

Abstract
ATP-binding cassette (ABC) transmembrane proteins evidently decrease the intracellular accumulation of substrate chemotherapeutic drugs by extruding them against a concentration gradient, thereby inducing drug resistance. Here we reported the effect of WHI-P154, an irreversible inhibitor of Janus kinase 3 and epidermal growth factor receptor tyrosine kinases, on reversing ABC transporters-mediated drug resistance. We found that WHI-P154 significantly enhanced the sensitivity of ABCG2-overexpressing cells to its substrates. WHI-P154 moderately sensitized ABCB1-overexpressing KB-C2 cells to its substrates whereas showed no sensitizing effect on ABCC1-, ABCC2 or ABCC10-mediated drug resistance. Moreover, WHI-P154 produced a significant increase in the intracellular accumulation of [³H]-mitoxantrone in ABCG2-overexpressing cells. The expression levels nor the localization of the ABCG2 protein was altered after treatment of ABCG2-overexpressing cells with WHI-P154. Further studies indicated that WHI-P154 enhanced the ATPase activity of ABCG2 at low concentrations (<10 μM). Additionally, a docking model predicted the binding conformation of WHI-P154 within the transmembrane region of homology-modeled human ABCG2 transporter. Collectively, these findings highlighted WHI-P154 could significantly reverse ABCG2-mediated multidrug drug resistance by directly blocking the efflux function.
Keywords: ABCG2 protein, ATP-binding cassette transporters, multidrug resistance, tyrosine kinase, WHI-P154
Multidrug drug resistance (MDR) remains a major obstacle towards attaining a successful chemotherapeutic outcome. Among several factors responsible for development of MDR, the overexpression of ATP-binding cassette (ABC) transporters is a major concern.(1) The human genome, which encodes 48 ABC transporter members has been divided into seven subfamilies (A–G) based on sequence and structure similarities.(2,3) ABC transporters are also known to influence oral absorption and disposition of a wide variety of drugs, but each transporter has its own particular substrate selectivity.(2) The ABCB1 (P-glycoprotein/MDR1) transports toxic endogenous substances, chemotherapeutic drugs such as vinca alkaloids, anthracyclines, epipodophyllotoxins and taxanes across the cell membrane.(4,5) The spectrum of chemotherapeutic drugs transported by ABCG2 (BCRP/MXR) includes mitoxantrone (MX), topotecan, irinotecan, flavopiridol and methotrexate (MTX).(5,6) ABCC1 (MRP1) confers resistance to a broad spectrum of anticancer drugs such as vincristine, camptothecin, doxorubicin and etoposide.(7) ABCC2 (MRP2/cMOAT) can transport various anticancer drugs such as anthracyclines, camptothecins, vinca alkaloids, MTX, etoposide, cisplatin and irinotecan.(8) Another important member of C subfamily is ABCC10 (MRP7); it is a broad-acting transporter of xenobiotics, including drugs such as taxanes and epothilone B.(9)
Therefore, identifying high-affinity drugs that can either block the key signaling pathway regulating the expression of ABC transporters or inhibit their function might be a promising approach to overcome MDR in cancer chemotherapy. Enormous efforts have been devoted to the development of inhibitors of ABC transporters. Previously, we reported several tyrosine kinase inhibitors (TKIs) such as erlotinib, lapatinib, vandetanib and tandutinib that could reverse ABCB1-, ABCG2-, ABCC1- or ABCC10- mediated MDR.(10)– (13)
WHI-P154 was synthesized based on the structure of dimethoxyquinazoline compounds with potent inhibitory activity against JAK3 and STAT3, and it was then proven to be an effective inhibitor for other kinases including epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), Src, Abl, MAPK and PI3K.(14)– (16) This study is to investigate the reversal effect of WHI-P154 on MDR mediated by ABC transporters.
Materials and Methods
Materials
[3H]-MX (4 Ci/mmol) was purchased from Moravek Biochemicals (Brea, CA, USA). BXP-21 (anti-ABCG2) antibody was purchased from Signet Laboratories (Ded-Ham, MA, USA). Anti-STAT3, p-STAT3, ERK1/2, p-ERK1/2, AKT and p-AKT antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-β-actin monoclonal antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). WHP-P154 was a gift from Selleck Chemicals (Houston, TX, USA). PAK-104P was a gift from Professor Shin-Ichi Akiyama (Kagoshima University, Kagoshima, Japan) from Nissan Chemical (Chiba, Japan). Fumitremorgin C (FTC) was kindly provided by Drs Susan E Bates and Robert W Robey (NCI, NIH, Bethesda, MD, USA). Cepharanthine was generously provided by Kakenshoyaku (Tokyo, Japan). MX, SN-38, cisplatin, colchicine, vincristine, verapamil, paclitaxel, penicillin/streptomycin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethylsulfoxide, and other chemicals were purchased from Sigma Chemical (St. Louis, MO, USA).
Cell lines and cell culture
Human non-small cell lung carcinoma cells H460 and H460/MX20 cells were kindly provided by Drs Susan E Bates and Robert W. Robey (NCI, NIH, Bethesda, MD, USA). H460/MX20 cells were cultured in DMEM with addition of 20 nM MX.(17) HEK293/pcDNA3.1, wild-type ABCG2-482-R2, mutant ABCG2-482-G2, and mutant ABCG2-482-T7 cells were provided by Drs Susan E Bates and Robert W Robey (NCI, NIH, Bethesda, MD, USA) and maintained in the medium with 2 mg/mL of G418.(18) Mutant ABCG2 (Phe489Leu) plasmid was sequenced in Innovative Mutagenesis Service (Hillsborough, NJ, USA) as shown in Fig. S1. ABCG2-489-Leu was generated by transient transfection of the mutant ABCG2 (Phe489Leu) plasmid into HEK293 cells (Fig. S2). Similarly, HEK293/ABCC1 and HEK293/ABCC10 cells were generated by transfecting ABCC1 or ABCC10 expression vectors into HEK293 cells.(9,19) LLC/CMV, LLC/cMOAT, KB-3-1 and KB-C2 cells were kindly provided by Dr Shin-Ichi Akiyama (Kagoshima University, Kagoshima, Japan).(20) All cells were grown as adherent monolayers in DMEM supplemented with 10% fetal bovine serum at 37°C in a humidified incubator containing 5% CO2.
Cytotoxicity assays
Cell sensitivity to drugs was analyzed using an MTT colorimetric assay as described previously.(11) The concentrations required to inhibit the growth by 50% (IC50) were calculated from survival curves.
[3H]-MX accumulation assay
The effect of WHI-P154 on the intracellular accumulation of [3H]-MX in ABCG2-overexpressing cells was determined by measuring the intracellular accumulation of [3H]-MX in ABCG2-transfected HEK293 cells as described previously.(21,22)
[3H]-MX efflux assay
For the efflux study, the cells were treated with 4 μM WHI-P154 and all the samples were placed in scintillation fluid and radioactivity were measured as described previously.(23)
Western blot analysis
The cells were washed two times with cold-PBS, and protein lysates were isolated and prepared for Western blot analysis as previously described.(24)
Immunofluorescence
The immunofluorescence was conducted to test the localization of ABCG2 protein after treatment with WHI-P154 for 72 h as described previously.(23)
ATPase assay
The vanadate (Vi)-sensitive ATPase activity of ABCG2 in the membrane vesicles of High Five insect cells was measured as previously described.(21)
Molecular modeling of ABCG2
WHI-P154 structure and human ABCG2 homology model along with various grids and docking simulations were carried out as our previous protocols.(22,25) All computations were carried out on a Dell Precision 490n dual processor with the Linux OS (Ubuntu 12.04 LTS).
Statistical analysis
All experiments were repeated at least three times. Microsoft Office Excel 2010 (Microsoft Corp. Redmond, WA, USA), and Image J (NIH, Bethesda, MD, USA) were used in data processing and analyzing. The data were analyzed using student's two-tailed t-test. Statistical significance was determined at p < 0.05.
Results
Cytotoxicity of WHI-P154 on MDR cells and their parental cells
We investigated the cytotoxicity of WHI-P154 in different cells by MTT assay. As shown in Figure 1, approximately 85% of the cells survived at the concentration of 4 μM WHI-P154 (Fig. 1b–g). Therefore, WHI-P154 at a concentration of 4 μM was chosen as a maximum concentration for combination treatment with known ABCB1, ABCG2, ABCC1, ABCC2 or ABCC10 substrate anticancer drugs.
Fig. 1.
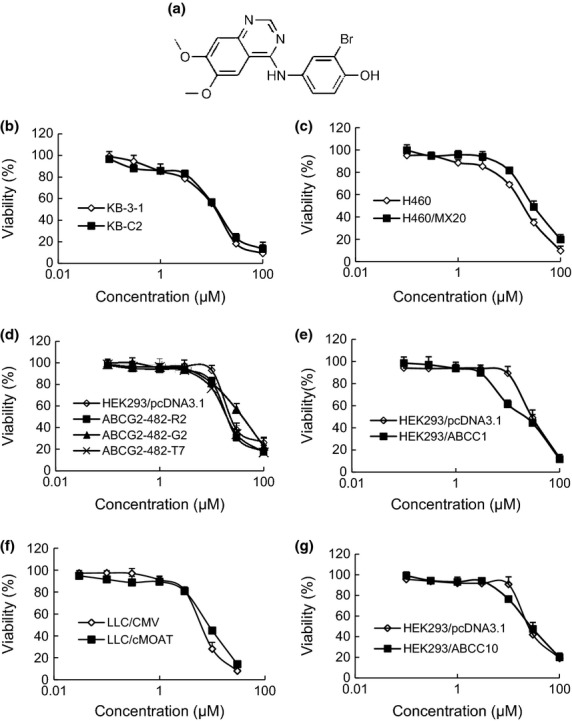
Cytotoxicity of WHI-P154 in the drug-resistant and parental sensitive cells. The chemical structure of WHI-P154 (a), MTT cytotoxicity assay was assessed in KB-3-1 and KB-C2 cells (b), H460 and H460/MX20 cells (c), HEK293/pcDNA3.1 and ABCG2 transfected cells (d), HEK293/pcDNA3.1 and HEK293/ABCC1 cells (e), HEK293/pcDNA3.1 and HEK293/ABCC10 cells (f), LLC/CMV and LLC/cMOAT cells (g). All the cells were exposed to various concentrations of WHI-P154 for 72 h. Each point represents the mean ± standard deviations (SDs) for three determinations. Each experiment was performed in three replicate wells.
Effect of WHI-P154 on cells overexpressing ABCG2
The ABCG2-overexpressing cells H460/MX20 showed much higher resistance to ABCG2 substrate chemotherapeutics than parental H460 cells (Table 1). WHI-P154 significantly sensitized H460/MX20 cells to the ABCG2 substrates, such as MX and SN-38. It also had a moderate effect on the parental H460 cells (1.6-fold). However, this effect was modest as compared to that of H460/MX20 cells. FTC is a well-known ABCG2 inhibitor and is used as a positive control of ABCG2. Moreover, the IC50 value of cisplatin, a non-ABCG2 substrate, was not affected when combined with WHI-P154. It has been reported that mutations at amino acid 482 in ABCG2 altered the substrate and antagonist specificity of ABCG2.(18,26) Therefore, we investigated whether WHI-P154 would reverse ABCG2-mediated resistance to MX in cells transfected with either the wild-type (Arg482) or mutant (Arg482Gly and Arg482Thr) forms of ABCG2. As shown in Table 2, WHI-P154 at nontoxic concentrations significantly enhanced the cytotoxic effect of MX and SN-38 in three ABCG2-transfected cells ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7 but not in HEK293/pcDNA3.1 cells. In addition, the single nucleotide polymorphism variant of ABCG2 (Phe489Leu) was reported to affect drug resistance toward its substrates.(27) To determine the reversal effect of WHI-P154 on the variant of ABCG2 (Phe489Leu), we tested the cytotoxicity of MX in the cells transfected with mutant ABCG2 (Phe489Leu) plasmid. Our results indicated that WHI-P154 can notably reverse mutant Phe489Leu ABCG2-mediated drug resistance (Table 3). These results suggested that WHI-P154 may enhance the cytotoxicity of ABCG2 substrates in cells expressing either wild-type or mutant ABCG2 (Arg482Gly/Thr or Phe489Leu).
Table 1.
WHI-P154 reverses ABCG2-mediated drug resistance in ABCG2-overexpressing cancer cells
| Treatments | IC50 ± SD† (nM) (Resistance fold) |
|
|---|---|---|
| H460 | H460/MX20 | |
| MX | 78.68 ± 4.19 (1.00)‡ | 2420.95 ± 72.19 (30.77) |
| +WHI-P154 (1 μM) | 77.94 ± 4.79 (0.99) | 690.64 ± 108.88 (8.78)* |
| +WHI-P154 (4 μM) | 54.65 ± 5.22 (0.69)* | 181.03 ± 21.06 (2.30)* |
| +FTC (2.5 μM) | 46.98 ± 6.03 (0.60)* | 82.96 ± 6.10 (1.05)* |
| SN-38 | 28.03 ± 3.49 (1.00)‡ | 2899.48 ± 289.60 (103.44) |
| +WHI-P154 (1 μM) | 20.14 ± 1.47 (0.72) | 473.50 ± 58.00 (16.89)* |
| +WHI-P154 (4 μM) | 16.71 ± 2.17 (0.60)* | 66.76 ± 7.31 (2.38)* |
| +FTC (2.5 μM) | 14.98 ± 0.62 (0.53)* | 38.27 ± 4.76 (1.37)* |
| Cisplatin | 1443.04 ± 163.44 (1.00)‡ | 2169.08 ± 237.06 (1.50) |
| +WHI-P154 (1 μM) | 1427.16 ± 121.94 (0.99) | 2089.10 ± 153.74 (1.45) |
| +WHI-P154 (4 μM) | 1373.42 ± 123.73 (0.95) | 2137.03 ± 240.22 (1.48) |
| +FTC (2.5 μM) | 1309.20 ± 147.27 (0.91) | 2047.32 ± 172.09 (1.42) |
P < 0.01 versus that obtained in the absence of WHI-P154.
Values represent mean ± SD of at least three independent experiments, each performed in triplicate.
Resistance fold was determined by dividing the IC50 values of substrate in H460/MX20 cells in the absence or presence of reversal agents, or H460 cells with reversal agents, by the IC50 of substrate H460 cells without reversal agents. The resistance fold for HEK293/pcDNA3.1 and its resistance cell was obtained in a similar manner.
Table 2.
WHI-P154 reverses the ABCG2-mediated drug resistance in transfected cells
| Treatments | IC50 ± SD† (nM) (Resistance fold) |
|||
|---|---|---|---|---|
| HEK293/pcDNA3.1 | ABCG2-482-R2 | ABCG2-482-G2 | ABCG2-482-T7 | |
| MX | 16.30 ± 1.49 (1.00)‡ | 185.38 ± 15.30 (11.37) | 205.55 ± 18.93 (12.61) | 247.07 ± 23.88 (15.16) |
| +WHI-P154 (1 μM) | 15.76 ± 1.59 (0.97) | 57.68 ± 6.08 (3.53)* | 45.12 ± 5.21 (2.77)* | 107.81 ± 11.60 (6.61)* |
| +WHI-P154 (4 μM) | 15.25 ± 1.66 (0.94) | 19.54 ± 1.70 (1.20)* | 18.91 ± 1.04 (1.16)* | 28.11 ± 3.21 (1.72)* |
| +FTC (2.5 μM) | 14.92 ± 1.32 (0.92) | 11.72 ± 0.71 (0.72)* | 13.71 ± 0.49 (0.84)* | 10.07 ± 1.03 (0.62)* |
| SN-38 | 7.15 ± 0.81 (1.00)‡ | 252.85 ± 20.19 (35.26) | 121.07 ± 10.92 (16.93) | 159.61 ± 10.22 (22.32) |
| +WHI-P154 (1 μM) | 8.27 ± 0.63 (1.16) | 90.62 ± 8.40 (12.67)* | 15.95 ± 2.03 (2.23)* | 40.78 ± 3.36 (5.70)* |
| +WHI-P154 (4 μM) | 7.44 ± 0.92 (1.03) | 17.46 ± 2.06 (2.44)* | 4.55 ± 0.38 (0.63)* | 10.60 ± 0.91 (1.48)* |
| +FTC (2.5 μM) | 7.73 ± 0.77 (1.08) | 10.24 ± 1.14 (1.43)* | 4.16 ± 0.55 (0.58)* | 7.00 ± 0.83 (0.98)* |
| Cisplatin | 2003.93 ± 224.99 (1.00)‡ | 1693.30 ± 180.43 (0.84) | 1631.22 ± 130.55 (0.81) | 1300.08 ± 90.22 (0.65) |
| +WHI-P154 (1 μM) | 1896.89 ± 177.47 (0.95) | 1651.81 ± 150.29 (0.82) | 1564.43 ± 201.93 (0.78) | 1258.31 ± 115.53 (0.63) |
| +WHI-P154 (4 μM) | 2158.71 ± 190.98 (1.08) | 1615.06 ± 90.20 (0.81) | 1494.24 ± 160.21 (0.75) | 1241.25 ± 108.94 (0.62) |
| +FTC (2.5 μM) | 2075.02 ± 210.61 (1.04) | 1538.93 ± 129.39 (0.77) | 1588.88 ± 120.34 (0.79) | 1236.91 ± 140.12 (0.62) |
P < 0.01 versus that obtained in the absence of WHI-P154.
Values represent mean ± SD of at least three independent experiments, each performed in triplicate.
Resistance fold was determined by dividing the IC50 values of substrate in resistant cells ABCG2-482-R2, ABCG2-482-G2 or ABCG2-482-T7 in the absence or presence of reversal agents, or HEK293/pcDNA3.1 cells with reversal agents, by the IC50 of substrate HEK293/pcDNA3.1 cells without reversal agents.
Table 3.
Effect of WHI-P154 on cells transfected with mutant ABCG2 (Phe489Leu)
| Treatments | IC50 ± SD† (nM) (Resistance fold) |
|
|---|---|---|
| HEK293/pcDNA3.1 | ABCG2-489-Leu | |
| MX | 18.22 ± 2.30 (1.00)‡ | 55.12 ± 6.02 (3.03) |
| +WHI-P154 (1 μM) | 19.30 ± 3.08 (1.06) | 32.70 ± 4.11 (1.79)* |
| +WHI-P154 (4 μM) | 16.26 ± 2.66 (0.89) | 22.03 ± 3.22 (1.21)* |
| +FTC (2.5 μM) | 16.90 ± 1.70 (0.93) | 20.12 ± 2.94 (1.10)* |
| Cisplatin | 2423.35 ± 290.10 (1.00) | 1737.74 ± 200.66 (0.72) |
| +WHI-P154 (1 μM) | 2268.60 ± 330.21 (0.94) | 1667.89 ± 190.40 (0.69) |
| +WHI-P154 (4 μM) | 2350.28 ± 150.65 (0.97) | 1857.01 ± 251.43 (0.77) |
| +FTC (2.5 μM) | 2205.04 ± 209.38 (0.91) | 1602.44 ± 220.30 (0.66) |
P < 0.01 versus that obtained in the absence of WHI-P154.
Values represent mean ± SD of at least three independent experiments, each performed in triplicate.
Resistance fold was determined by dividing the IC50 values of substrate in ABCG2-489-Leu cells in the absence or presence of reversal agents, or HEK293/pcDNA3.1cells with reversal agents, by the IC50 of substrate HEK293/pcDNA3.1 cells without reversal agents.
Effects of WHI-P154 on cells overexpressing ABCB1, ABCC1, ABCC2 and ABCC10
In order to confirm whether WHI-P154 could modulate the activity of ABCB1, we treated parental KB-3-1 and ABCB1-overexpressing KB-C2 cells with 4 μM WHI-P154 and used paclitaxel as a substrate. Our results indicated that WHI-P154 shows moderate inhibitory effect on ABCB1 (Table 4). Our results also showed that WHI-P154 could not reverse ABCC1-, ABCC2- or ABCC10- mediated MDR (Table 4). Overall, our data suggested that WHI-P154 mainly modulate ABCG2-mediated drug efflux.
Table 4.
The effect of WHI-P154 on cells overexpress ABCB1, ABCC1, ABCC2 or ABCC10
| Treatments | IC50 ± SD† (nM) (Resistance fold) |
|
|---|---|---|
| KB-3-1 | KB-C2 | |
| Paclitaxel | 0.62 ± 0.05 (1.00)‡ | 78.71 ± 8.20 (126.95) |
| +WHI-P154 (4 μM) | 0.68 ± 0.07 (1.10) | 34.52 ± 3.34 (55.68)* |
| +Verapamil (4 μM) | 0.65 ± 0.07 (1.05) | 2.46 ± 0.21 (3.97)* |
| HEK293/pcDNA3.1 | HEK293/ABCC1 | |
| Vincristine | 1.19 ± 0.14 (1.00)‡ | 20.48 ± 2.66 (17.47) |
| +WHI-P154 (4 μM) | 1.16 ± 0.08 (0.97) | 17.68 ± 2.35 (14.85) |
| +PAK-104P (2.5 μM) | 0.49 ± 0.06 (0.41)* | 2.81 ± 0.31 (2.35)* |
| LLC/CMV | LLC/cMOAT | |
| Vincristine | 214.65 ± 23.22 (1.00)‡ | 888.19 ± 100.93 (4.14) |
| +WHI-P154 (4 μM) | 256.42 ± 31.89 (1.19) | 714.98 ± 139.43 (3.33) |
| +PAK-104P (2.5 μM) | 211.75 ± 24.90 (0.99) | 253.14 ± 22.04 (1.18)* |
| HEK293/pcDNA3.1 | HEK293/ABCC10 | |
| Paclitaxel | 8.38 ± 1.66 (1.00)‡ | 77.80 ± 4.21 (9.28) |
| +WHI-P154 (4 μM) | 7.02 ± 1.19 (0.84) | 73.69 ± 8.65 (8.79) |
| +Cepharanthine (2.5 μM) | 6.46 ± 1.30 (0.77) | 6.53 ± 1.01 (0.78)* |
P < 0.01 versus that obtained in the absence of WHI-P154.
Values represent mean ± SD of at least three independent experiments, each performed in triplicate.
Resistance fold was determined by dividing the IC50 values of substrate in KB-C2 cells in the absence or presence of reversal agents, or KB-3-1 cells with reversal agents, by the IC50 of substrate KB-3-1 cells without reversal agents; the resistance fold for HEK293/pcDNA3.1, HEK293/ABCC1, LLC/cMOAT and HEK293/ABCC10 cells was obtained in the similar manner.
Effect of WHI-P154 on intracellular accumulation of [3H]-MX in ABCG2- overexpressing cells
To investigate the potential reversal mechanism of ABCG2-mediated MDR by WHI-P154, we determined the effect of WHI-P154 on the accumulation of [3H]-MX in ABCG2-overexpressing cells. The intracellular level of [3H]-MX was measured in the presence or absence of WHI-P154 in ABCG2-482-R2 and ABCG2-482-G2 cells. The intracellular accumulation of [3H]-MX was lower in the drug resistant cells than that in the parental HEK293/pcDNA3.1 cells. In the presence of WHI-P154 (1 and 4 μM) and FTC (2.5 μM), the intracellular level of [3H]-MX was significantly increased in ABCG2-overexpressing cells (Fig. 2a). However, neither WHI-P154 nor FTC increased the accumulation of [3H]-MX in HEK293/pcDNA3.1 cells (Fig. 2a).
Fig. 2.
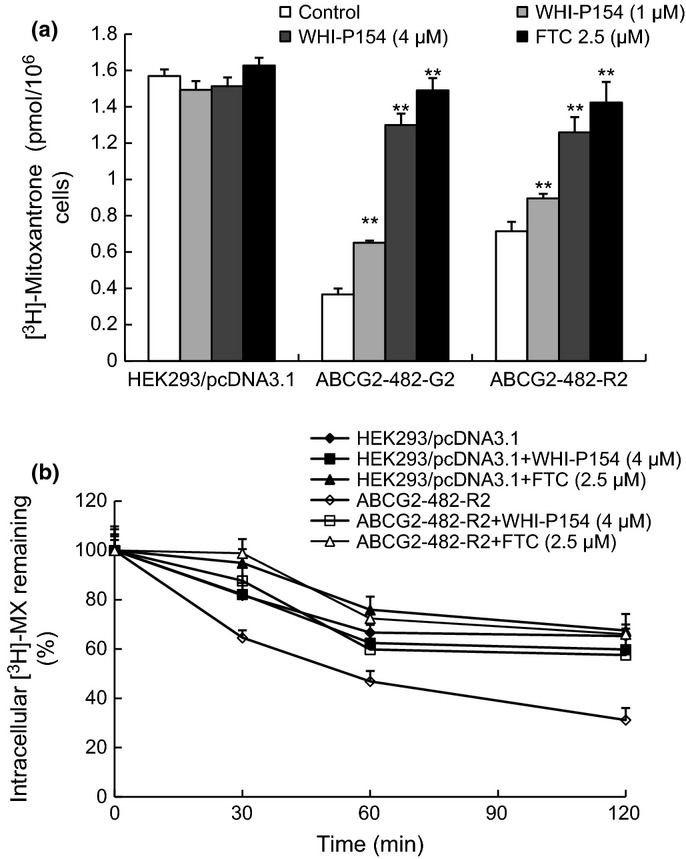
Effect of WHI-P154 on the accumulation and efflux of [3H]-MX. The accumulation of [3H]-MX was determined in both wild-type and mutant ABCG2-overexpressing ABCG2-482-R2 and ABCG2-482-G2 cells with or without WHI-P154. Columns are the mean of triplicate determinations; bars represent SD (a). *P < 0.05, **P < 0.01 versus the control group. The effects of WHI-P154 on the efflux of [3H]-MX from HEK293/pcDNA3.1 and ABCG2-482-R2 cells were measured as described in the “Materials and Methods” section (b). Data shown are means ± SD for independent determinations in triplicate.
Effect of WHI-P154 on efflux of [3H]-MX in ABCG2-overexpressing cells
In order to confirm the increased accumulation of [3H]-MX was due to inhibition of drug efflux, a time course study was performed to measure intracellular [3H]-MX levels in the presence or absence of WHI-P154. In the absence of WHI-P154, the intracellular [3H]-MX was less in ABCG2-482-R2 cells than the parental cells due to the active ABCG2 efflux. Treatments of 4 μM WHI-P154 in both cells, lead to the blocking of efflux function of ABCG2 thereby the intracellular accumulation of [3H]-MX increased at different time points (30, 60 and 120 min) (Fig. 2b). FTC, at a concentration of 2.5 μM, showed a similar effect. These results herein further validated the ability of WHI-P154 to inhibit the efflux activity of ABCG2.
Effect of WHI-P154 on the expression levels and localization of ABCG2
The reversal effect of ABCG2-mediated MDR could be achieved by either inhibiting ABCG2 function or decreasing the expression levels of ABCG2 protein. Therefore, Western blot analysis was performed to further ascertain the effect of WHI-P154 on the expression levels of ABCG2. Our results showed the expression levels of ABCG2 protein (Fig. 3) were not significantly altered in H460/MX20 cells after treatment with 4 μM WHI-P154 for 0, 24, 48 and 72 h, and the data also showed low-level expression of ABCG2 in parent H460 cells (Fig. 3a, c). The grayscale ratios of ABCG2/β-actin were proportional to the ABCG2 protein levels (Fig. 3c). Similarly, WHI-P154 did not significantly alter the expression levels of ABCG2 in ABCG2-482-R2 cells (Fig. 3b, d). In addition, we performed immunofluorescence to determine the effect of WHI-P154 on the cellular localization of ABCG2. As shown in Figure 3(e), WHI-P154 did not alter the localization of ABCG2. There results indicated that neither the expression levels nor localization of ABCG2 was significantly altered by WHI-P154.
Fig. 3.

The effect of WHI-P154 on the expression levels and localization of ABCG2, and on the blockade of STAT3, AKT and ERK1/2 phosphorylation in ABCG2-overexpressing cells. Western blot showed the effect of WHI-P154 on the expression level of ABCG2 after cells were treated with 4 μM of WHI-P154 for 0, 24, 48, and 72 h (a). Grayscale ratios of BCRP/β-actin were analyzed with Image J. The differences were statistically not significant (P > 0.05) (c). Similarly, WHI-P154 did not significantly alter the expression level of ABCG2 in ABCG2-482-R2 cells (b, d). Figure 3(a, c) representative result is shown from at least three independent experiments. Immunofluorescence staining showed membranous cellular localization of ABCG2 in H460/MX20 cells after treatment with WHI-P154 for 72 h. ABCG2 staining is shown in green. Fluorescent DAPI (blue) counterstains the nuclei (e). Western blot showed the effect of various concentrations of WHI-P154 on the phosphorylation of STAT3, AKT and ERK1/2 (f). Representative results are shown and similar results were obtained in two other independent trials.
Effects of WHI-P154 on the blockade of STAT3, AKT and ERK1/2 phosphorylation
WHI-P154 is a potent inhibitor of JAK3 and EGFR, which regulate two important downstream STAT3 and PI3K/AKT pathways. We treated H460/MX20 cells with various concentrations of WHI-P154 for 72 h. As shown in Figure 3(f), the phosphorylation of STAT3, AKT or ERK1/2 was not significantly decreased after treatment with low concentrations of WHI-P154 (1, 2 μM). In addition, WHI-P154 at 4 μM could slightly inhibit phosphorylation of STAT3, AKT or ERK1/2. The phosphorylation of STAT3, AKT and ERK1/2 was not affected by treatment with WHI-P154 at low concentrations that produced a significant reversal effect. These indicate that the blockade of JAK3 or EGFR signaling is unlikely to play a significant role in the sensitizing effect of WHI-P154 on the ABCG2-overexpressing cells.
Effect of WHI-P154 on the ATPase activity of ABCG2
The drug efflux function of ABCG2 is coupled with ATP hydrolysis, which is stimulated in the presence of ABCG2 substrates. To assess the effect of WHI-P154 on the ATPase activity of ABCG2, the membrane vesicles of High Five insect cells overexpressing ABCG2 were used in the presence of various concentrations of WHI-P154 under conditions that suppressed the activity of other major membrane ATPases. As shown in Figure 4, WHI-P154 produced a significantly stimulation of ABCG2 ATPase activity at low concentrations (<10 μM). The results suggested that WHI-P54 may be a substrate for ABCG2.
Fig. 4.
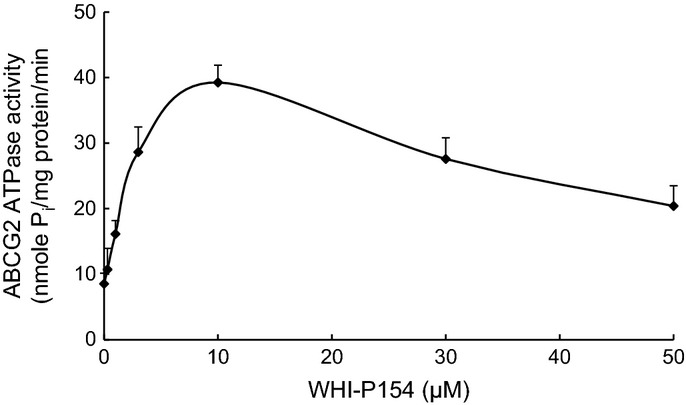
Effect of WHI-P154 on the ATPase activity of ABCG2. Crude membranes (10–20 μg protein/reaction) from High-five cells expressing ABCG2 were incubated with increasing concentrations of WHI-P154 (0–50 μM), in the presence and absence of 10 mM vanadate, in ATPase assay buffer as described in Materials and Methods. The mean values are plotted and error bars depict SD (n = 3).
WHI-P154 docking analysis with human ABCG2 homology models
The Glide predicted docked model of WHI-P154 at Arg482 centroid-based grid of ABCG2 is shown in Figure 5. The aniline ring of WHI-P154 formed hydrophobic interactions with the side chains of Phe489, Ile573, and Pro574. The hydroxyl substituent of the aniline ring may form a hydrogen bond with the side chain of Tyr570 (HO···HO-Tyr570, 2.9 Å). This hydroxyl group was involved in hydrogen bonding interaction with the side chains of Ser486 (OH···OH-Ser486, 1.8 Å). The bromine atom present on the aniline ring entered into electrostatic interaction with the hydroxyl group of Tyr464 (Br···HO-Tyr464, 2.9 Å). The 6,7-dimethoxyquinazoline ring was stabilized into a large hydrophobic cavity formed by Phe489, Phe507, Phe511, Ala580, Leu581, Trp627 and Val631. The oxygen atom of the 7-methoxy group formed electrostatic contacts with the imidazole ring NH of His630 (CH3O···HN-His630, 4.1 Å) and side chain amino group of Asn629 (CH3O···H2N-Asn629, 4.0 Å). The N1 of the quinazoline ring was stabilized by electrostatic interaction with the side chain -NH of His630 (-N1···HN-His630, 3.7 Å).
Fig. 5.
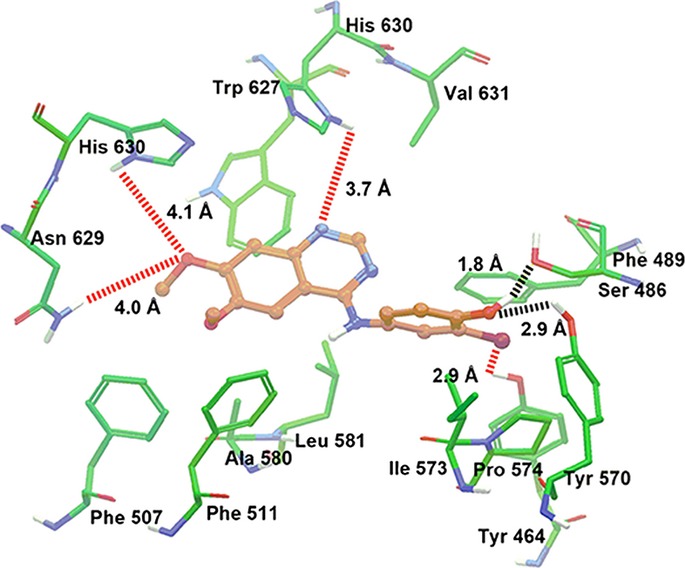
XP Glide predicted binding mode of WHI-P154 with homology modeled ABCG2. The docked conformation of WHI-P154 as ball and stick model is shown within the large cavity of ABCG2. Important amino acids are depicted as sticks with the atoms colored as carbon–green, hydrogen–white, nitrogen–blue, oxygen–red, whereas WHI-P154 is shown with the same color scheme as above except carbon atoms are represented in orange. Dotted black lines indicate hydrogen-bonding interactions, whereas dotted red lines indicate electrostatic interactions.
Discussion
ABCG2 is a known molecular cause of MDR in various cancer cells.(26,28) Therefore, strategies to block ABCG2-mediated active efflux may provide a therapeutic benefit for overcoming MDR. WHI-P154, a potent inhibitor of JAK3 and EGFR, displays antitumor activity by directly inhibiting tumor cell proliferation and survival, causing apoptotic cell death at low concentrations.(29) WHI-P154 was proven to be an effective agent against acute lymphoblastic leukemia, and it inhibited glioblastoma cell adhesion and migration in the context of ECM.(30)– (32)
Here we reported for the first time that WHI-P154 could specifically inhibit the function of ABCG2 transporter in ABCG2 overexpressing cells. As demonstrated by MTT assay, WHI-P154 enhanced the cytotoxicity of MX and SN-38 in ABCG2-overexpressing cells, and also moderately increased the cytotoxicity of these two drugs in H460 cells with low level of ABCG2 as seen in Figure 3(a,c). As reported that amino acid at position 482 is a hot spot for mutation in ABCG2,(18) Arg482 to mutant Gly482 or Thr482 may result in differences in substrate specificities as well as the efficacy of ABCG2 modulators. Our results further validated that WHI-P154 significantly increased the cytotoxicity of anticancer drugs in both wild-type and mutant ABCG2 transfected cells. In addition, we tested the effect of WHI-P154 on the mutant ABCG2 (Phe489Leu). As shown in Table 3, WHI-P154 could significantly enhance the cytotoxicity of MX in the cells transfected with mutant ABCG2 (Phe489Leu). Additionally, WHI-P154 also moderately increased the sensitivity of ABCB1-overexpressing cells to its substrate paclitaxel. In contrast, it had no effect on ABCC1-, ABCC2-, or ABCC10-mediated resistance. These data indicated that WHI-P154 acted as a potent inhibitor of ABCG2.
To investigate the mechanism of the reversal effect, we examined the effects of WHI-P154 via the accumulation assay of [3H]-MX. Our results showed that WHI-P154 could significantly enhance the intracellular concentration of [3H]-MX in ABCG2-482-R2 cells and ABCG2-482-G2 cells, but had no effect on the parental cells. This was confirmed by a time course efflux study of [3H]-MX cellular levels where WHI-P154 at 4 μM significantly reduced the efflux of [3H]-MX from ABCG2-482-R2 cells. Moreover, our results showed that WHI-154 did not alter the expression level or the cellular localization of ABCG2 protein in MDR cells. These results collectively indicated that WHI-P154 could inhibit the transport function of ABCG2, thereby increasing the intracellular accumulation of its substrates.
A previous report showed that the phosphorylation of AKT and ERK1/2 pathways might be related to the sensitivity of chemotherapeutic agents in cancer cells.(33,34) In addition, JAK3 was involved in the downstream activation of STAT3, which often correlated with chemotherapy drug resistance.(35) However, low concentrations of WHI-P154 (1, 2 μM) did not block the phosphorylation of STAT3, AKT or ERK1/2 (Fig. 3f) in H460/MX20 cells. WHI-P154 at 4 μM could moderately inhibit the phosphorylation of STAT3, AKT and ERK1/2). The phosphorylation of STAT3, AKT and ERK1/2 was not affected by treatment with low concentrations of WHI-P154. Therefore, the antagonism of JAK3 or EGFR receptors is unlikely to play a significant role in the sensitizing effect of WHI-P154 in ABCG2-overexpressing cells.
ABC transporters use the binding and hydrolysis of ATP to power the translocation of diverse substrates, thus ATPase activity is directly proportional to the transport activity.(36) In our previous studies, several reversal agents such as apatinib, lapatinib, erlotinib could stimulate ATPase activity of ABCG2 at extremely low concentrations.(10,11,24,37) Thus we investigated the interaction of WHI-P154 with ABCG2 by ATPase assay. Our results showed that WHI-P154 could stimulate the ABCG2 ATPase activity at low concentrations. These results suggested that WHI-P154 interact directly with ABCG2 and may be a substrate of the transporter.
To identify the molecular binding of WHI-P154 with ABCG2 transporter, we performed docking studies at various sites of human ABCG2. WHI-P154 showed QikProp value of 3.2 and then its inhibition activity on ABCG2 might be explained based on its ability to distribute with biomembrane from which ABCG2 extracts it.(38) Moreover, WHI-P154 appeared to exhibit all of the pharmacophoric features such as hydrophobic groups, aromatic ring centers, hydrogen bond donors and acceptors that had been identified as critical for ABCG2 inhibition.(39) Overall, this docking model of WHI-P154 will form the basis for further optimization of dimethoxyquinazoline derivatives as inhibitors of ABCG2.
In conclusion, here we report for the first time that WHI-P154 could significantly reverse wild-type and mutant ABCG2-mediated MDR by inhibiting the efflux function without affecting expression level or localization of ABCG2. Further studies are warranted to confirm whether WHI-P154 could contribute to improving clinical outcomes in patients receiving chemotherapy.
Acknowledgments
This work was supported by St. John's University Research Seed grant (No. 579-1110-7002) to Z.S. Chen, the Major science and technology project of the National National Natural Sciences Foundation of China grant (No. 81072669 and No. 81061160507) for L.W. Fu, and Sun Yat-Sen University Ph. D. visiting scholar abroad program and international collaborative research project of Guangdong Province. We thank Dr Shinichi Akiyama (Kagoshima University, Japan) for the LLC/CMV, LLC/cMOAT, KB-3-1 and KB-C2 cell lines; Drs Susan E. Bates and Robert W. Robey (NIH, Bethesda, MD) for FTC, NCI-H460, NCI-H460/MX20, HEK293/pcDNA3.1 and the ABCG2 transfectant cell lines; the late Dr Gary D. Kruh for the the MRP7 cDNA (University of Illinois, Chicago); Dr Mark F. Rosenberg (University of Manchester, Manchester, UK) and Dr Zsolt Bikádi (Virtua Drug, Budapest, Hungary) for providing coordinates of ABCG2 homology model. We thank Selleck Chemicals for supplying us free WHI-P154 sample.
HZ, YKZ, YJW, RJ.K, KS, HZ performed research and analyzed data. HZ, AP, TTT, SVA wrote the paper. ZSC and LWF designed the research and revised the paper.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1 Basic sequencing to confirm target mutation, the sequence alignment of wild type ABCG2 and mutant ABCG2 (Phe489Leu), TAA is a stop codon.
Fig. S2 Western blot analysis of ABCG2 (Phe489Leu) in HEK293 cells transfected with pcDNA3.1 plasmids, pcDNA3.1-ABCG2 (Phe489Leu) plasmids, β-actin was used as equal loading control.
Table S1 Binding energies of WHI P154 within each of the predicted binding sites of ABCG2.
References
- 1.Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv. 2004;1:27–42. doi: 10.2174/1567201043480036. [DOI] [PubMed] [Google Scholar]
- 2.Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat. 2003;6:71–84. doi: 10.1016/s1368-7646(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 3.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–17. [PubMed] [Google Scholar]
- 4.Fardel O, Lecureur V, Guillouzo A. The P-glycoprotein multidrug transporter. Gen Pharmacol. 1996;27:1283–91. doi: 10.1016/s0306-3623(96)00081-x. [DOI] [PubMed] [Google Scholar]
- 5.Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007;26:39–57. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- 6.Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 7.Morrow CS, Peklak-Scott C, Bishwokarma B, Kute TE, Smitherman PK, Townsend AJ. Multidrug resistance protein 1 (MRP1, ABCC1) mediates resistance to mitoxantrone via glutathione-dependent drug efflux. Mol Pharmacol. 2006;69:1499–505. doi: 10.1124/mol.105.017988. [DOI] [PubMed] [Google Scholar]
- 8.Chen ZS, Tiwari AK. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011;278:3226–45. doi: 10.1111/j.1742-4658.2011.08235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen ZS, Hopper-Borge E, Belinsky MG, Shchaveleva I, Kotova E, Kruh GD. Characterization of the transport properties of human multidrug resistance protein 7 (MRP7, ABCC10) Mol Pharmacol. 2003;63:351–8. doi: 10.1124/mol.63.2.351. [DOI] [PubMed] [Google Scholar]
- 10.Dai CL, Tiwari AK, Wu CP, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–14. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Z, Peng XX, Kim IW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–20. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- 12.Zheng LS, Wang F, Li YH, et al. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS ONE. 2009;4:e5172. doi: 10.1371/journal.pone.0005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng W, Dai CL, Chen JJ, et al. Tandutinib (MLN518) reverses multidrug resistance by inhibiting the efflux activity of the multidrug resistance protein 7 (ABCC10) Oncol Rep. 2013;29:2479–85. doi: 10.3892/or.2013.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marzec M, Kasprzycka M, Ptasznik A, et al. Inhibition of ALK enzymatic activity in T-cell lymphoma cells induces apoptosis and suppresses proliferation and STAT3 phosphorylation independently of Jak3. Lab Invest. 2005;85:1544–54. doi: 10.1038/labinvest.3700348. [DOI] [PubMed] [Google Scholar]
- 15.Sudbeck EA, Ghosh S, Liu XP, Zheng Y, Myers DE, Uckun FM. Tyrosine kinase inhibitors against EGF receptor-positive malignancies. Methods Mol Biol. 2001;166:193–218. doi: 10.1385/1-59259-114-0:193. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh S, Jennissen JD, Liu XP, Uckun FM. 4-[3-Bromo-4-hydroxyphenyl)amino]-6,7-dimethoxyquinazolin-1-ium chloride methanol solvate and 4-[(3-hydroxyphenyl)amino]-6,7-dimethoxy-1-quinazolinium chloride. Acta Crystallogr C. 2001;57:76–8. doi: 10.1107/s0108270100013561. [DOI] [PubMed] [Google Scholar]
- 17.Henrich CJ, Bokesch HR, Dean M, et al. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J Biomol Screen. 2006;11:176–83. doi: 10.1177/1087057105284576. [DOI] [PubMed] [Google Scholar]
- 18.Robey RW, Honjo Y, Morisaki K, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–8. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller M, Yong M, Peng XH, Petre B, Arora S, Ambudkar SV. Evidence for the role of glycosylation in accessibility of the extracellular domains of human MRP1 (ABCC1) Biochemistry. 2002;41:10123–32. doi: 10.1021/bi026075s. [DOI] [PubMed] [Google Scholar]
- 20.Akiyama S, Fojo A, Hanover JA, Pastan I, Gottesman MM. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somat Cell Mol Genet. 1985;11:117–26. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- 21.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–14. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 22.Tiwari AK, Sodani K, Dai CL, et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013;328:307–17. doi: 10.1016/j.canlet.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Kathawala RJ, Wang YJ, et al. Linsitinib (OSI-906) antagonizes ATP-binding cassette subfamily G member 2 and subfamily C member 10-mediated drug resistance. Int J Biochem Cell Biol. 2014;51:111–1119. doi: 10.1016/j.biocel.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mi YJ, Liang YJ, Huang HB, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010;70:7981–91. doi: 10.1158/0008-5472.CAN-10-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun YL, Kathawala RJ, Singh S, et al. Zafirlukast antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Anticancer Drugs. 2012;23:865–73. doi: 10.1097/CAD.0b013e328354a196. [DOI] [PubMed] [Google Scholar]
- 26.Honjo Y, Hrycyna CA, Yan QW, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–9. [PubMed] [Google Scholar]
- 27.Tamura A, Wakabayashi K, Onishi Y, et al. Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 2007;98:231–9. doi: 10.1111/j.1349-7006.2006.00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Herwaarden AE, Jonker JW, Wagenaar E, et al. The breast cancer resistance protein (Bcrp1/Abcg2) restricts exposure to the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Res. 2003;63:6447–52. [PubMed] [Google Scholar]
- 29.Narla RK, Liu XP, Myers DE, Uckun FM. 4-(3′-Bromo-4′hydroxylphenyl)-amino-6,7-dimethoxyquinazoline: a novel quinazoline derivative with potent cytotoxic activity against human glioblastoma cells. Clin Cancer Res. 1998;4:1405–14. [PubMed] [Google Scholar]
- 30.Sudbeck EA, Liu XP, Narla RK, et al. Structure-based design of specific inhibitors of Janus kinase 3 as apoptosis-inducing antileukemic agents. Clin Cancer Res. 1999;5:1569–82. [PubMed] [Google Scholar]
- 31.Wu W, Sun XH. Janus kinase 3: the controller and the controlled. Acta Biochim Biophys Sin (Shanghai) 2012;44:187–96. doi: 10.1093/abbs/gmr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narla RK, Liu XP, Klis D, Uckun FM. Inhibition of human glioblastoma cell adhesion and invasion by 4-(4′-hydroxylphenyl)-amino-6,7-dimethoxyquinazoline (WHI-P131) and 4-(3′-bromo-4′-hydroxylphenyl)-amino-6,7-dimethoxyquinazoline (WHI-P154) Clin Cancer Res. 1998;4:2463–71. [PubMed] [Google Scholar]
- 33.Oshiro MM, Landowski TH, Catlett-Falcone R, et al. Inhibition of JAK kinase activity enhances Fas-mediated apoptosis but reduces cytotoxic activity of topoisomerase II inhibitors in U266 myeloma cells. Clin Cancer Res. 2001;7:4262–71. [PubMed] [Google Scholar]
- 34.Knuefermann C, Lu Y, Liu B, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–12. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- 35.Fang B. Genetic interactions of STAT3 and anticancer drug development. Cancers (Basel) 2014;6:494–525. doi: 10.3390/cancers6010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–27. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Z, Tiwari AK, Shukla S, et al. Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011;71:3029–41. doi: 10.1158/0008-5472.CAN-10-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klepsch F, Jabeen I, Chiba P, Ecker GF. Pharmacoinformatic approaches to design natural product type ligands of ABC-transporters. Curr Pharm Des. 2010;16:1742–52. doi: 10.2174/138161210791163992. [DOI] [PubMed] [Google Scholar]
- 39.Nicolle E, Boumendjel A, Macalou S, et al. QSAR analysis and molecular modeling of ABCG2-specific inhibitors. Adv Drug Deliv Rev. 2009;61:34–46. doi: 10.1016/j.addr.2008.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Basic sequencing to confirm target mutation, the sequence alignment of wild type ABCG2 and mutant ABCG2 (Phe489Leu), TAA is a stop codon.
Fig. S2 Western blot analysis of ABCG2 (Phe489Leu) in HEK293 cells transfected with pcDNA3.1 plasmids, pcDNA3.1-ABCG2 (Phe489Leu) plasmids, β-actin was used as equal loading control.
Table S1 Binding energies of WHI P154 within each of the predicted binding sites of ABCG2.