

Effects of p62-silencing on morphological formation of autophagosomes. (a,b) PC9 cells were transfected with Control siRNA (upper left), sip62-1 (upper right and (b)) or sip62-2 (Lower right). Representative electron microscopical images of low and high magnifications were shown. N, nucleus. Triangles indicated autophagic vesicles. Scale bars, 5 μm (low magnification) or 0.5 μm (high magnification). (c) PC9 cells were transfected with indicated siRNAs for 3 days. After transfection, protein homogenates were isolated and immunoblotting was used to determine the levels of p62, Atg5 and β-actin. Representative electron microscopical images were also shown. N, nucleus. Triangles indicated multilayer vesicles. Number of multilayer vesicles per each cell (at least 30 cells) was counted. Scale bars, 2 μm. *P < 0.05.
Autophagy inhibition restored p62 silencing-mediated suppression of cell viability
To determine the impacts of p62 silencing-induced formation of autophagosomes with multilayer membranes upon carcinoma cell proliferation, we evaluated the cell viability and protein expression of the cells transfected with siRNA targeting p62 and/or autophagy-related genes. Silencing of the autophagy-related gene Atg5 or Atg7 (siAtg5 or aiAtg7, respectively) successfully inhibited the development of autophagy through the suppression of LC3B-II expression (Fig.4a, lower panel). This suppressed cell viability of p62-silenced cells was markedly restored by siAtg5 or siAtg7 transfection in both PC9 and A549 cells (Fig.4a, upper panel). We also confirmed these results using another sip62s purchased from Ambion. Two siRNAs targeting p62 were transfected in PC9 cells, and p62 expression was successfully suppressed by sip62-3. Cell viability was decreased by the transfection of sip62-3 and restored by siAtg5 or siAtg7 transfection (Fig. S2). In addition, we examined whether pharmacological inhibition of autophagy could also prevent p62 inhibition-induced decreased cell viability. The suppressed cell viability of p62-silenced cells was restored by autophagy inhibitor 3-methyladenine (3MA, PI3K III inhibitor), but not chloroquine (autophagy-lysosomal inhibitor) (Fig.4b). These findings above all indicated that p62 inhibition-induced autophagy was mis-regulated and caused autophagic cell death.
Figure 4.
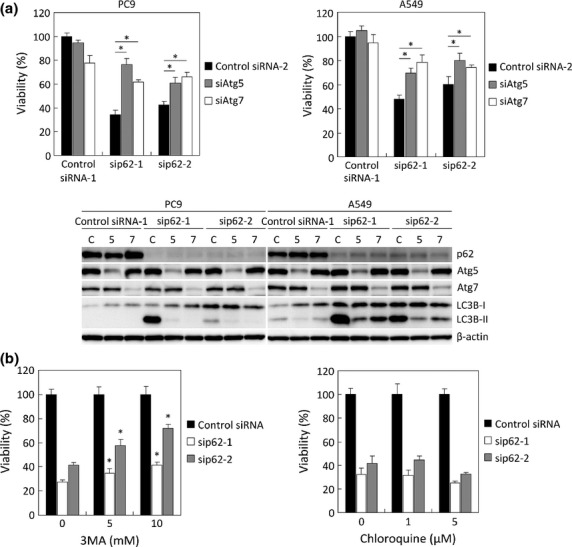
Effects of autophagy induced by p62-silencing on cell viability. (a) PC9 cells (left panel) and A549 cells (right panel) were transfected with indicated siRNAs for 3 days. After transfection, cell viability was measured, and protein homogenates were isolated and immunoblotting was used to determine the levels of p62, Atg5, Atg7, LC3B and β-actin expression. C, Control siRNA-2; 5, siAtg5; 7, siAtg7. *P < 0.05 (n = 6). (b) PC9 cells were pre-treated with 3-methyladenine (3MA, left panel) or chloroquine (right panel) for 30 min before transfection with indicated siRNAs. On 3 days after transfection, cell viability was measured. *P < 0.05 (n = 6) compared with cells transfected with same siRNAs but not treated with these drugs.
p62-silencing causes carcinoma cell death but not apoptosis
Previous reports revealed that p62 activates several signal transduction pathways regulating apoptosis. Therefore, in this study, we examined the potential contribution of apoptosis to reduced cell viability induced by p62 inhibition. Both sip62s markedly increased cell death in PC9 and A549 cells in a siRNAs concentration dependent manner (Fig.5a, Fig. S3). The cleavage of two apoptosis markers, Caspase-3 and PARP (poly ADP ribose polymerase), was slightly increased in the cells transfected with sip62s (Fig.5b). However, pan-caspase inhibitor Z-VAD-FMK did not necessarily restore cell viability decreased by p62-silencing (Fig.5c). These results indicated that p62 inhibition strongly induced cell death, mainly as a result of autophagic cell death.
Figure 5.

Effects of p62-silencing upon cell death. (a) Both PC9 and A549 cells were transfected with indicated siRNAs for 3 days. After transfection, the cells were stained with trypan blue, and the ratio of dead cells was measured. *P < 0.05 (n = 3) compared with control cells. (b) Both PC9 and A549 cells were transfected with indicated siRNAs for 3 days. After transfection, protein homogenates were isolated and immunoblotting was used to measure the levels of p62, Caspase-3, PARP and β-actin expression. (c) Both PC9 and A549 cells were pre-treated with 20 or 40 μM pan-caspase inhibitor (Z-VAD-FMK) for 30 min before transfection with indicated siRNAs. On 3 days after transfection, cell viability was measured.
Exogenous p62-overexpression in p62 low-expressed and p62-silenced cells
We then examined the roles of p62 using p62 low-expressing (Fig.1a) H23 cells. p62-silencing slightly suppressed cell viability of H23 cells (Fig.6a). We then constructed p62 expression vector and transfected in H23 cells. p62 expression was increased by the transfection of p62 expression vector in a concentration dependent manner, but cell viability was not changed (Fig.6b,c). Because p62 inhibition also induced autophagy via mTOR inactivation (Fig.2a), we then hypothesized that exogenous p62-overexpression could influence the autophagy-activated cells. Figure6(d) demonstrated that the starvation-induced cell death was attenuated by p62-overexpression, which suggested that starvation-induced autophagy caused autophagic cell death in p62 low-expressing H23 cells, and exogenous p62-overexpression contributed to autophagy maturation. In addition, exogenous p62-overexpression decreased the effects of p62-silencing in PC9 cells (Fig.6e).
Figure 6.
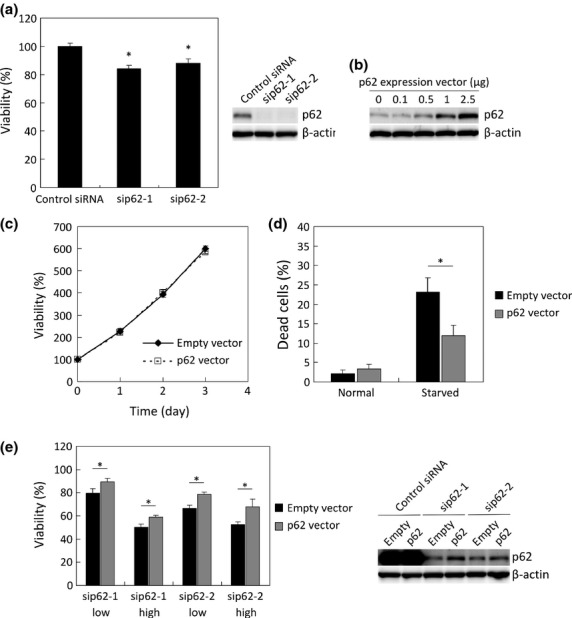
Effects of exogenous p62-overexpression. (a) H23 cells were transfected with indicated siRNAs for 3 days. After transfection, cell viability was measured, and protein homogenates were isolated and immunoblotting was used to determine the levels of p62 and β-actin expression. *P < 0.05 (n = 6) compared with control cells. (b) H23 cells were cultured in 6-well plates and transfected with indicated volumes of p62 expression vector. After transfection, protein homogenates were isolated and immunoblotting was used to determine the levels of p62 and β-actin expression. (c) H23 cells were transfected with indicated vectors, and cell viability was measured. (d) H23 cells were transfected with indicated vectors. After transfection, these cells were treated with HBSS for 24 h. The cells were stained with trypan blue, and the ratio of dead cells was measured. *P < 0.05 (n = 3). (e) PC9 cells were transfected with indicated vectors and siRNAs. After transfection, cell viability was measured, and protein homogenates were isolated and immunoblotting was used to determine the levels of p62 and β-actin expression. *P < 0.05 (n = 6).
Autophagic cell death occurred in various carcinoma cell types
We examined the effects of autophagic cell death on cell viability and cell death of various carcinoma cells other than lung adenocarcinoma cell lines in order to confirm whether the above were specific in lung adenocarcinoma or not. Autophagic cell death induced by p62-silencing was also detected in various carcinoma cells; PANC-1 (pancreatic adenocarcinoma), ECGI-10 (esophageal squamous cell carcinoma (SCC)), HSC-4 (oral SCC) and HSC-1 (skin SCC) (Fig.7a). Restoration of cell viability by autophagy inhibition in p62-silenced cells were also detected in all these carcinoma cell lines examined (Fig.7b).
Figure 7.

Effects of autophagy induced by p62-silencing in various carcinoma cells. (a) Cells were transfected with indicated siRNAs for 3 days. After transfection, the cells were stained with trypan blue, and the ratio of dead cells was measured. *P < 0.05 (n = 3). (b) Cells were transfected with indicated siRNAs for 3 days. After transfection, cell viability was measured, and protein homogenates were isolated and immunoblotting was used to determine the levels of p62, Atg5, Atg7, LC3B and β-actin expression. *P < 0.05 (n = 6). C, Control siRNA-2; 5, siAtg5; 7, siAtg7.
Discussion
To the best of our knowledge, this is the first study to demonstrate a pivotal role of p62 as an autophagy mediator during carcinoma progression. Results of our present study demonstrated that p62-silencing induced the formation of autophagosomes with multilayer membranes and resulted in the development of autophagic carcinoma cell death, as in vitro analysis of p62-overexpressed carcinoma cells derived from lung adenocarcinomas demonstrated. In addition, the results above was also detected in adenocarcinomas and squamous cell carcinoma cell lines. p62-overexpression was recently reported in various human carcinoma cells compared with normal cells using immunohistochemistry; non-small cell lung carcinomas, breast carcinoma, hepatocellular carcinoma and several other carcinomas.8,12,13,21 Therefore, the results of our present study could be applicable to various human carcinomas, and further studies into the significance of p62 expression and its functions during carcinoma progression are warranted.
An inhibition of p62 by RNA interference markedly induced both LC3B-II expression and autophagy. Results of previously reported studies revealed that p62 directly bound to mTOR as a component of mTOR complex 1 and activated mTOR pathway in prostate carcinoma CaP2 cells,16 but an association between p62 and autophagy induction has remained largely unknown. In our present study, we also confirmed mTOR inactivation induced by p62-silencing in PC9 and A549 cells, and p62 silencing-induced higher LC3B-II expression, possibly resulting in mTOR inactivation.
One of our important findings in our present study was the formation of multilayer vesicles induced by p62-silencing. The presence of multilayer body has been reported in the cytoplasm, for instance, mutant human tau expressed culture Aplysia neurons and mutant human β-synuclein transfected culture Rat neuroblastoma cells.22,23 These reports indicated the association between accumulation of autophagosomes with multilayer membranes and neurodegenerative diseases such as Alzheimer's and Parkinson's disease, widely recognized as impaired autophagy associated diseases.24–27 Therefore, multilayer autophagosomes detected in our present study could be induced and accumulated by p62-silencing.
p62 is considered to act as a cargo receptor for degradation of damaged or long-lived proteins via autophagy.5–7 In our present study, some p62-silenced cells had large autophagosomes with multilayer membranes involving other autophagosomes with multilayer membranes. Therefore, it is reasonably postulated that autophagosomes could not recognize the proteins that are required to be degraded in p62-silenced cells, although autophagy was induced. In addition, increased autophagosomes may contain other autophagosomes that were formed through an interaction with autophagosome-binding proteins, such as Atg5 or LC3B-II. However, further investigations are required to clarify the mechanisms of formation and maturation of autophagosomes with multilayer membranes.
Genomic and pharmacological inhibition of autophagy resulted in the restoration of cell viability reduced by p62-silencing in various cell types, suggesting that the formation of multilayer autophagosomes is mis-regulated and cause carcinoma cells into autophagic cell death. It is true that p62-silencing significantly increased the rate of dying cells because Z-VAD-FMK did not rescue carcinoma cells from enhanced cell death, but not an induction of apoptosis. In addition, an autophagy inhibition, itself, did not reduce cell proliferation in our present study, suggesting that non-degradation or accumulation of damaged organelles or proteins were not necessarily critical for carcinoma cells. Therefore, non-damaged organelles or proteins could be involved in mis-regulated autophagosomes with multilayer membranes, which subsequently disturbed the cellular homeostasis.
Recently, p62 has become of enormous interest because an increasing number of the reports had indicated p62-overexpression and its multifunctionality in carcinoma cells.5,6,14–16 For example, p62 activated anti-oxidant Nrf2 pathway,14 NF-kB pathway in lung carcinoma cells15 and mTOR pathway in pancreatic carcinoma cells.16 In our present study, p62-silencing reduced expression of NQO1 (indicative of Nrf2 pathway) but not IL-6 and COX-2 (indicative of NF-kB pathway) in carcinoma cells (data not shown). These results all suggest that p62 could function through several different pathways, and reduction of cell proliferation by p62-silencing could be partly due to an inhibition of these pathways above.
Cell proliferation was not changed by exogenous p62-overexpression, suggesting that exogenous p62 did not activate several pathways known to be associated with p62 and acceleration of carcinoma cell proliferation. Of interest, exogenous p62-overexpression resulted in the suppression of cell death induced by starvation. Starved conditions have been largely considered as an autophagy inducer, and autophagic cell death could therefore occur when autophagy was markedly activated in p62 low-expressing cells. Recently, autophagy was also reported to be activated to protect carcinoma cells from hypoxia, metabolic stress and anti-neoplastic agents.17,18 Therefore, p62-overexpressing carcinoma cells are considered more resistant to these conditions, and p62 expression levels could be a prognostic marker of anti-neoplastic treatment.
In summary, we demonstrated that an inhibition of p62 induced the formation of mis-regulated multilayer autophagosomes in various human carcinoma cells. This perturbation resulted in an autophagic cell death and markedly suppressed carcinoma cell proliferation. These results all indicated that an inhibition of p62 could be a novel strategy for carcinoma treatment in terms of crucial mediator of autophagy. However, the reasons why p62-silencing promoted the formation of multilayer autophagosomes, and why the formation of mis-regulated autophagosomes with multilayer membranes causes autophagic carcinoma cell death should be clarified by further investigations.
Disclosure Statement
The authors have no conflict of interest.
Funding information
None declared.
Supporting Information
Additional supporting information may be found in the online version of this article:
Effects of control siRNAs on cell proliferation.
Fig. S2 Effects of sip62s purchased from Ambion.
Fig. S3 siRNAs concentration-dependent induction of autophagic cell death.
Table S1 Cell lines used in this study.
References
- 1.Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–97. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 2.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med. 2008;8:78–91. doi: 10.2174/156652408783769616. [DOI] [PubMed] [Google Scholar]
- 5.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–4. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moscat J, Diaz-Meco MT, Albert A, Campuzano S. Cell signaling and function organized by PB1 domain interactions. Mol Cell. 2006;23:631–40. doi: 10.1016/j.molcel.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 8.Su Y, Qian H, Zhang J, Wang S, Shi P, Peng X. The diversity expression of p62 in digestive system cancers. Clin Immunol. 2005;116:118–23. doi: 10.1016/j.clim.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Biol Chem. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rolland P, Madjd Z, Durrant L, Ellis IO, Layfield R, Spendlove I. The ubiquitin-binding protein p62 is expressed in breast cancers showing features of aggressive disease. Endocr Relat Cancer. 2007;14:73–80. doi: 10.1677/erc.1.01312. [DOI] [PubMed] [Google Scholar]
- 13.Kitamura H, Torigoe T, Asanuma H, et al. Cytosolic overexpression of p62 sequestosome 1 in neoplastic prostate tissue. Histopathology. 2006;48:157–61. doi: 10.1111/j.1365-2559.2005.02313.x. [DOI] [PubMed] [Google Scholar]
- 14.Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 15.Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–46. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res. 2010;70:3431–4. doi: 10.1158/0008-5472.CAN-09-4027. [DOI] [PubMed] [Google Scholar]
- 18.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy Nature reviews. Clin Oncol. 2011;8:528–39. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 19.Dunn WA. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–45. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 21.Inoue D, Suzuki T, Mitsuishi Y, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–6. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei J, Fujita M, Nakai M, et al. Enhanced lysosomal pathology caused by beta-synuclein mutants linked to dementia with Lewy bodies. J Biol Chem. 2007;282:28904–14. doi: 10.1074/jbc.M703711200. [DOI] [PubMed] [Google Scholar]
- 23.Shemesh OA, Spira ME. Hallmark cellular pathology of Alzheimer's disease induced by mutant human tau expression in cultured Aplysia neurons. Acta Neuropathol. 2010;120:209–22. doi: 10.1007/s00401-010-0689-7. [DOI] [PubMed] [Google Scholar]
- 24.Nixon RA, Cataldo AM, Mathews PM. The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res. 2000;25:1161–72. doi: 10.1023/a:1007675508413. [DOI] [PubMed] [Google Scholar]
- 25.Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009) Neurology. 2009;72:S1–136. doi: 10.1212/WNL.0b013e3181a1d44c. [DOI] [PubMed] [Google Scholar]
- 26.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 27.Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131:1969–78. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of control siRNAs on cell proliferation.
Fig. S2 Effects of sip62s purchased from Ambion.
Fig. S3 siRNAs concentration-dependent induction of autophagic cell death.
Table S1 Cell lines used in this study.