

Abstract
Diffuse-type gastric carcinomas (DGC) exhibit more aggressive progression and poorer prognosis than intestinal-type and other gastric carcinomas. To identify potential therapeutic targets, we examined protein tyrosine phosphorylation in a panel of DGC and other gastric cancer cell lines. Protein tyrosine phosphorylation was significantly enhanced or altered in DGC cell lines compared with that in other gastric cancer cell lines. Affinity purification and mass spectrometry analysis of tyrosine-phosphorylated proteins identified Met as a protein that is preferentially expressed and phosphorylated in DGC cell lines. Unexpectedly, Met inhibitors blocked cell growth, Met downstream signaling and peritoneal dissemination in vivo in only a subset of cell lines that exhibited remarkable overexpression of Met. Likewise, only cell lines with overexpression of fibroblast growth factor receptor 2 (FGFR2) or phosphorylation of FRS2 were sensitive to an FGFR2 inhibitor. A Src inhibitor saracatinib impaired growth in cell lines that are insensitive to both Met and FGFR2 inhibitors. Saracatinib also effectively impaired peritoneal dissemination of Met-independent and FGFR2-independent SGC cells. Moreover, DGC cell lines exhibited nearly mutually exclusive susceptibility to Met, FGFR and Src inhibitors. These results suggest that DGC have distinct sensitivities to molecular target drugs and that targeting Src is beneficial in the treatment of DGC insensitive to Met and FGFR inhibition.
Keywords: c-Met, c-src, neoplasm metastasis, saracatinib, stomach neoplasms
Gastric adenocarcinomas are histologically classified into two major subtypes according to the Laurén classification: intestinal and diffuse.1 Diffuse-type gastric carcinoma (DGC) is also called poorly differentiated gastric carcinoma and often exhibits aggressive progression.2–4 Scirrhous gastric carcinoma (SGC) is a subtype of DGC characterized by rapid infiltrative growth accompanied by massive stromal fibrosis and frequent metastasis to lymph nodes and the peritoneum.5 These aggressive characteristics contribute to the extremely poor prognosis of patients with SGC.6,7 Several genetic alterations have been implicated in DGC, including gene amplification of c-met and FGFR2/k-sam.8–10
The oncogene c-met encodes Met receptor type tyrosine kinase whose ligand is hepatocyte growth factor (HGF). Met signaling regulates multiple aspects of cancer malignancies, including cell migration and invasion, cell proliferation and survival, and angiogenesis.11 Amplification and germline and somatic mutations of c-met have been found in a wide spectrum of human cancers.12 Therefore, Met is considered to be a promising therapeutic target, and dozens of Met inhibitors are being evaluated in clinical trials.12–14 Met amplification is correlated with poor prognosis in gastric cancer patients.10,15,16
FGFR2/k-sam encodes fibroblast growth factor receptor (FGFR) type 2, a member of the FGFR receptor tyrosine kinase family, and its mutation and amplification have been detected and correlated with poor prognosis in several human cancers, including gastric cancers.17 Similar to Met, FGFR2 signaling regulates many cellular functions that contribute to cancer progression, including cell proliferation, survival and migration.17 Accordingly, FGFR inhibitors are being tested in clinical trials.18
Several studies have revealed that gastric cancer cell lines exhibiting Met amplification are sensitive to Met inhibitors.16,19–24 Likewise, FGFR2 inhibitors have been shown to block cell growth and peritoneal dissemination of SGC cells with FGFR gene amplification.25–27 However, amplification of c-met and FGFR2 occurs only in a limited fraction: approximately 2–20% and 10% of all gastric cancers, respectively.10,15,19,28–30 Therefore, a molecular target remains to be determined for the treatment of the fraction of DGC with neither c-met nor FGFR2 amplification/activating mutation.
In this study, we performed a detailed analysis of tyrosine-phosphorylated proteins in a panel of gastric cancer cell lines to identify signaling pathways or molecules that could be molecular targets for DGC chemotherapy.
Materials and Methods
Cell culture
The human gastric cancer cell lines used, that is, HSC-39, HSC-43, HSC-59, HSC-60, HSC-64, HSC-44PE, 58As9, 58As1, 44As3 and 44As3Luc, have been described previously.31–34 MKN1, MKN7, MKN74, NUGC-4, KATO-III, MKN45 and IM95 were obtained from the Health Science Research Resources Bank. AGS, NCI-N87 and SNU-5 were obtained from the American Type Culture Collection (ATCC). GCIY, ECC12, AZ521 and KE-97 were provided by the RIKEN Bio-Resource Center through the National Bio-Resource Project of the MEXT, Japan. These cells were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS, 10 units/mL of penicillin and 10 μg/mL of streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Reagents and antibodies
Antibodies, including phospho-specific antibodies, against Met, Src, ERK, Akt, FRS2α and Stat3 were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Met and FRS2 were also purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against FGFR2α and phospho-FGFR1-4 were purchased from R&D Systems (Minneapolis, MN, USA). Anti-phosphotyrosine (4G10) antibody was obtained from Merck Millipore (Billerica, MA, USA). PHA-665752, crizotinib (PF-2341066), saracatinib (AZD0530) and JNJ-38877605 were purchased from Selleck Chemicals (Houston, TX, USA). Saracatinib was also obtained from Adooq BioScience (Irvine, CA, USA). PD-173074 was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Immunoblotting
Immunoblotting was carried out as described previously.35 ImageJ software (version 1.41o; National Institute of Health, Bethesda, MD, USA) was used to quantify the band intensity from immunoblot data.
Affinity purification and identification of tyrosine-phosphorylated proteins
58As9 cells were lysed in a buffer containing 50 mM Hepes-NaOH (pH 7.0), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 1 mM Na3VO4 and protease inhibitors. The lysates were incubated with 4G10 coupled with cyanogen bromide-activated Sepharose 4B beads (GE Healthcare, Little Chalfont, UK). The beads were washed with lysis buffer, and 4G10-associated proteins were eluted using 0.1 M phenylphosphate. The purified proteins were subjected to SDS-PAGE, stained using a Silver Stain MS kit (Wako, Osaka, Japan), excised, digested with trypsin and subjected to liquid chromatography-tandem mass spectrometry analysis. Proteins were identified using a Mascot search.
Cell proliferation assay
Cells were plated onto 96-well plates at 1–2 × 103 per well and cultured for 4 days in the presence or absence of inhibitors. Cell growth was determined using a Premix WST-1 Cell Proliferation Assay System (Takara, Shiga, Japan) according to the manufacturer's instructions. Absorbance at 450 nm was measured with an iMark microplate reader (Bio-Rad Laboratories, Hercules, CA, USA), and the measurement was conducted in quadruplicate.
Peritoneal dissemination assay
44As3 (5 × 105) or 58As9 cells (4 × 106) were inoculated intraperitoneally into BALB/c nude mice purchased from CLEA Japan (Tokyo, Japan). Inhibitors (PHA-665752, 10 mg/kg; saracatinib, 50 mg/kg) were administrated via intraperitoneal injection, thrice per week, starting 1 day after inoculation. In the case of 44As3Luc, the progression of metastasis was monitored via bioluminescence analysis using an IVIS system (Xenogen, Alameda, CA, USA) as described previously.32 At 9–11 days for 44As3 and 16–18 days for 58As9 after inoculation, the mice were killed and dissected to evaluate peritoneal dissemination, liver metastasis, omental tumor weight and ascites formation. These experiments were approved by the Committee for Ethics of Animal Experimentation of the National Cancer Center and conducted in accordance with the guidelines for animal experiments of the National Cancer Center.
Statistical analysis
The data are representative of at least three independent experiments. Statistical analysis and calculation of median inhibitory concentration (IC50) values were performed using graphpad prism version 6.0 (GraphPad Software, San Diego, CA, USA).
Results
Diffuse-type gastric carcinoma cells exhibit different patterns of protein tyrosine phosphorylation
We first examined protein tyrosine phosphorylation in a panel of gastric cancer cell lines (Table S1) by immunoblotting with an anti-phosphotyrosine antibody 4G10 (Fig.1a). The amount of phosphotyrosine-containing proteins mainly distributed from 37 to 250 kDa was greatly elevated in several DGC cell lines, including MKN45, KATO-III, 58As1, 58As9 and SNU-5. Other cell lines exhibited moderate or only a modest level of tyrosine-phosphorylated proteins. Notably, the band pattern of phosphotyrosine-containing proteins in DGC cell lines, which apparently varied among lines, was significantly distinct from those in differentiated gastric cancer cells. These observations indicate that DGC display specific activation of some intracellular signaling pathways involving tyrosine phosphorylation.
Figure 1.
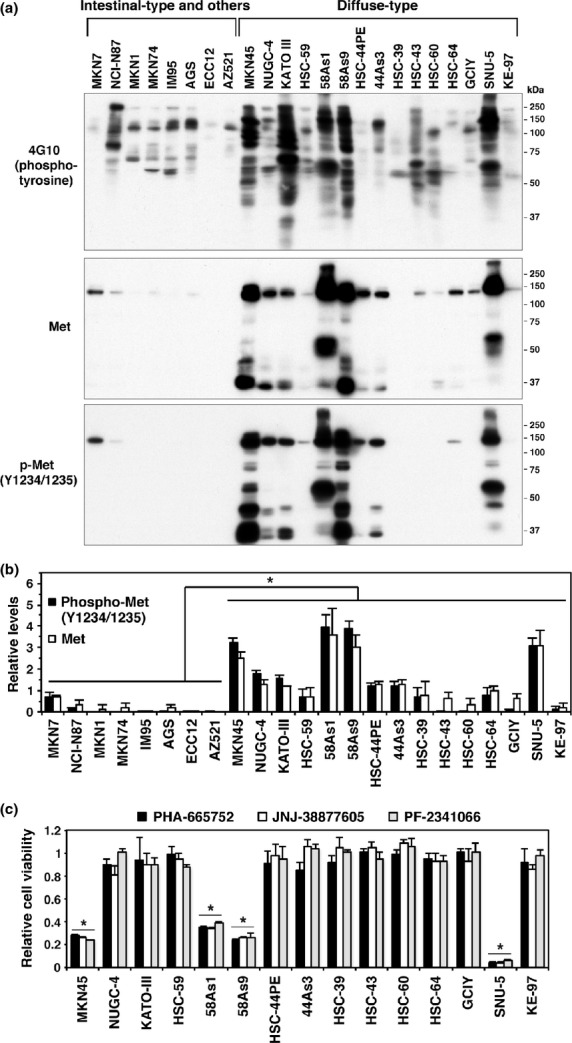
Diffuse-type gastric carcinoma (DGC) cell lines with robust overexpression of Met are sensitive to Met inhibitors. (a) DGC and other gastric carcinoma cell lines were subjected to immunoblot analysis with anti-phosphotyrosine (4G10) and anti-Met antibodies. (b) The relative levels of phospho-Met and total Met were quantified from immunoblot data as described in the Materials and Methods. Bars, SD (n = 4 for phospho-Met and 3 for total Met). *P < 0.01, by Student's t-test. (c) DGC cell lines were treated with 300 nM of Met inhibitors for 4 days, and cell viability was assessed. Bars, SD (n = 4). *P < 0.0001 versus other cell lines, by anova with Tukey's test.
Met is overexpressed and highly phosphorylated in diffuse-type gastric carcinoma cells
To identify molecules that account for increased tyrosine phosphorylation in DGC cell lines, phosphotyrosine proteins were affinity purified from 58As9 cells and subjected to mass spectrometry. As a result, the most prominent band at approximately 140 kDa was identified as Met, a receptor tyrosine kinase for HGF ligand (Fig. S1). Many of the DGC cell lines showed significant increases in both expression and phosphorylation of Met compared with those in other gastric cancer cell lines (Fig.1a,b). Notably, MKN45, 58As1, 58As9 and SNU-5 cells have higher amounts of Met than those in other DGC cell lines. This observation is consistent with previous studies showing that these cell lines display amplification of c-met.23,24
Different sensitivities to Met inhibitors in diffuse-type gastric carcinoma cell lines
The effects of Met inhibitors (PHA-665752, JNJ-38877605 and PF-2341066 [crizotinib]) on the growth of DGC cell lines were determined (Fig.1c). These Met inhibitors effectively suppressed the growth of MKN45, 58As1, 58As9 and SNU-5 cells that showed the highest expression of Met. By contrast, Met inhibitors had no effect on the growth of other cell lines. These results are consistent with those of other studies that have used similar but different sets of gastric cancer cell lines and Met inhibitors.20,23,24 Met inhibitor-insensitive 44As3 and Met inhibitor-sensitive 58As9 cells were chosen for further analyses, as both are highly metastatic in a mouse peritoneal dissemination model.33
The IC50 values of Met inhibitors in 44As3 and 58As9 cells were approximately 1500 and 19 nM for PHA-665752, >10 000 and 6.9 nM for JNJ-38877605, and 2600 and 21 nM for PF-2341066, respectively (Fig. S2a). Met inhibitors significantly blocked phosphorylation of Met in both 44As3 and 58As9 cells (Fig.2a). Nonetheless, overall protein tyrosine phosphorylation and phosphorylation of ERK, Akt and Stat3 were significantly decreased only in 58As9 cells (Fig.2a,b). Interestingly, Src phosphorylation was rather increased in 58As9 cells upon Met inhibition, whereas it was unchanged in 44As3 cells (Fig.2a). Similar results were obtained with PF-2341066 (Fig. S2b). Met inhibitor treatment also induced marked changes in cell morphology in 58As9 but not in 44As3 cells: rounded morphology was converted to flattened and adherent phenotypes (Fig.2c). Serum starvation reduced and following HGF stimulation increased Met phosphorylation in 44As3 cells, whereas Met was constitutively phosphorylated in 58As9 cells (Fig.2d). Consequently, growth of 44As3 but not of 58As9 cells was sensitive to serum starvation (Fig.2e). These results indicate that overexpressed Met proteins confer the capability for serum-independent growth to 58As9 cells via activation of downstream signaling pathways, and, therefore, that 58As9 cells are susceptible to Met inhibitors.
Figure 2.

44As3 and 58As9 scirrhous gastric carcinoma (SGC) cells have different sensitivities to Met inhibitors. (a, b) 44As3 and 58As9 cells were treated with 100 or 300 nM of Met inhibitors PHA-665752 (PHA) or JNJ-3887760 (JNJ) for 2 h and subjected to immunoblot analyses. (c) Phase contrast micrographs of cells treated with Met inhibitors for 1 day. (d) Cells were cultured in the presence of 10 or 0.1% serum for 1 day and then stimulated with 100 ng/mL hepatocyte growth factor (HGF) for 10 min. The cells were then analyzed with immunoblotting. (e) Viability of cells cultured in the presence of various concentrations of serum for 3 days. Bars, SD (n = 4). *P < 0.005 and **P < 0.0005 by Student's t-test. (f) 44As3 and 58As9 cells were inoculated intraperitoneally into nude mice, and DMSO (vehicle) or the Met inhibitor PHA was administered. Representative macroscopic views of mesentery tumor nodules are shown. (g) The number of mesentery tumor nodules (>1 mm in diameter). Bars, SEM (n = 11 for DMSO and 13 for PHA in 44As3; n = 10 for DMSO and PHA in 58As9). *P < 0.001 by the Mann–Whitney test.
Next, the effect of Met inhibition on peritoneal dissemination of 44As3 and 58As9 cells was examined. Met inhibition remarkably reduced the formation of ascites and peritoneal dissemination in mice injected with 58As9 cells (Fig.2f,g; Table1). By contrast, Met inhibition had minimal effects on peritoneal dissemination of 44As3 cells; only a modest decrease in ascites formation was observed (Table1). Taken together, Met inhibition seems to be extraordinarily effective against a portion of DGC that express high levels of Met proteins, although it is ineffective against other DGC.
Table 1.
Effect of Met inhibition on ascites formation and dissemination of 44As3 and 58As9 cells
| Cell line | Treatment | Ascites | Metastasis |
|||
|---|---|---|---|---|---|---|
| Omentum | Mesentery | Diaphragm | Liver | |||
| 44As3 | DMSO | 6/11 | 11/11 | 11/11 | 5/11 | 5/11 |
| PHA | 3/13 | 12/13 | 12/13 | 4/13 | 6/13 | |
| 58As9 | DMSO | 8/10 | 8/10 | 8/10 | 8/10 | 8/10 |
| PHA | 0/10 | 6/10 | 1/10 | 0/10 | 0/10 | |
Number of mice bearing ascites or tumors at the indicated site per total number of mice bearing tumors.
Src inhibitor blocks growth of Met/fibroblast growth factor receptor-independent diffuse-type gastric carcinoma cell lines
The effects of several inhibitors targeted to signaling molecules other than Met were then examined. PD-173074, a pan-FGFR inhibitor, treatment strongly inhibited the growth of KATO-III and HSC-39 and moderately inhibited HSC-43 and HSC-64 (Fig.3a). These sensitive cell lines displayed increased phosphorylation of FRS2α, an adaptor protein for FGFR (Fig.3b). KATO-III and HSC-43 cells also showed robust overexpression and phosphorylation of FGFR2α, which is consistent with k-sam amplification in those cell lines.36 It is unclear why FGFR2α expression was not detected in HSC-39 cells, which also have k-sam amplification.36 Taken together, phosphorylation of FRS2α and overexpression of FGFR2α seem to be correlated with sensitivity to FGFR inhibition.
Figure 3.
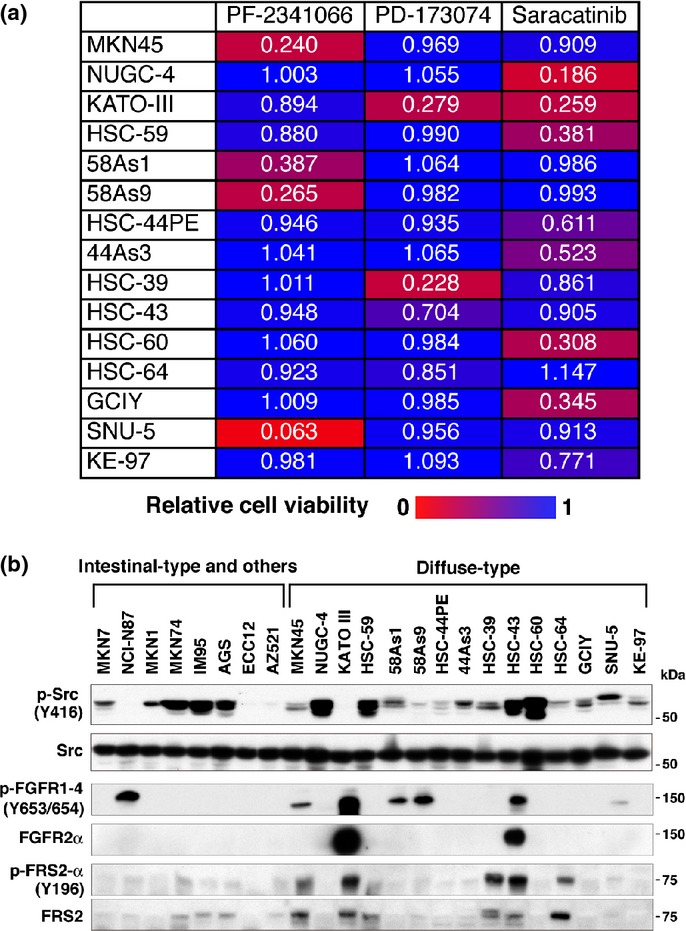
Diffuse-type gastric carcinoma (DGC) cell lines have different sensitivities to Met, fibroblast growth factor receptor (FGFR) and Src inhibitors. (a) Viability of DGC cell lines cultured in the presence of Met inhibitor PF-2341066 (300 nM), FGFR inhibitor PD-173074 (300 nM), or Src inhibitor saracatinib (3 μM) for 4 days. Numbers shown are mean values of relative cell viability (n = 4). The sensitivity of each cell line to each inhibitor is indicated by the increasing intensity of the red signal on a blue background. (b) Immunoblot analysis for expression and phosphorylation of Src, FGFR and FRS2.
Because Met inhibitor treatment upregulated Src phosphorylation in 58As9 cells, we next tested the effect of the Src inhibitor saracatinib. Treatment with saracatinib significantly blocked the growth of several DGC cell lines, including NUGC-4, KATO-III, HSC-59, HSC-44PE, 44As3, HSC-60, GCIY and KE-97, but it had no effect on Met-dependent cell lines (Fig.3a). These cell lines, with the exception of KATO-III, also lacked sensitivity to PD-173074. We also tested whether the combination of Met inhibitor and saracatinib has a synergistic effect on the proliferation of 58As9 cells. However, simultaneous addition of the both inhibitors had a minimal effect (Fig. S3). These observations clearly suggest that DGC have different, and nearly mutually exclusive, sensitivities to Met, FGFR and Src inhibitors.
Immunoblot analysis showed that Src is equally expressed in all gastric cancer cell lines tested, and its phosphorylation also occurred in several DGC and other cell lines (Fig.3b). Notably, however, the amount of phosphorylated Src was inversely correlated with that of phosphorylated Met in some DGC cell lines: cell lines with Met overexpression (MKN45, 58As1, 58As9 and SNU-5) have lower amounts of phosphorylated Src, whereas those with low expression of Met (NUGC-4, HSC-59 and HSC-60) have relatively high amounts of phosphorylated Src. As expected, saracatinib, but not Met and FGFR inhibitors, decreased overall tyrosine phosphorylation and ERK and Akt phosphorylation in saracatinib-responsive cell lines GCIY and HSC-60 (Fig. S4). Similarly, Met inhibitors, but not saracatinib or PD-173074, blocked overall tyrosine phosphorylation, as well as ERK, and Akt phosphorylation in MKN45 and 58As1. In HSC-39 and KATO-III cells, FGFR inhibition significantly suppressed phosphorylation of these signaling molecules.
Saracatinib impairs peritoneal dissemination of Met/fibroblast growth factor receptor-independent scirrhous gastric carcinoma cells
The IC50 values of saracatinib for the cell growth of 44As3 and 58As9 cells were 8.2 and 30 μM, respectively (Fig.4a). Saracatinib treatment significantly decreased overall protein tyrosine phosphorylation in 44As3 but not in 58As9 cells (Fig.4b). Interestingly, saracatinib did not affect the phosphorylation of Met, ERK and Akt (Fig.4c). Saracatinib administration reduced the incidence of ascites formation and tumor dissemination to diaphragm and liver in 44As3 cells expressing luciferase (44As3Luc; Table2). Although the incidence of dissemination to mesentery was not affected much by saracatinib, a marked reduction in the number of mesentery nodules was observed (Fig.4d,e). In vivo imaging analysis confirmed that saracatinib treatment significantly reduces the growth and dissemination of 44As3Luc cells (Fig.4f,g). In contrast, neither the incidence of omental metastasis nor the growth of omental tumors was affected by saracatinib treatment (Table2 and Fig.4h). Although saracatinib tended to suppress the formation of ascites and peritoneal dissemination by 58As9 cells, the effects were more moderate (Table2 and Fig.4d,e). These data indicate that saracatinib can suppress peritoneal dissemination of SGC cells that are insensitive to Met and FGFR inhibitors.
Figure 4.

Saracatinib impairs peritoneal dissemination of 44As3 cells. (a) Viability of 44As3 and 58As9 cells treated with various concentrations of saracatinib for 4 days. (b, c) Cells were treated with saracatinib for 2 h and subjected to immunoblot analyses with indicated antibodies. (d) 44As3Luc and 58As9 cells were intraperitoneally injected into nude mice and DMSO (vehicle) or saracatinib was administered. Representative macroscopic views of mesentery tumor nodules are shown. (e) The number of mesentery tumor nodules. Bars, SEM (n = 16 for DMSO and 18 for saracatinib in 44As3Luc; n = 8 for DMSO and 7 for saracatinib in 58As9). P < 0.001; and **P < 0.0001 by the Mann–Whitney test. (f) Representative images of mice obtained via in vivo imaging. (g) Quantitative analysis of luminescence photon counts. Bars, SD (n = 8 for DMSO and 9 for saracatinib). *P < 0.05; and **P < 0.0005 by the Mann–Whitney test. (h) Omental tumor weight in mice inoculated with 44As3Luc cells. Bars, SEM (n = 10 for DMSO and 9 for saracatinib). P = 0.3451 by the Mann–Whitney test.
Table 2.
Effect of saracatinib on ascites formation and dissemination of 44As3Luc and 58As9 cells
| Cell line | Treatment | Ascites | Metastasis |
|||
|---|---|---|---|---|---|---|
| Omentum | Mesentery | Diaphragm | Liver | |||
| 44As3Luc | DMSO | 14/16 | 14/16 | 16/16 | 4/16 | 9/16 |
| Saracatinib | 3/18 | 16/18 | 15/18 | 0/18 | 4/18 | |
| 58As9 | DMSO | 8/8 | 8/8 | 8/8 | 8/8 | 6/8 |
| Saracatinib | 3/7 | 7/7 | 7/7 | 5/7 | 5/7 | |
Number of mice bearing ascites or tumors at the indicated site per total number of mice bearing tumors.
Discussion
This study showed that significant increases and changes in protein tyrosine phosphorylation occur in DGC cell lines. In particular, several cell lines showed remarkable increases in the levels of protein tyrosine phosphorylation mainly due to Met overexpression. Although Met was overexpressed and phosphorylated in a large portion of DGC cell lines, only a subset of cell lines with remarkable amounts of Met protein through gene amplification was sensitive to Met inhibitors. One such cell line, 58As9, exhibited constitutive activation of Met and resistance to serum starvation. These results are consistent with those of previous studies showing that genetic alteration of Met drives addiction to Met activity and predicts effective treatment outcome.12,14,15,19,24,37
Two previous studies have reported that Met inhibitors block peritoneal dissemination of MKN45 poorly differentiated gastric adenocarcinoma cells.16,21 The present study is the first to show that Met inhibition also markedly reduces peritoneal dissemination of 58As9 SGC cells. Importantly, Met inhibition had little effect on dissemination of 44As3 SGC cells that have moderate levels of Met expression and phosphorylation. Likewise, FGFR inhibition had growth-inhibitory effects only in cell lines with a high FGFR or FRS2 activation status. Therefore, the activation status of the receptor signaling pathway may need to be considered in the clinical use of these molecular targeted drugs. We demonstrated that the amount of Met and FRS2 phosphorylation are closely correlated with sensitivity to Met and FGFR2 inhibitors, respectively. At least in the case of Met, immunohistochemical studies have demonstrated that Met is expressed and phosphorylated in human DGC with high Met gene copy number.15,37 Thus, our results imply that the phosphorylation status of Met and FRS2 may be a good marker for predicting the therapeutic efficacy of Met and FGFR inhibitors.
To our knowledge, this study is the first to show that saracatinib, an Src inhibitor, suppresses peritoneal dissemination of SGC cells. More importantly, saracatinib reduced cell growth of DGC that are insensitive to both Met and FGFR inhibitors. We also found that sensitivities to Met, FGFR and Src inhibitors are almost mutually exclusive in DGC cell lines. Similar results regarding different sensitivities to Met and Src inhibitors in vitro have been reported in a different set of gastric carcinoma cell lines.23 Our data show that SGC cell lines lacking amplification of Met and FGFR but displaying relatively high levels of Src phosphorylation tend to be responsive to saracatinib. Immunohistochemical study has shown that DGC cells display activation of Src that increases with the depth of invasion and correlates with the progression of DGC.38 Thus, the activation status of Src may predict a good outcome for anti-Src therapy in DGC. Saracatinib treatment also effectively suppressed the growth of MKN74 and IM95 intestinal-type gastric carcinoma cells with high levels of Src phosphorylation (Figs S5 and 3b). Therefore, Src inhibitors may also be applicable to intestinal-type gastric carcinomas.
Administration of saracatinib did not affect the growth of omental tumors in mice inoculated with 44As3 cells, while it effectively blocked the formation of mesentery nodules. It is reported that the omentum is the first implantation site for malignant cells in peritoneal dissemination.39 Therefore, saracatinib may mainly inhibit secondary spread of SGC cells from omental tumors. Alternatively, saracatinib may inhibit cell functions that are more important for colonization in mesenterium than in omentum. Src is a well-established regulator of cell migration and invasion, contributing to metastatic spread of malignant tumors.40 This raises the possibility that saracatinib targets phosphoproteins involved in cell migration and invasion to exert such inhibitory effects on peritoneal dissemination of SGC cells.
In summary, our results demonstrate that Met and FGFR inhibitors are effective in subsets of DGC but not in other DGC. We found that the Src inhibitor saracatinib blocks cell growth and peritoneal dissemination of SGC cells that are insensitive to both Met and FGFR inhibitors. These results may provide a novel approach for molecular targeted therapy in DGC patients.
Acknowledgments
We thank Emi Saito for technical assistance. This work was supported by the National Cancer Center Research and Development Fund (23-A-9); by Grants-in-Aid for Scientific Research by the Japan Society for the Promotion of Science; by Grants-in-Aid for Scientific Research on Innovative Areas by the Ministry of Education, Culture, Sports, Science and Technology of Japan; and by a Grant-in-Aid from the Ministry of Health, Labour and Welfare of Japan for the 3rd Term Comprehensive 10-year Strategy for Cancer Control. This work was also supported in part by The Mochida Memorial Foundation for Medical and Pharmaceutical Research and The Sagawa Foundation for Promotion of Cancer Research.
Disclosure Statement
The authors have no conflict of interest.
Funding information
National Cancer Center Research and Development Fund (23-A-9). Japan Society for the Promotion of Science. Ministry of Education, Culture, Sports, Science and Technology of Japan. Ministry of Health, Labour and Welfare of Japan. The Mochida Memorial Foundation for Medical and Pharmaceutical Research. The Sagawa Foundation for Promotion of Cancer Research.
Supporting Information
Additional supporting information may be found in the online version of this article:
Purification and identification of tyrosine-phosphorylated proteins in 58As9 cells.
Effects of Met inhibitors on cell growth and signaling in 44As3 and 58As9 cells.
Effect of simultaneous addition of Met and Src inhibitors on proliferation of 58As9 cells.
Effects of Met, fibroblast growth factor receptor (FGFR), and Src inhibitors on intracellular signaling pathways in diffuse-type gastric carcinoma (DGC) cell lines.
Effect of saracatinib on proliferation of intestinal-type gastric carcinoma cell lines.
List of cell lines used in this study.
References
- 1.Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31–49. doi: 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- 2.Jinawath N, Furukawa Y, Hasegawa S, et al. Comparison of gene-expression profiles between diffuse- and intestinal-type gastric cancers using a genome-wide cDNA microarray. Oncogene. 2004;23:6830–44. doi: 10.1038/sj.onc.1207886. [DOI] [PubMed] [Google Scholar]
- 3.Ming SC. Cellular and molecular pathology of gastric carcinoma and precursor lesions: a critical review. Gastric Cancer. 1998;1:31–50. doi: 10.1007/s101200050053. [DOI] [PubMed] [Google Scholar]
- 4.Sipponen P. Gastric cancer: pathogenesis, risks, and prevention. J Gastroenterol. 2002;37(Suppl 13):39–44. doi: 10.1007/BF02990098. [DOI] [PubMed] [Google Scholar]
- 5.Yashiro M, Hirakawa K. Cancer-stromal interactions in scirrhous gastric carcinoma. Cancer Microenviron. 2010;3:127–35. doi: 10.1007/s12307-010-0036-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeguchi M, Miyake T, Matsunaga T, et al. Recent results of therapy for scirrhous gastric cancer. Surg Today. 2009;39:290–4. doi: 10.1007/s00595-008-3860-1. [DOI] [PubMed] [Google Scholar]
- 7.Otsuji E, Kuriu Y, Okamoto K, et al. Outcome of surgical treatment for patients with scirrhous carcinoma of the stomach. Am J Surg. 2004;188:327–32. doi: 10.1016/j.amjsurg.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Hattori Y, Itoh H, Uchino S, et al. Immunohistochemical detection of K-sam protein in stomach cancer. Clin Cancer Res. 1996;2:1373–81. [PubMed] [Google Scholar]
- 9.Hattori Y, Odagiri H, Nakatani H, et al. K-sam, an amplified gene in stomach cancer, is a member of the heparin-binding growth factor receptor genes. Proc Natl Acad Sci USA. 1990;87:5983–7. doi: 10.1073/pnas.87.15.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuniyasu H, Yasui W, Kitadai Y, Yokozaki H, Ito H, Tahara E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem Biophys Res Commun. 1992;189:227–32. doi: 10.1016/0006-291x(92)91548-5. [DOI] [PubMed] [Google Scholar]
- 11.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–48. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 12.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 13.Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer. 2010;46:1260–70. doi: 10.1016/j.ejca.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Newton RC, Scherle PA. Development of c-MET pathway inhibitors. Expert Opin Investig Drugs. 2011;20:1225–41. doi: 10.1517/13543784.2011.600687. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Seo JW, Jun HJ, et al. Impact of MET amplification on gastric cancer: possible roles as a novel prognostic marker and a potential therapeutic target. Oncol Rep. 2011;25:1517–24. doi: 10.3892/or.2011.1219. [DOI] [PubMed] [Google Scholar]
- 16.Toiyama Y, Yasuda H, Saigusa S, et al. Co-expression of hepatocyte growth factor and c-Met predicts peritoneal dissemination established by autocrine hepatocyte growth factor/c-Met signaling in gastric cancer. Int J Cancer. 2012;130:2912–21. doi: 10.1002/ijc.26330. [DOI] [PubMed] [Google Scholar]
- 17.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–29. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 18.Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3:264–79. doi: 10.1158/2159-8290.CD-12-0362. [DOI] [PubMed] [Google Scholar]
- 19.Kawakami H, Okamoto I, Arao T, et al. MET amplification as a potential therapeutic target in gastric cancer. Oncotarget. 2013;4:9–17. doi: 10.18632/oncotarget.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDermott U, Sharma SV, Dowell L, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007;104:19936–41. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagawa T, Tohyama O, Yamaguchi A, et al. E7050: a dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010;101:210–5. doi: 10.1111/j.1349-7006.2009.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamoto W, Okamoto I, Arao T, et al. Antitumor action of the MET tyrosine kinase inhibitor crizotinib (PF-02341066) in gastric cancer positive for MET amplification. Mol Cancer Ther. 2012;11:1557–64. doi: 10.1158/1535-7163.MCT-11-0934. [DOI] [PubMed] [Google Scholar]
- 23.Okamoto W, Okamoto I, Yoshida T, et al. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol Cancer Ther. 2010;9:1188–97. doi: 10.1158/1535-7163.MCT-10-0002. [DOI] [PubMed] [Google Scholar]
- 24.Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci USA. 2006;103:2316–21. doi: 10.1073/pnas.0508776103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunii K, Davis L, Gorenstein J, et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008;68:2340–8. doi: 10.1158/0008-5472.CAN-07-5229. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura K, Yashiro M, Matsuoka T, et al. A novel molecular targeting compound as K-samII/FGF-R2 phosphorylation inhibitor, Ki23057, for Scirrhous gastric cancer. Gastroenterology. 2006;131:1530–41. doi: 10.1053/j.gastro.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 27.Yashiro M, Shinto O, Nakamura K, et al. Synergistic antitumor effects of FGFR2 inhibitor with 5-fluorouracil on scirrhous gastric carcinoma. Int J Cancer. 2010;126:1004–16. doi: 10.1002/ijc.24763. [DOI] [PubMed] [Google Scholar]
- 28.Hara T, Ooi A, Kobayashi M, Mai M, Yanagihara K, Nakanishi I. Amplification of c-myc, K-sam, and c-met in gastric cancers: detection by fluorescence in situ hybridization. Lab Invest. 1998;78:1143–53. [PubMed] [Google Scholar]
- 29.Nessling M, Solinas-Toldo S, Wilgenbus KK, Borchard F, Lichter P. Mapping of chromosomal imbalances in gastric adenocarcinoma revealed amplified protooncogenes MYCN, MET, WNT2, and ERBB2. Genes Chromosom Cancer. 1998;23:307–16. doi: 10.1002/(sici)1098-2264(199812)23:4<307::aid-gcc5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 30.Sakakura C, Mori T, Sakabe T, et al. Gains, losses, and amplifications of genomic materials in primary gastric cancers analyzed by comparative genomic hybridization. Genes Chromosom Cancer. 1999;24:299–305. doi: 10.1002/(sici)1098-2264(199904)24:4<299::aid-gcc2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 31.Yanagihara K, Seyama T, Tsumuraya M, Kamada N, Yokoro K. Establishment and characterization of human signet ring cell gastric carcinoma cell lines with amplification of the c-myc oncogene. Cancer Res. 1991;51:381–6. [PubMed] [Google Scholar]
- 32.Yanagihara K, Takigahira M, Takeshita F, et al. A photon counting technique for quantitatively evaluating progression of peritoneal tumor dissemination. Cancer Res. 2006;66:7532–9. doi: 10.1158/0008-5472.CAN-05-3259. [DOI] [PubMed] [Google Scholar]
- 33.Yanagihara K, Takigahira M, Tanaka H, et al. Development and biological analysis of peritoneal metastasis mouse models for human scirrhous stomach cancer. Cancer Sci. 2005;96:323–32. doi: 10.1111/j.1349-7006.2005.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yanagihara K, Tanaka H, Takigahira M, et al. Establishment of two cell lines from human gastric scirrhous carcinoma that possess the potential to metastasize spontaneously in nude mice. Cancer Sci. 2004;95:575–82. doi: 10.1111/j.1349-7006.2004.tb02489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaguchi H, Yoshida S, Muroi E, et al. Phosphoinositide 3-kinase signaling pathway mediated by p110alpha regulates invadopodia formation. J Cell Biol. 2011;193:1275–88. doi: 10.1083/jcb.201009126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueda T, Sasaki H, Kuwahara Y, et al. Deletion of the carboxyl-terminal exons of K-sam/FGFR2 by short homology-mediated recombination, generating preferential expression of specific messenger RNAs. Cancer Res. 1999;59:6080–6. [PubMed] [Google Scholar]
- 37.Catenacci DV, Henderson L, Xiao SY, et al. Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence. Cancer Discov. 2011;1:573–9. doi: 10.1158/2159-8290.CD-11-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humar B, Fukuzawa R, Blair V, et al. Destabilized adhesion in the gastric proliferative zone and c-Src kinase activation mark the development of early diffuse gastric cancer. Cancer Res. 2007;67:2480–9. doi: 10.1158/0008-5472.CAN-06-3021. [DOI] [PubMed] [Google Scholar]
- 39.Hagiwara A, Takahashi T, Sawai K, et al. Milky spots as the implantation site for malignant cells in peritoneal dissemination in mice. Cancer Res. 1993;53:687–92. [PubMed] [Google Scholar]
- 40.Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010;223:14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Purification and identification of tyrosine-phosphorylated proteins in 58As9 cells.
Effects of Met inhibitors on cell growth and signaling in 44As3 and 58As9 cells.
Effect of simultaneous addition of Met and Src inhibitors on proliferation of 58As9 cells.
Effects of Met, fibroblast growth factor receptor (FGFR), and Src inhibitors on intracellular signaling pathways in diffuse-type gastric carcinoma (DGC) cell lines.
Effect of saracatinib on proliferation of intestinal-type gastric carcinoma cell lines.
List of cell lines used in this study.