

Abstract
The c-MET receptor tyrosine kinase is the receptor for hepatocyte growth factor. Recently, activation of the c-MET/hepatocyte growth factor signaling pathway was associated with poor prognosis in various solid tumors and was one of the mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitor, gefitinib. But the link between c-MET activation and the cytotoxic anticancer drug has not been fully examined. Here, we found that the enhanced expression and activation of c-MET in cytotoxic anticancer agent-resistant small-cell lung cancer cells. Downregulation of c-MET expression by siRNA against the c-MET gene or inhibition of c-MET activation by SU11274, a c-MET inhibitor, in the resistant cells altered resistance to the cytotoxic anticancer agent. These results indicated that c-MET overexpression might play an important role in acquired resistance to cytotoxic anticancer drugs. Furthermore, the number of c-MET gene loci was increased in the resistant cells compared to the parental cells. In conclusion, increased c-Met expression through an increase in the number of c-MET gene loci is one of the mechanisms of acquired resistance to cytotoxic anticancer drugs. Our results add a new strategy, the targeting of c-MET, for overcoming resistance to cytotoxic agents in small-cell lung cancer.
Keywords: c-MET inhibitor, c-MET overexpression, drug resistance, gene amplification, lung cancer
Approximately 12% of all lung cancer patients have SCLC, of whom 75% are staged as having ED at the time of diagnosis.(1,2) Several therapeutic agents have been tested during the last three decades in ED-SCLC. With the first-line chemotherapy, usually etoposide or irinotecan plus platinum compound, response rates are 70–80% and median survival time is 9–12 months.(3,4) Topotecan or amrubicin are active agents for the treatment of refractory and relapsed SCLC.(5,6) But the prognosis of ED-SCLC patients is very poor and less than 5% of them remain alive at 2 years.(2) The problem is the rapid development of drug resistance, which results in the failure of the first- or second-line therapy. Therefore, new therapeutic strategies to overcome the drug resistance are urgently needed.
The c-MET receptor tyrosine kinase is the receptor for HGF.(7) The extracellular sema domain of c-MET mediates binding to HGF, resulting in stimulating phosphorylation of Y1234/1235 and activating c-MET's autocatalytic domain.(8) Activation of c-MET carboxy-terminal binding domain (Y1349/1356) might stimulate many downstream genes in the c-MET pathways, including members of the matrix metalloproteinase family, plasminogen activator, and integrins, all associated with an invasive growth phenotype.(9)– (11) Missense germline mutations in the tyrosine kinase domain lead to constitutive activation of the c-MET protein in hereditary papillary renal carcinomas.(12) Activation of the HGF/c-MET signaling pathway was associated with poor prognosis in various solid tumors.(13)– (16) Therefore, agents targeting HGF/c-MET signaling have been developed, which inhibit downstream signaling and biological events typical to c-MET activity, such as oncogenesis, cancer metastasis, and drug resistance.(17)
Recently, activation of HGF/c-MET signaling was reported to be a new mechanism of acquired resistance to gefitinib (EGFR-TKI). Amplification of the c-MET gene leads to gefitinib resistance by transactivation of ERBB3.(18) Hepatocyte growth factor-mediated c-MET activation was also a novel mechanism of gefitinib resistance in lung adenocarcinoma with EGFR-activating mutations.(19) However, it was not fully clarified whether there were fundamental linkages between HGF/c-MET signaling activation and resistance to the cytotoxic anticancer drugs. c-MET receptor activation by scatter factor/HGF protects certain glioblastoma cells from DNA-damaging agents by activating PI3K-dependent and AKT-dependent antiapoptotic pathways.(20) In addition, HGF induced cisplatin resistance through c-MET to activate FAK and downregulate apoptosis-inducing factor expression in lung cancer cells.(21) However, HGF-secreting cells did not show altered proliferation rates or survival but were strongly sensitized to death triggered by CDDP and TXL in ovarian cancer.(22) c-MET overexpression increased the sensitivity to SN-38, compared through upregulation of topo I activities resulting from increased topo I mRNA and protein expression in non-SLCL.(23)
We here found that levels of c-MET expression were significantly increased in cytotoxic anticancer drug-resistant lung cancer cells. These data prompted us to determine whether THE HGF/c-MET signaling pathway has an important role in acquired resistance to cytotoxic anticancer agents. Therefore, we examined the significance of c-MET overexpression in drug-resistant cells.
Materials and Methods
Cell lines and chemicals
We used the SN-38-, TXL-, and CDDP-resistant cell lines PC-6/SN-38, PC-6/TXL, and PC-6/CDDP that were derived from the human SCLC cell line PC-6.(20,21) The human SCLC cell lines NCI-H69 and cells from the TXL-resistant human lung SCLC cell lines NCI-H69/TXL were used as described previously.(24) PC-6/SN-38 cells were approximately 4500-fold more resistant to SN-38, PC-6/TXL and NCI-H69/TXL cells were approximately 460-fold and 460-fold more resistant to TXL, respectively, and PC-6/CDDP cells were approximately 1800-fold more resistant to CDDP than each parental cell line (Table 1). SU11274 was purchased from Calbiochem (Darmstadt, Germany), SN-38 from Daiichi-Sankyo (Tokyo, Japan), and CDDP and TXL from Bristol Myers (Tokyo, Japan).
Table 1.
Inhibitory concentrations (50%) of 7-ethyl-10-hydroxycamptothesin (SN-38), paclitaxel (TXL), and cisplatin (CDDP) in PC-6 and NCI-H69 small-cell lung cancer cells
| PC-6 | PC-6/SN-38 | RR | |
| SN-38 | 0.98 pM | 4.48 nM | 4571.42 |
| PC-6 | PC-6/TXL | RR | |
| TXL | 23.75 pM | 11.05 nM | 465.26 |
| PC-6 | PC-6/CDDP | RR | |
| CDDP | 8.34 nM | 15.02 μM | 1800.95 |
| NCI-H69 | NCI-H69/TXL | RR | |
| TXL | 0.028 pM | 13.12 pM | 468.57 |
RR, relative rate.
Quantitative real-time PCR
Total RNA was extracted using an RNeasy mini kit (Qiagen, Chatsworth, CA, USA). Quantitative real-time PCR was carried out with a TaqMan One-Step RT-PCR Master Mix Reagents kit (Applied Biosystems, Foster City, CA, USA) using the Step One Plus Real Time PCR System (Applied Biosystems) according to the manufacturer's instructions. The PCR program was carried out as described previously.(22) The primer and TaqMan probe sets (TaqMan Gene Expression Assays, Inventoried) for c-MET (Hs00179845_m1), HGF (Hs0030159_m1), and GAPDH (Hs99999905_m1) were purchased from Applied Biosystems (sequences not disclosed).
Protein extraction and WB analysis
Protein extraction was carried out as described previously.(25) Lysates were electrophoresed on 10% Ready Gel Tris-HCl Gel (Bio-Rad Laboratories, Hercules, CA, USA) and transferred to Immobilon-P filters (Millipore, Billerica, MA, USA). The filters were first incubated with primary antibodies for 2 h (c-MET, p-MET, p-AKT, p-ERK1/2, and cleaved-PARP) and 1 h (α-tubulin) at room temperature and then with HRP-conjugated secondary antibodies. The following antibodies were used: c-MET, p-MET (Tyr1234/1235), p-AKT (se473), p-ERK1/2 (Cell Signaling Technology, Danvers, MA, USA), cleaved-PARP (BD PharMingen, San Jose, CA, USA), α-tubulin (Sigma-Aldrich, St. Louis, MO, USA) and HRP-conjugated secondary antibody (GE Healthcare Bioscience, Little Chalfont, UK). α-Tubulin was used as a loading control. The relative band intensities were determined by densitometry using NIH Image (U. S. National Institutes of Health, Bethesda, MD, USA).
Cell viability assay
Cells were cultured at 5000 per well in 96-well tissue culture plates. To assess cell viability, stepwise 10-fold dilutions of the anticancer drug were added 2 h after plating, and the cultures were incubated at 37°C for 96 h. At the end of the culture period, 20 μL MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) solution (CellTiter 96 Aqueous One Solution Cell Proliferation Assay; Promega, Madison, WI, USA) was added, the cells were incubated for a further 4 h, and the absorbance was measured at 490 nm using an ELISA plate reader. Chemosensitivity is expressed as the drug concentration producing 50% growth inhibition.
Inhibition of c-MET activation by SU11274
At 2 h after cells (1 × 106) were exposed to DMSO or SU11274 at 0.5 or 2.0 μM concentration, total protein was extracted, or the cells were cultured at 5000 per well in 96-well tissue culture plates for 2 h, and the cultures were incubated at 37°C for 72 h after adding stepwise dilutions of SN-38 or TXL to assess cell viability by the MTS assay.
Apoptosis
Cells (1 × 106) were cultured for 2 h after exposure to DMSO or SU11274 at 2.0 μM concentration, the cells were incubated at 37°C for 72 h after adding stepwise dilutions of SN-38 or TXL, and total protein was extracted. We examined the levels of cleaved-PARP expression by WB.
Transfection and siRNA experiments
Cells (1 × 106) were transfected with siRNA oligonucleotides to result in a final RNA concentration of 30 nM. We used using siPORT NeoFX Transfection Agent (Ambion, Austin, TX, USA) according to the manufacturer's instructions. Total RNA or protein at 24 or 48 h after transfection was extracted, or the cells were cultured at 5000 per well in 96-well tissue culture plates for 2 h, and the cultures were incubated at 37°C for 72 h after adding stepwise dilutions of SN-38 or TXL to assess cell viability by MTS assay. The siRNA oligonucleotides for c-MET (silencer select siRNA, ID s8701) and the negative control siRNA (silencer select siRNA) were purchased from Ambion.
c-MET copy number assays
The c-MET gene copy number was analyzed by quantitative real-time PCR, carried out on StepOnePlus (Applied Biosystems) by TaqMan Copy Number Assays (Applied Biosystems), as described previously.(26)– (28) The PCR program was 40 cycles at 95°C for 15 s and 60°C for 1 min. The primer for c-MET (predesigned copy number assays ID, Hs 01432482_cn) was purchased from Applied Biosystems. We used the ribonuclease P RNA component H1 gene as an endogenous control.
Fluorescence in situ hybridization
The c-MET probe was labeled with Cy3 by the nick translation method using the RP11-163C9 BAC clone (Chromosome Science Labo, Sapporo, Japan). We used the chromosome 7 centromere probe (CEP7), manufactured by Chromosome Science Labo, as a control. Cells were collected by centrifugation and trypsinization, fixed with methanol and acetic acid (3:1 solution), and expanded on a slide glass. The probe mixture (c-MET and CEP7) was applied to the fixed cell specimens, which were denatured on a hot plate at 70°C for 5 min and hybridized overnight at 37°C. One hundred cells from each fixed cell specimen were analyzed and the number of c-MET and CEP7 signals were determined.
Hepatocyte growth factor immunoassay
A total of 1 × 106 cells were seeded on 60-mm dishes. The next day, cells were washed twice with PBS and incubated for 48 h with 4 mL Roswell Park Memorial Institute medium (RPMI) medium. Cell culture supernatants were then collected and cleared by centrifugation. The secretion of HGF was quantified by quantitative ELISA according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
The differences between samples were evaluated with Student's unpaired t-test. The level of significance was set at 5% using two-sided analysis.
Results
Levels of c-MET expression increased in drug-resistant SCLC cells
The levels of c-MET gene expression in PC-6/SN-38, PC-6/CDDP, PC-6/TXL, and NCI-H69/TXL cells were significantly increased relative to the parental cells (Fig. 1a). c-MET protein expression in the drug-resistant cells was also upregulated (Fig. 1b). To investigate activation of c-MET in the drug-resistant cells, we confirmed the levels of p-MET protein. The expression levels of p-MET in these resistant cells were also significantly increased relative to the parental cells (Fig. 1b,c).
Fig. 1.
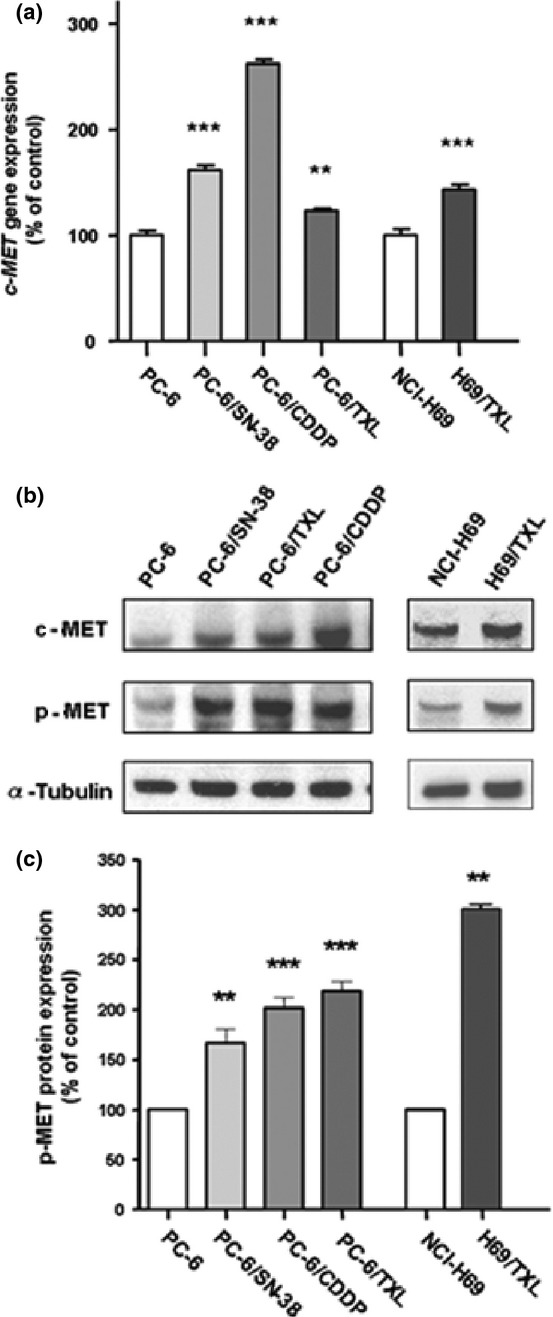
Increased c-MET expression and activation in PC-6 human small-cell lung cancer cells resistant to 7-ethyl-10-hydroxycamptothesin (PC-6/SN-38), paclitaxel (PC-6/TXL), and cisplatin (PC-6/CDDP), as well as the NCI-H69 and TXL-resistant NCI-H69 (NCI-H69/TXL) small-cell lung cancer cell lines. (a) c-MET gene expression was examined using quantitative real-time PCR. (b) c-MET and p-MET protein expression was determined by Western blot analysis. (c) Relative band intensities of p-MET protein were determined by densitometry using NIH Imaging. **P < 0.01.***P < 0.001.
Inhibition of c-MET activation by c-MET inhibitor (SU11274) improved resistance to cytotoxic anticancer drugs
We examined the growth inhibition of PC-6, PC-6/SN-38, and PC-6/TXL cells by SU11274, a specific c-MET TKI.(15,29,30) SU11274 significantly inhibited the growth of the drug-resistant cells relative to the parental cells (Fig. 2a). Next, we exposed PC-6/SN-38 cells to SN-38 alone, or in combination with DMSO or SU11274 (0.5 or 2 μM concentrations). Although PC-6/SN-38 cells were resistant to SN-38 alone or in combination with DMSO, the combined treatment of SN-38 with SU11274 resulted in alteration of cytotoxicity in a dose-dependent manner (Fig. 2b). We obtained the same results in PC-6/TXL cells (Fig. 2b). We then examined downstream in the c-MET pathway. SU11274 inhibited p-MET and p-ERK1/2 in PC-6/SN-38 and PC-6/TXL cells, but not p-AKT at the 2 μM concentration (Fig. 2c). We confirmed the protein levels of cleaved-PARP in PC-6, PC-6/SN-38, and PC-6/TXL cells. The levels of cleaved-PARP protein by treatment with SN-38 and SU11274 were increased relative to that by treatment with SN-38 alone in PC-6/SN-38 cells (Fig. 2d). The cleaved-PARP levels in PC-6/SN38 cells with SU11274 alone were higher than PC6 cells (Fig. 2d). We obtained the same results in PC-6/TXL cells (Fig. 2d).
Fig. 2.
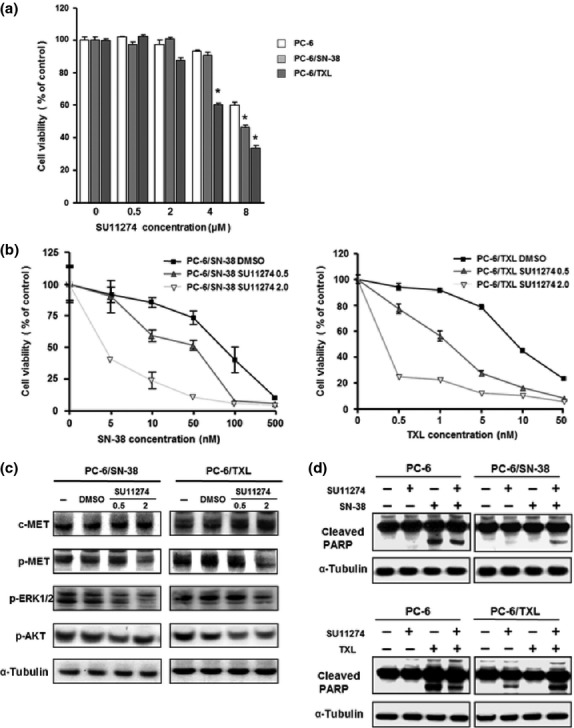
Inhibition of c-MET activation by SU11274. Growth inhibition induced by DMSO or SU11274 (0.5, 2.0, 4.0, and 8.0 μM) in small-cell lung cancer PC-6 (□), 7-ethyl-10-hydroxycamptothesin-resistant (PC-6/SN-38) (▪) and paclitaxel-resistant (PC-6/TXL) (▪) cell lines was examined with the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) assay (a). The growth inhibition of SN-38 or TXL to PC-6/SN-38 or PC-6/TXL was examined with the MTS assay (b), the protein levels of c-MET, p-MET, p-ERK1/2, and p-AKT in PC-6/SN-38 and PC-6/TXL were examined (c), and at 72 h after adding none, SN-38 (5 nM), or TXL (10 nM), the levels of cleaved poly(ADP-ribose) polymerase (cleaved-PARP) were examined by Western blotting (d). *P < 0.05
Downregulation of c-MET expression by siRNA against c-MET gene
We knocked down c-MET expression by siRNA against the c-MET gene, to confirm whether c-MET overexpression activated c-MET signaling resulting in resistance to the cytotoxic anticancer drugs. The levels of c-MET gene expression in PC-6/SN-38 or PC-6/TXL cells transfected with siRNA against the c-MET gene were significantly decreased by 30% or 35% relative to the cells with negative-control (Fig. 3a). The levels of c-MET and p-MET protein in PC-6/SN-38 or PC-6/TXL cells with siRNA against the c-MET gene were also downregulated (Fig. 3b). In addition, the growth inhibition of SN-38 in PC-6/SN-38 cells with c-MET siRNA was improved relative to the cells with negative-control (Fig. 3c). We also obtained the same result in PC-6/TXL cells with c-MET siRNA (Fig. 3c).
Fig. 3.
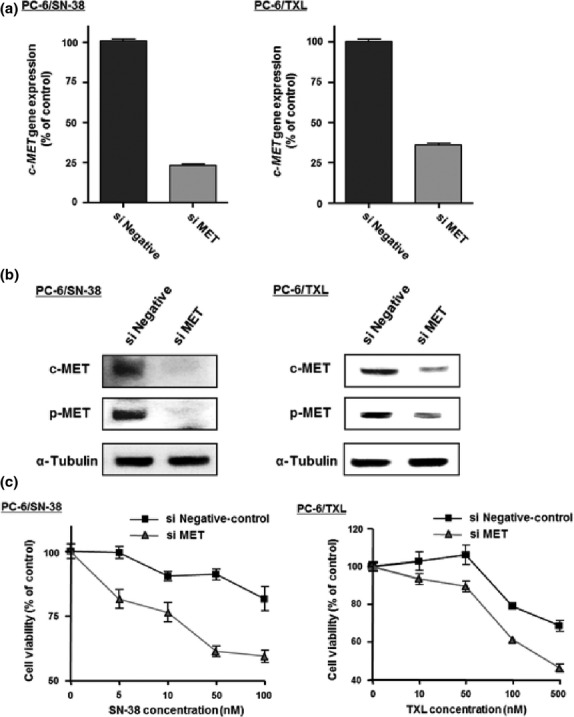
Knockdown of c-MET expression by siRNA in small-cell lung cancer cells. (a) c-MET gene expression was examined using real-time RT-PCR. (b) c-MET and p-MET protein expression was determined using Western blotting. (c) Growth inhibition of stepwise 7-ethyl-10-hydroxycamptothesin (SN-38) or paclitaxel (TXL) concentration in SN-38-resistant (PC-6/SN-38) or TXL-resistant (PC-6/TXL) cells transfected with c-MET siRNA (si MET; ▴) or negative-control (▪) was examined by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) assay.
c-MET gene amplification in cytotoxic anticancer drug-resistant cells
We examined the c-MET DNA copy number in the cytotoxic anticancer drug-resistant cells compared with the parental cells by PCR. The relative c-MET DNA copy numbers of the resistant cells was significantly increased compared with the parental cells (Fig. 4a). In addition, we examined the c-MET gene amplification in the PC-6 cells at 7 days after exposure to SN-38 (5 nM) or TXL (10 nM). The c-MET gene amplification in the PC-6 cells exposed to SN-38 or TXL was significantly increased relative to the cells not exposed (Fig. 4b).
Fig. 4.
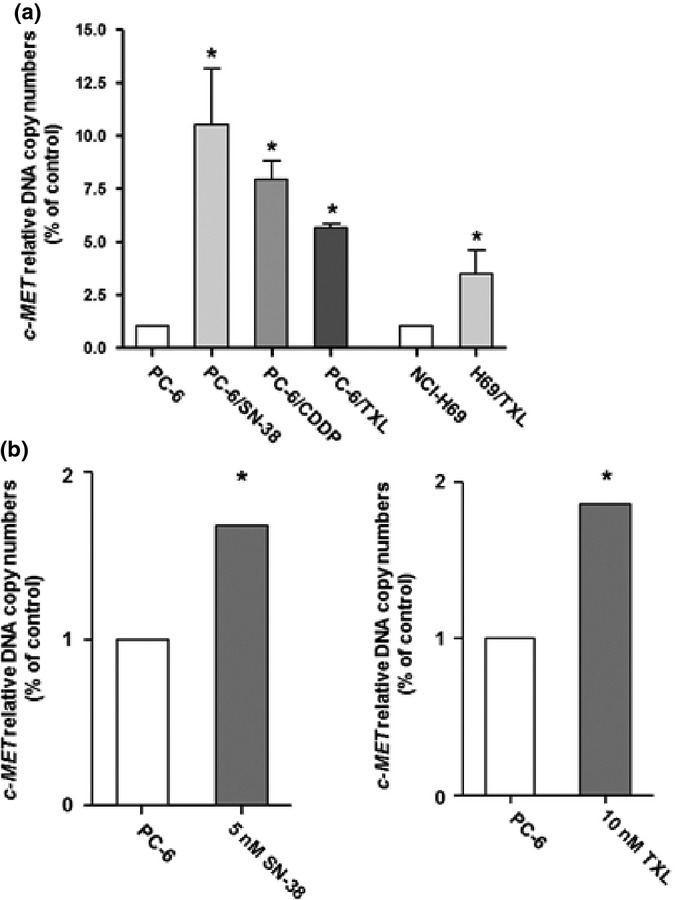
Increased c-MET DNA copy numbers in PC-6 human small-cell lung cancer cells resistant to 7-ethyl-10-hydroxycamptothesin (PC-6/SN-38), paclitaxel (PC-6/TXL), and cisplatin (PC-6/CDDP), as well as the NCI-H69 and TXL-resistant NCI-H69 (H69/TXL) small-cell lung cancer cell lines. (a) c-MET DNA copy numbers in drug-resistant cells were examined relative to the parental cells by quantitative real-time PCR. (b) c-MET DNA copy numbers in PC-6 cells, exposed to 7-ethyl-10-hydroxycamptothesin (SN-38, 5 nM) or paclitaxel (TXL, 10 nM) for 7 days, were examined by quantitative real-time PCR. *P < 0.05.
Chromosomal instability in PC-6/SN-38 cells
We examined c-MET gene amplification or chromosomal instability in PC-6 and PC-6/SN-38 cells by FISH analysis. Diploid cells accounted for 95% (95 cells/100 cells) and tetraploid only 1% (1 cell/100 cells) of all PC-6 cells (Fig. 5a,c), whereas diploid cells accounted for 35% (35 cells/100 cells), triploid 7% (7 cells/100 cells), and tetraploid 48% (48 cells/100 cells) of all PC-6/SN-38 cells (Fig. 5b,c). In addition, the copy number of the c-MET gene increased by one as compared with CEP7 in 6% of PC-6/SN-38 cells relative to PC-6 cells (Fig. 5c).
Fig. 5.
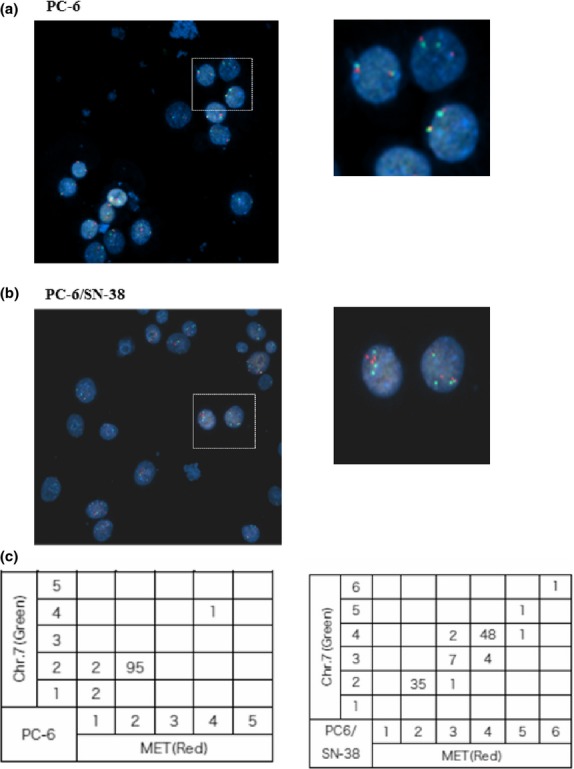
Chromosomal instability in PC-6 small-cell lung cancer cells and 7-ethyl-10-hydroxycamptothesin-resistant (PC-6/SN-38) cells. Chromosomal instability was detected in PC-6/SN-38 and compared to that in PC-6. Dual-color FISH (CEP7 [green]/c-MET [red]) was carried out on fixed cell specimens from PC-6 (a) and PC-6/SN-38 (b) cells. The number of CEP7 and c-MET signals in 100 cells was listed (c). 20× maginification.
Levels of HGF expression in drug-resistant cells
Hepatocyte growth factor-mediated MET activation was a novel mechanism of the acquired resistance to gefitinib in lung adenocarcinoma.(19,31) Furthermore, HGF has been shown to accelerate the emergence of c-MET amplification in gefitinib-resistant lung cancer cells.(31) In this study, we examined the levels of endogenous HGF expression in PC-6/SN-38, PC-6/TXL, PC-6/CDDP, and NCI-H69/TXL cells by WB. Hepatocyte growth factor protein expression in the resistant cells was upregulated compared with the parental cells (Fig. 6a). The concentration of HGF in cell culture supernatants was significantly higher from PC-6/SN-38, PC-6/TXL, and PC-6/CDDP cells than from PC-6 cells (Fig. 6b). We examined the sensitivity of PC-6 cells to SN-38 after cultured with HGF for 2 weeks. PC-6 cells with HGF were significantly resistant to SN-38 relative to PC-6 cells without HGF (Fig. 6c).
Fig. 6.
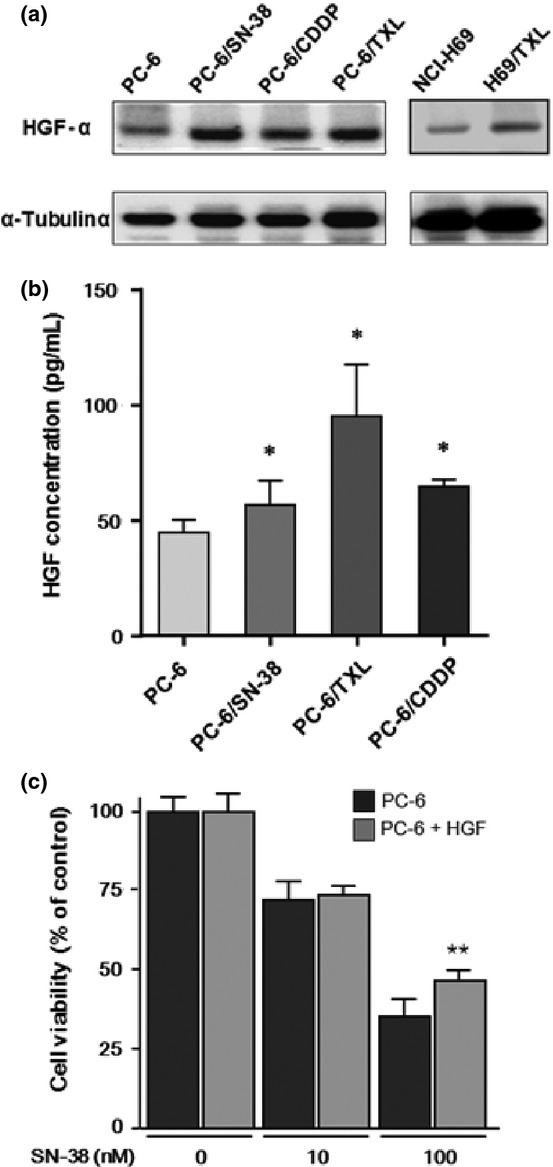
Hepatocyte growth factor (HGF) protein expression in cytotoxic anticancer drug-resistant small-cell lung cancer cells, including PC-6 and NCI-H69 parental cells and those resistant to 7-ethyl-10-hydroxycamptothesin (PC-6/SN-38), paclitaxel (PC-6/TXL and H69/TXL), and cisplatin (PC-6/CDDP). (a) Levels of HGF protein expression in cytotoxic anticancer drug-resistant cells were examined by Western blotting. (b) Secretion of HGF in resistant cells was quantified by quantitative ELISA and compared to that in parental cells. (c) Growth inhibition of SN-38 to PC-6 cultured with or without HGF (50 ng/mL) for 2 weeks was examined with the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) assay. *P < 0.05; **P < 0.01.
Discussion
We here found that increased c-Met expression through an increase in the number of c-MET gene loci is one of the mechanisms acquired resistance to cytotoxic anticancer drugs. Our results add a new strategy for overcoming the resistance to cytotoxic agents in SCLC.
Activation of the c-MET/HGF signaling pathway through the overexpression of HGF and/or c-MET is identified in many lung cancer patient samples.(15,16,32) The functional mutations of c-MET were identified within the semaphoring domain and the juxtamembrane domain of SCLC.(33) In addition, a functional link between downstream signaling of the c-MET/HGF pathway and tumor invasion was shown by phosphoantibody-based immunohistochemical analysis of SCLC tumor tissue and microarray.(15) Enhanced therapeutic efficacy could potentially be achieved through inhibiting c-MET activation and its known downstream signaling intermediates in SCLC.(17) Therefore, c-MET would be an attractive therapeutic target to be inhibited in SCLC to expand the therapeutic armamentarium. In this study, we showed that downregulation of c-MET expression by siRNA against the c-MET gene or inhibition of c-MET activation by SU11274 in anticancer-resistant SCLC cells altered resistance to the cytotoxic anticancer agent. These results showed that c-MET overexpression might play an important role in acquired resistance to the cytotoxic anticancer drugs. Therefore, c-MET inhibitor in combination with cytotoxic anticancer agents could be attractive treatments for patients with SCLC, to overcome the rapid development of drug resistance.
There are two general mechanisms of resistance of human tumors to anticancer agents. The impaired ability of drugs to penetrate tumor tissue and to reach all of the tumor cells in a potentially lethal concentration has an important role in resistance to anticancer agents.(34,35) Another important determinant of anticancer drug resistance is most often ascribed to epigenetic alternations that affect the expression of genes encoding proteins that influence the uptake, metabolism, and export of drugs from the tumor cell itself.(36) Recently, amplification of the c-MET gene was induced by clonal selection after exposure to gefitinib, which caused gefitinib resistance by driving ERBB3-dependent activation of PI3K, a pathway thought to be specific to EGFR/ERBB family receptors in lung adenocarcinoma with EGFR-activating mutations.(18,31) In this study, we found that c-MET gene amplification in the resistant cells was significantly increased compared with the parental cells, by PCR. In addition, c-MET gene amplification in the PC-6 cells exposed to SN-38 or TXL was significantly increased relative to the cells not exposed. However, although the number of cells in which c-MET gene amplification was observed among PC-6/SN-38 cells was increased by only 6%, FISH analysis revealed that the number of polyploidy cells was markedly higher in PC-6/SN-38 cells than in PC-6 cells. A clinical study previously reported that chromosomal instability was a risk factor for poor prognosis in patients with lung adenocarcinoma.(37) Furthermore, MDR-associated protein overexpression in non-SCLC may be a consequence of the reassortment of chromosome 16, which is known to be catalyzed by aneuploidy.(38) Chromosomal instability including polyploidy has been associated with cytotoxic anticancer-drug resistance including TXL and CDDP.(39)– (41) Our results are in agreement with the findings of these previous studies. Differences in the experimental results obtained between FISH analysis and quantitative real-time PCR may be explained by differences in the controls used. We used CEP7 as a control in the FISH analysis, and the ribonuclease P RNA component H1 gene mapped to chromosome 14q just below the centromere as a control in PCR. We speculate that the mechanisms responsible for the overexpression of c-MET in drug-resistant cells may be induced by an increase in c-MET DNA copy numbers and/or polyploidy in the resistant cells.
Hepatocyte growth factor was first identified as a mitogenic protein for hepatocytes and enhanced pleiotropic biological phenomena in a wide variety of cells, including antiapoptotic activities.(42) Tumor cell-derived HGF could induce gefitinib resistance of lung adenocarcinoma cells with EGFR-activating mutations.(19) In addition, HGF accelerates the emergence of c-MET amplification in gefitinib-resistant lung cancer cells.(31) In this study, we showed that endogenous HGF protein expression in cytotoxic anticancer drug-resistant SCLC cells was increased relative to the parental cells. PC-6 cells cultured with HGF were resistant to SN-38 relative to cells without HGF. Therefore, increased endogenous HGF expression in the resistant cells might be needed to obtain antiapoptotic activation and induce the selection of cells with a higher number of c-MET gene loci in the process of obtaining resistance to the cytotoxic anticancer drugs.
Downstream signaling of the c-MET/HGF pathway is activated in a variety of manners, including activating PI3K/AKT, ERK1/2, and FAK signaling, which resistant to the cytotoxic anticancer drugs.(21,20,43) In this study, SU11274 at 2 μM concentration inhibited ERK1/2 activities in PC-6/SN-38 and PC-6/TXL, but not AKT activities. Inhibition of ERK1/2 activities enhances a stimulation of PARP cleavage, a programmed cell death by apoptosis and chemosensitivity.(44)– (47) Therefore, HGF/c-MET signaling activation may protect SCLC cells from cytotoxic anticancer drugs by activating the ERK1/2-dependent antiapoptotic pathway. In addition, c-MET activation by gene amplification and HGF can independently rescue not only PI3K/AKT but also ERK signaling in the presence of gefitinib and lead to drug resistance.(18,19,31) c-MET activation might induce cross-resistance between the cytotoxic anticancer drugs and EGFR-TKI.
Our current findings provide insight into future therapeutic strategies for the treatment of SCLC. Recent progress in the understanding of the biology of SCLC has led to the identification of crucial signaling pathways and the subsequent development of targeted therapies.(48) This is the first report that activation of the c-MET signaling pathway through an increase in the number of c-MET gene loci is one of the novel mechanisms contributing to resistance to cytotoxic anticancer drugs. c-MET inhibitor may overcome the rapid development of drug resistance, which results in poor prognosis for ED-SCLC patients. Therefore, c-MET inhibitor alone or in combination with cytotoxic anticancer agents could be attractive treatments for patients with SCLC.
Disclosure Statement
The authors have no conflicts of interest.
Glossary
- AKT
serine/threonine protein kinase
- CDDP
cisplatin
- ED
extensive disease
- EGFR-TKI
epidermal growth factor receptor tyrosine kinase inhibitor
- FAK
Focal Adhesion Kinase
- HGF
hepatocyte growth factor
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- p-
phosphorylated
- PARP
poly(ADP-ribose) polymerase
- PI3K
Phosphoinositide 3-kinase
- SCLC
small-cell lung cancer
- SN-38
7-ethyl-10-hydroxycamptothesin
- TXL
paclitaxel
- WB
Western blot
References
- 1.Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell, lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24:4539–44. doi: 10.1200/JCO.2005.04.4859. [DOI] [PubMed] [Google Scholar]
- 2.Lally BE, Urbanic JJ, Blackstock AW, Miller AA, Perry MC. Small cell lung cancer: have we made any progress over the last 25 years? Oncologist. 2007;12:1096–104. doi: 10.1634/theoncologist.12-9-1096. [DOI] [PubMed] [Google Scholar]
- 3.Noda K, Nishiwaki Y, Kawahara M, et al. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med. 2002;346:85–91. doi: 10.1056/NEJMoa003034. [DOI] [PubMed] [Google Scholar]
- 4.Lara PN, Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530–5. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Brien M, Eckardt J, Ramlau R. Recent advances with topotecan in the treatment of lung cancer. Oncologist. 2007;12:1194–204. doi: 10.1634/theoncologist.12-10-1194. [DOI] [PubMed] [Google Scholar]
- 6.Inoue A, Sugawara S, Yamazaki K, et al. Randomized phase II trial comparing amrubicin with topotecan in patients with previously treated small-cell lung cancer: North Japan Lung Cancer Study Group Trial 0402. J Clin Oncol. 2008;26:5401–6. doi: 10.1200/JCO.2008.18.1974. [DOI] [PubMed] [Google Scholar]
- 7.Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 8.Ferracini R, Longati P, Naldini L, Vigna E. Comoglio PM. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J Biol Chem. 1991;266:19558–64. [PubMed] [Google Scholar]
- 9.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2:289–300. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- 10.Zhang YW, Vande Woude GF. HGF/SF-met signaling in the control of branching morphogenesis and invasion. J Cell Biochem. 2003;88:408–17. doi: 10.1002/jcb.10358. [DOI] [PubMed] [Google Scholar]
- 11.Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309–25. doi: 10.1023/a:1023768811842. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 13.Lee WY, Chen HH, Chow NH, Su WC, Lin PW, Guo HR. Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin Cancer Res. 2005;11:2222–8. doi: 10.1158/1078-0432.CCR-04-1761. [DOI] [PubMed] [Google Scholar]
- 14.Sawada K, Radjabi AR, Shinomiya N, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67:1670–99. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
- 15.Ma PC, Tretiakova MS, Nallasura V, Jagadeeswaran R, Husain AN, Salgia R. Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: implications for tumour invasion. Br J Cancer. 2007;97:368–77. doi: 10.1038/sj.bjc.6603884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masuya D, Huang C, Liu D, et al. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br J Cancer. 2004;90:1555–62. doi: 10.1038/sj.bjc.6601718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toschi L, Jänne PA. Single-agent and combination therapeutic strategies to inhibit hepatocyte growth factor/MET signaling in cancer. Clin Cancer Res. 2008;14:5941–6. doi: 10.1158/1078-0432.CCR-08-0071. [DOI] [PubMed] [Google Scholar]
- 18.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 19.Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–87. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 20.Bowers DC, Fan S, Walter KA, et al. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res. 2000;60:4277–83. [PubMed] [Google Scholar]
- 21.Chen JT, Huang CY, Chiang YY, et al. HGF increases cisplatin resistance via down-regulation of AIF in lung cancer cells. Am J Respir Cell Mol Biol. 2008;38:559–65. doi: 10.1165/rcmb.2007-0001OC. [DOI] [PubMed] [Google Scholar]
- 22.Bardella C, Dettori D, Olivero M, Coltella N, Mazzone M, Di Renzo MF. The therapeutic potential of hepatocyte growth factor to sensitize ovarian cancer cells to cisplatin and paclitaxel in vivo. Clin Cancer Res. 2007;13:2191–8. doi: 10.1158/1078-0432.CCR-06-1915. [DOI] [PubMed] [Google Scholar]
- 23.Sakai A, Kasahara K, Ohmori T, et al. MET increases the sensitivity of gefitinib-resistant cells to SN-38, an active metabolite of irinotecan, by up-regulating the topoisomerase I activity. J Thorac Oncol. 2012;7:1337–44. doi: 10.1097/JTO.0b013e31825cca4c. [DOI] [PubMed] [Google Scholar]
- 24.Oguri T, Ozasa H, Uemura T, et al. MRP7/ABCC10 expression is a predictive biomarker for the resistance to paclitaxel in non-small cell lung cancer. Mol Cancer Ther. 2008;7:1150–5. doi: 10.1158/1535-7163.MCT-07-2088. [DOI] [PubMed] [Google Scholar]
- 25.Ozasa H, Oguri T, Uemura T, et al. Significance of thymidylate synthase for resistance to pemetrexed in lung cancer. Cancer Sci. 2010;101:161–6. doi: 10.1111/j.1349-7006.2009.01358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasai D, Ozasa H, Oguri T, et al. Thymidylate synthase gene copy number as a predictive marker for response to pemetrexed treatment of lung adenocarcinoma. Anticancer Res. 2013;33:1935–40. [PubMed] [Google Scholar]
- 27.Brønstad I, Wolff AS, Løvås K, Knappskog PM, Husebye ES. Genome-wide copy number variation (CNV) in patients with autoimmune Addison's disease. BMC Med Genet. 2011;12:111. doi: 10.1186/1471-2350-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horlitz M, Lucas A, Sprenger-Haussels M. Optimized quantification of fragmented, free circulating DNA in human blood plasma using a calibrated duplex real-time PCR. PLoS ONE. 2009;4:e7207. doi: 10.1371/journal.pone.0007207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sattler M, Pride YB, Ma P, et al. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003;63:5462–9. [PubMed] [Google Scholar]
- 30.Arena S, Pisacane A, Mazzone M, Comoglio PM, Bardelli A. Genetic targeting of the kinase activity of the Met receptor in cancer cells. Proc Natl Acad Sci USA. 2007;104:11412–7. doi: 10.1073/pnas.0703205104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 33.Ma PC, Kijima T, Maulik G, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272–81. [PubMed] [Google Scholar]
- 34.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441–54. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 35.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 36.Choi CM, Seo KW, Jang SJ, et al. Chromosomal instability is a risk factor for poor prognosis of adenocarcinoma of the lung: fluorescence in situ hybridization analysis of paraffin-embedded tissue from Korean patients. Lung Cancer. 2009;64:66–70. doi: 10.1016/j.lungcan.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 37.Doubre H, Césari D, Mairovitz A, et al. Multidrug resistance-associated protein (MRP1) is overexpressed in DNA aneuploid carcinomatous cells in non-small cell lung cancer (NSCLC) Int J Cancer. 2005;113:568–74. doi: 10.1002/ijc.20617. [DOI] [PubMed] [Google Scholar]
- 38.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;18:338–41. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 39.McClelland SE, Burrell RA, Swanton C. Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle. 2009;8:3262–6. doi: 10.4161/cc.8.20.9690. [DOI] [PubMed] [Google Scholar]
- 40.Shen H, Perez RE, Davaadelger B, Maki CG. Two 4N cell-cycle arrests contribute to cisplatin-resistance. PLoS ONE. 2013;8:e59848. doi: 10.1371/journal.pone.0059848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto K, Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int J Cancer. 2006;119:477–83. doi: 10.1002/ijc.21808. [DOI] [PubMed] [Google Scholar]
- 43.Tang MK, Zhou HY, Yam JW, Wong AS. c-Met overexpression contributes to the acquired apoptotic resistance of nonadherent ovarian cancer cells through a cross talk mediated by phosphatidylinositol 3-kinase and extracellular signal-regulated kinase 1/2. Neoplasia. 2010;12:128–38. doi: 10.1593/neo.91438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boucher MJ, Morisset J, Vachon PH, Reed JC, Lainé J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–69. [PubMed] [Google Scholar]
- 45.Lunghi P, Tabilio A, Dall'Aglio PP, et al. Downmodulation of ERK activity inhibits the proliferation and induces the apoptosis of primary acute myelogenous leukemia blasts. Leukemia. 2003;17:1783–93. doi: 10.1038/sj.leu.2403032. [DOI] [PubMed] [Google Scholar]
- 46.Su YJ, Tsai MS, Kuo YH, et al. Role of Rad51 down-regulation and extracellular signal-regulated kinases 1 and 2 inactivation in emodin and mitomycin C-induced synergistic cytotoxicity in human non-small-cell lung cancer cells. Mol Pharmacol. 2010;77:633–43. doi: 10.1124/mol.109.061887. [DOI] [PubMed] [Google Scholar]
- 47.Huang G, Mills L, Worth LL. Expression of human glutathione S-transferase P1 medi9ates the chemosensitivity of osteosarcoma cells. Mol Cancer Ther. 2007;6:1610–9. doi: 10.1158/1535-7163.MCT-06-0580. [DOI] [PubMed] [Google Scholar]
- 48.Puglisi M, Dolly S, Faria A, Myerson JS, Popat S, O'Brien ME. Treatment options for small cell lung cancer - do we have more choice? Br J Cancer. 2010;102:629–38. doi: 10.1038/sj.bjc.6605527. [DOI] [PMC free article] [PubMed] [Google Scholar]