

Abstract
Epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) are validated molecular targets in cancer therapy. Dual blockade has been explored and one such agent, lapatinib, is in clinical practice but with modest activity. Through chemical screening, we discovered a novel EGFR and HER2 inhibitor, S-222611, that selectively inhibited both kinases with IC50s below 10 nmol/L. S-222611 also inhibited intracellular kinase activity and the growth of EGFR-expressing and HER2-expressing cancer cells. In addition, S-222611 showed potent antitumor activity over lapatinib in a variety of xenograft models. In evaluations with two patient-oriented models, the intrafemoral implantation model and the intracranial implantation model, S-222611 exhibited excellent activity and could be effective against bone and brain metastasis. Compared to neratinib and afatinib, irreversible EGFR/HER2 inhibitors, S-222611 showed equivalent or slightly weaker antitumor activity but a safer profile. These results indicated that S-222611 is a potent EGFR and HER2 inhibitor with substantially better antitumor activity than lapatinib at clinically relevant doses. Considering the safer profile than for irreversible inhibitors, S-222611 could be an important option in future cancer therapy.
Keywords: Antitumor activity, epidermal growth factor receptor, human epidermal growth factor receptor 2, kinase inhibitor, S-222611
Epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) belong to the HER receptor tyrosine kinase family together with two other members (HER3 and HER4).(1) The signal stemming from the HER dimer is transduced to the cellular signaling network and often potentiates the growth and/or survival of cancer cells. Genetic alteration and aberrational expression of the HER family are frequently observed in a variety of cancers.(1)– (4)
Both EGFR and HER2 are appropriate molecular targets for cancer therapy as evidenced by the development of appropriate antagonists. Several anti-EGFR agents (gefitinib, erlotinib, cetuximab and panitumumab) have been approved for selective cancers, including non-small lung, pancreatic, colon, and head and neck cancers.(5) Trastuzumab, an anti-HER2 mAb, is widely used for the treatment of HER2-positive breast cancer patients.(1) Because many of these cancers express both EGFR and HER2, dual targeting of both molecules has been approached to improve efficacy. Lapatinib, an orally active EGFR and HER2 dual kinase inhibitor, was the first such agent and it was approved by the FDA for the treatment of advanced or metastatic breast cancer in 2007.(6) Although lapatinib has shown significant activity in some solid cancers in clinical trials,(6)– (8) its efficacy is limited and the approved indications have not been extended beyond breast cancer. In the search for a more potent agent, irreversible EGFR/HER2 dual inhibitors, including neratinib and afatinib, have been in development.(9)– (11) Recently, afatinib was approved worldwide for the treatment of non-small cell lung cancer having mutated EGFR.(11) These irreversible inhibitors have shown substantial toxicity, with a resultant requirement for dose reduction in phase 2 and 3 trials.(11)– (15) Thus, there is still strong demand for more effective and safer EGFR/HER2 dual inhibitors.
We conducted chemical screening to find a powerful EGFR/HER2 dual inhibitor superior to lapatinib and discovered a novel quinazoline derivative, S-222611.(16)– (18) Herein we describe the nonclinical pharmacological properties of S-222611. This agent has now proceeded to clinical trials.(19,20)
Materials and Methods
Cells and reagents
The human cell lines used in this study were as follows: NCI-N87 (gastric cancer), BT-474, SK-BR-3, MDA-MB-453, MDA-MB-175VII, MDA-MB-361 (breast cancer), HT115 (colon cancer), Calu-3 (non-small cell lung cancer), A-431 (epidermoid carcinoma), fR2 (SV40-transformed breast epithelial cell) and MRC-5 (normal lung fibroblast). All these cells were purchased from ATCC (Manassas, VA, USA) and the European Collection of Cell Cultures (Salisbury, Wiltshire, UK). The cells were maintained in DMEM supplemented with 10–20% FBS, 100 units/mL penicillin and 100 units/mL streptomycin.
Animals
Nude mice (BALB/cAJcl-nu/nu, CLEA Japan, Tokyo, Japan) were used for most of the in vivo studies, except SCID mice (C.B-17/Icr-scid/scidJcl, CLEA Japan) for the HT115 model and the intrafemoral implantation model of BT-474-luc (luciferase-expressing BT-474), SCID Beige mice (CB17.Cg-PrkdcscidLystbg-J/Crl; Charles River Laboratories Japan) for the BT-474 model, and hairless SCID mice (SHO-PrkdcscidHrhr; Charles River Laboratories Japan, Yokohama, Kanagawa, Japan) for the intracranial implantation model of BT-474-luc. All mice were female and 6–9 weeks old. All the mice used in the experiments were killed immediately after the last measurement except the survival study. All animal studies were conducted under the approval of the Institutional Animal Care and Use Committees of Shionogi Research Laboratories.
Reagents
S-222611, lapatinib, neratinib and afatinib were chemically synthesized in Shionogi Research Laboratories (Toyonaka, Osaka, Japan). Lapatinib, neratinib and afatinib were prepared according to the preceding references.(11,21)– (24) Their structures were identified by comparison of their 1H NMR and mass spectrum with the reported values. In addition, their purities were confirmed by liquid chromatography mass spectrometry to be over 95%.
In in vitro experiments, these agents were dissolved with DMSO (Nacalai Tesque, Kyoto, Kyoto, Japan) and the solutions were further diluted with an assay buffer or a culture medium. In in vivo experiments, these agents were suspended in 0.5 w/v% methylcellulose 400cP solution (Wako Pure Chemical Industries, Osaka, Osaka, Japan) to make dosing formulations.
In vitro kinase assay
Enzyme activities of EGFR, HER2, HER4, IGF1R, KDR, KIT, PDGFRβ and SRC were evaluated using the QSS Assist ELISA kit (Carna Bioscience, Kobe, Hyogo, Japan) following the manufacturer's protocol. The relative inhibition rate of each data point was calculated and used to obtain the IC50 value for each drug. Each experiment was carried out thrice, each time in triplicate.
Evaluation of phosphorylation of epidermal growth factor receptor and human epidermal growth factor receptor 2
Human gastric cancer cells, NCI-N87, were treated with serially diluted drug for 24 h. Total and phosphorylated EGFR/HER2 were quantitated using Human Total-EGFR, Total-ErbB2, Phospho-EGFR and Phospho-ErbB2 DuoSet IC ELISA kits (R&D Systems (Minneapolis, MN, USA)) following the manufacturer's protocol. First, the phosphorylation ratio (phosphorylated protein/total protein) of each of triplicate sample was calculated. Subsequently, the relative phosphorylation (mean phosphorylation ratio of treated sample/mean phosphorylation ratio of control sample) for each data point was calculated and used to obtain the IC50 value.
Growth inhibition assay
Cells were seeded at 3000 cells/well in 96-well plates and incubated overnight. Serially diluted drug was added to the well and the plates were incubated for 72 h. After chromogenic reaction with WST-8 (Kishida Chemicals, Osaka, Osaka, Japan), the OD450 (with reference of OD650) was measured using an Emax microplate reader (Molecular Devices, Sunnyvale, CA, USA) and used to obtain the IC50 value. Each experiment was carried out thrice, each time in triplicate.
The following two studies were performed with NCI-N87 cells. In the study with human serum protein, 2% serum albumin (Sigma-Aldrich, St. Louis, MO, USA) or 0.08% α1-acid glycoprotein (Sigma-Aldrich) was added to the culture containing the drug. In the study with short-time pulse treatment, 6000 cells/well were seeded in 96-well crystal glass plates. At 1, 6 or 24 h after addition of serially diluted drug to the well, the culture medium was removed and each well was washed three times with DMEM with 1% FBS. The plate was then reincubated for a total of 72 h.
Evaluation of epidermal growth factor receptor and human epidermal growth factor receptor 2 expression in cell lines
Two days after cell seeding at the density of 6.0 × 105 cells/100 mm cell culture dish, the cells were lysed with lysis buffer and the amounts of EGFR and HER2 in the lysate were quantitated using Human Total-EGFR and Total-ErbB2, DuoSet IC ELISA kits (R&D Systems). The protein content of the lysates was determined using a DC protein assay kit (Bio-Rad, Hercules, CA, USA).
In vivo antitumor assay
In in vivo studies, 4 × 106 to 3 × 107 cells were implanted subcutaneously into the back of mice; however, the two breast cancer cell lines, BT-474 and MDA-MB-361, were implanted orthotopically into the mammary fat pad of mice. The cell suspensions for implantation of these two cell lines and HT115 cells included 50 v/v% Matrigel (Beckton Dickinson, Franklin Lakes, NJ, USA). After randomization, vehicle or multiple doses of S-222611 or lapatinib were administered by oral gavage daily for 10–28 days. The length and width of tumors were measured using an electronic caliper twice or thrice weekly and the tumor volume was calculated using the following formula: (length × width2)/2.
In vivo evaluation of phosphorylation of epidermal growth factor receptor and human epidermal growth factor receptor 2
NCI-N87 cells were implanted subcutaneously into the back of mice as in the in vivo antitumor assay. Tumor xenografts were excised at 6 or 24 h after administration and homogenized in the lysis buffer. The composition of the lysis buffer and the methods hereafter are the same as those of the in vitro study.
In vivo patient-oriented models
Luciferase-expressing BT-474 (BT-474-luc) cells were established by transfection of firefly luciferase expression vector into BT-474 cells and subsequent cloning. In the intrafemoral implantation model, the cell suspension containing 1.05 × 106 BT-474-luc cells and 50 v/v% Matrigel (Beckton Dickinson) were surgically implanted into the left femur of mice. In the intracranial implantation model, the cell suspension containing 2 × 105 BT-474-luc cells was surgically implanted into the brain parenchyma of mice. For measurement of bioluminescence, 0.2 mL of 10 mg/mL luciferin solution was injected intravenously into each mouse via tail vein under anesthesia. Immediately after that, the photon emitted from tumors was measured using the IVIS Imaging System 200 (Caliper Life Sciences, Hopkinton, MA, USA).
In the survival model, a cell suspension containing 2.5 × 106 BT-474 cells was surgically implanted into the brain parenchyma of mice. Mice were observed for any health defects once a day and the body weights were measured at least twice weekly. Mice in a moribund state or reaching any humane endpoint were immediately killed. In addition, all the surviving mice were killed at the end of study (106 days after implantation).
Histopathological evaluation
The colon and the eyeball were excised from the mice treated with S-222611, neratinib or afatinib and fixed in 10% neutral buffered formalin. After routine processing of these fixed organs, the paraffin sections were stained with H&E and examined microscopically.
Results
In vitro inhibition of kinase activity by S-222611
In search of a powerful EGFR/HER2 kinase inhibitor, we screened thousands of compounds and found S-222611. S-222611 potently inhibited EGFR, HER2 and HER4 with IC50 values below 10 nmol/L. These values are comparable to those of lapatinib (Table 1a). The IC50 values of S-222611 for five other tyrosine kinases were more than 10 000 nmol/L. The selective kinase inhibition of S-222611 was further confirmed in profiling with 107 kinases (Table S1). These results showed that S-222611, like lapatinib, was a selective inhibitor for EGFR, HER2 and HER4.(25)
Table 1.
In vitro pharmacological activity of S-222611 and lapatinib
| IC50 (nmol/L) |
||||
|---|---|---|---|---|
| S-222611 | Lapatinib | |||
| (a) In vitro kinase inhibition | ||||
| EGFR | 1.48 ± 0.06 | 1.52 ± 0.07ns | ||
| HER2 | 7.15 ± 0.51 | 8.74 ± 0.24** | ||
| HER4 | 2.49 ± 0.10 | 4.62 ± 1.35ns | ||
| KDR, IGF1R, SRC, KIT, PDGFRβ | >10 000 | >10 000 | ||
| (b) Inhibition of relative phosphorylation of receptor kinases in NCI-N87 cells | ||||
| Phosphorylation of EGFR | 4.5 | 11.0 | ||
| Phosphorylation of HER2 | 1.6 | 5.4 | ||
| (c) Growth inhibition of cultured cells | EGFR | HER2 | ||
| NCI-N87 (stomach) | ++ | +++ | 8.3 ± 2.6 | 36.4 ± 5.2** |
| BT-474 (breast) | + | +++ | 9.9 ± 0.8 | 29.4 ± 2.2** |
| SK-BR-3 (breast) | ++ | +++ | 14.0 ± 3.6 | 56.9 ± 5.4** |
| MDA-MB-453 (breast) | − | ++ | 48.6 ± 3.1 | 169.6 ± 3.0** |
| MDA-MB-175VII (breast) | + | ++ | 21.6 ± 4.3 | 70.2 ± 10.2** |
| HT115 (colon) | ++ | + | 53.3 ± 8.6 | 100.2 ± 1.5** |
| Calu-3 (lung) | ++ | +++ | 241.5 ± 29.2 | 509.8 ± 85.8* |
| fR2 (breast) | ++ | + | 5366.7 ± 65.2 | 5987.0 ± 386.0ns |
| MRC-5 (lung) | ++ | + | 4964.6 ± 340.3 | 9138.9 ± 642.7** |
| (d) Effect of human serum protein | ||||
| Standard medium | 7.8 ± 1.4 | 35.2 ± 6.0 | ||
| +2% Serum albumin | 22.9 ± 0.8 | 372.7 ± 35.5 | ||
| +0.08% α1-Acid glycoprotein | 34.4 ± 3.2 | 448.5 ± 36.6 | ||
(a) Inhibition of enzyme activity. IGF1R, KDR, KIT, PDGFRβ and SRCB were not inhibited over 50% by both drugs at the highest concentration tested (10 000 nmol/L). (b) Inhibition of relative phosphorylation of EGFR and HER2 in NCI-N87 cells. (c) Inhibition of growth of cell lines. Expression levels of EGFR and HER2 for each cell line are indicated as scores (−: below 1, +: 1–10, ++: 10–100, +++: above 100, pg EGFR or HER2/μg total cellular protein). In a and c, the results of statistical analysis (ns P ≥ 0.05, *P < 0.05, **P < 0.01, S-222611 versus lapatinib, Welch's t-test) are presented in the right side of the date for lapatinib. (d) Inhibition of growth of NCI-N87 cells in the presence of human serum proteins. All the values are shown as mean ± SD of triplicate experiments except for inhibition of phospho-RTKs in NCI-N87 cells (b) where only IC50 values are shown.
Next we studied the effect of S-222611 on autophosphorylation of EGFR and HER2 in cells in order to examine whether S-222611 could be incorporated into cells and could inhibit EGFR and HER2 in intact cells. S-222611 decreased the relative phosphorylation of EGFR and HER2 in NCI-N87 cells with IC50 values below 10 nmol/L (Table 1b). These inhibitory activities of S-222611 on intracellular kinases were 2.4–3.4 times stronger than those of lapatinib.
Cancer cell growth inhibition by S-222611
Next, we evaluated the growth inhibitory activity of S-222611 for several cancer cell lines expressing EGFR and/or HER2 (Table 1c). The IC50 values for all the cancer cell lines except Calu-3 were below 1000 nmol/L, while those for SV40-transformed human breast epithelial cells, fR2, and normal human lung fibroblasts, MRC-5, were approximately 5000 nmol/L. These antiproliferative activities of S-222611 for cancer cell lines were 1.9–4.4 times higher than those of lapatinib. Thus, S-222611 can selectively inhibit the proliferation of a range of cancer cell lines expressing EGFR and/or HER2.
Growth inhibition by S-222611 in the presence of serum protein
The activity of a drug is commonly attenuated by the binding of serum proteins. S-222611 is highly bound (>99%) to serum albumin and α1-acid glycoprotein, as well as lapatinib.(26) Therefore, we evaluated the growth inhibitory activity of both agents in the presence of additional serum protein. Addition of serum proteins elevated the IC50 values of both S-222611 and lapatinib (2.9–4.4-fold and 10.6–12.7-fold, respectively), but the degree of attenuation was much smaller with S-222611 (Table 1d). This refractoriness of S-222611 to serum protein binding helps explain why S-222611 can exert advantageous activity over lapatinib in a cell-based test system which intrinsically contains serum protein and, thereby, can lead to potent antitumor activity in vivo.
In vivo antitumor activity of S-222611
The in vivo antitumor activity of S-222611 was evaluated. Nude mice bearing NCI-N87 xenograft were treated orally with S-222611 or lapatinib. The changes of tumor volume over time are shown in Figure 1(a) (Fig. S1). S-222611 significantly inhibited the tumor growth in a dose-dependent manner. Comparison of ED50 between S-222611 and lapatinib (10.2 and 57.7 mg/kg, respectively, Table S2) indicated that the in vivo antitumor activity of S-222611 was approximately six times more potent than that of lapatinib. No body weight loss and no macroscopic toxicity were observed in the S-222611-treated mice.
Fig. 1.
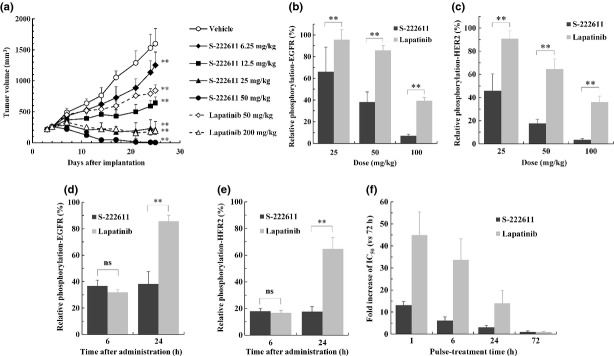
S-222611 inhibits the growth of NCI-N87 tumor and phosphorylation of epidermal growth factor receptor (EGFR) and human epidermal growth factor 2 (HER2) in the tumor. The animal model was prepared by subcutaneous implantation of human gastric cancer cells, NCI-N87 into the back of nude mice. (a) After randomization at 3 days after implantation (n = 9), vehicle or multiple doses of S-222611 or lapatinib were orally administered daily for 21 days. To avoid visual complexity, only two doses of lapatinib are depicted in this graph. The four full doses of lapatinib are presented in Figure S1. Tumor volume was measured twice or thrice weekly and the mean tumor volume with SD was represented at each data point. **P < 0.01 (versus vehicle, Dunnett's test). (b–e) After randomization at 12 days after implantation (n = 5), vehicle or multiple doses of S-222611 or lapatinib were administered orally. The relative phosphorylations of EGFR (b,d) and HER2 (c,e) were calculated and the mean with SD is presented in each bar graph. In b and c, the relative phosphorylations of EGFR (b) and HER2 (c) in tumor collected 24 h after administration of the indicated doses of S-222611 or lapatinib are shown. In d and e, the relative phosphorylation levels of EGFR (d) and HER2 (e) in the tumor collected 6 and 24 h after administration of 50 mg/kg of S-222611 or lapatinib are shown. ns: P ≥ 0.05, **: P < 0.01 (Tukey's multiple comparison). (f) The fold increase of the IC50 of each short-time pulse treatment compared to that of 72-h treatment was calculated and the mean with SD is presented in each bar graph (n = 3).
Inhibition of phosphorylation of epidermal growth factor receptor and human epidermal growth factor receptor 2 in tumor xenograft
To confirm the mechanism of the antitumor activity of S-222611, the relative phosphorylation of EGFR and HER2 in a tumor xenograft was evaluated in an NCI-N87 model. In a dose-dependent manner, S-222611 treatment reduced the relative phosphorylation of EGFR and HER2 in the tumor (Fig. 1b,c). The degree of reduction by S-222611 was significantly stronger than that of lapatinib at the same dose basis.
Comparison of the effect on phosphorylation of EGFR and HER2 between 6 and 24 h after a single administration revealed that the inhibitory activity of S-222611 persisted even at 24 h, while that of lapatinib had largely disappeared (Fig. 1d,e). Pharmacokinetic analysis revealed that the drug concentrations in the tumor xenograft of both S-222611 and lapatinib were nearly identical (Fig. S2a). Thus, the inhibitory activity of S-222611 on the phosphorylation of EGFR/HER2 in tumor xenograft was shown to be longer than that of lapatinib. Such sustained kinase inhibitory activity of S-222611 contributed to the superior antitumor activity over lapatinib in vivo.
Temporal evaluation of kinase inhibition and growth inhibitory activity of S-222611
To examine the time stability of kinase inhibition by S-222611, the kinetics of the enzyme inhibition was studied. Lapatinib was reported to bind to an inactive conformation of enzyme and showed a slower off-rate of dissociation from EGFR than erlotinib.(27) We compared the dissociation rates from EGFR and HER2 between S-222611 and lapatinib and found that the rates of dissociation of S-222611 were significantly slower than those of lapatinib (Fig. S3a,b). Importantly, these results showed that S-222611 was a reversible inhibitor like lapatinib.
Next we evaluated the growth inhibitory activity for NCI-N87 cells with short-time pulse treatment, a pattern which resembles the drug exposure in vivo. Generally, the shorter the pulse treatment time, the higher the concentration of drug needed to inhibit the growth. If the kinase inhibition by drug is more sustainable, it is reasoned that the increase of IC50 in the short-time pulse treatment would be smaller. The IC50 values of S-222611 and lapatinib were calculated and the increase of the IC50 in short-time pulse treatment is represented in Figure 1(f). Compared to 72-h treatment, the IC50 of lapatinib increased 44.9, 33.7 and 14.1-fold with 1, 6 and 24-h treatment, respectively. However, the IC50 value of S-222611 increased only 13.1, 6.2 and 3.1-fold with 1, 6 and 24-h treatment, respectively. In addition, the differences of IC50 between S-222611 and lapatinib were 17.0, 27.0 and 22.6 with 1, 6 and 24-h treatment, respectively. Thus, S-222611 was clearly superior to lapatinib with short-time pulse exposure. Considering these data along with sustained inhibition of phosphorylated EGFR and HER2 in the tumor in vivo, the slower dissociation rate of S-222611 should contribute to its superior in vivo antitumor activity over lapatinib.
In vivo antitumor activity in various types of cancer
As EGFR and HER2 are expressed in many types of cancer, S-222611 was tested for its antitumor activity in various cancer models in vivo (Fig. 2a–d, Table S2). In HER2-overexpressing human breast cancer models, S-222611 showed remarkable antitumor activity in the BT474 tumor model (Fig. 2a) and complete growth inhibition at 50 mg/kg even in the relatively less sensitive MDA-MB-361 tumor model (Fig. 2b). The antitumor activity of lapatinib for EGFR-dominant cancer was limited compared with that for HER2-expressing cancer.(28,29) S-222611 showed potent antitumor activity even in EGFR-dominant tumor models (Fig. 2c,d and Table S2). Moreover, the treatment with a higher dose of S-222611 (>50 mg/kg) completely inhibited the growth of tumors or caused marked tumor regression in most of the models tested. These results indicate that S-222611 should be an effective treatment in a variety of tumor types.
Fig. 2.
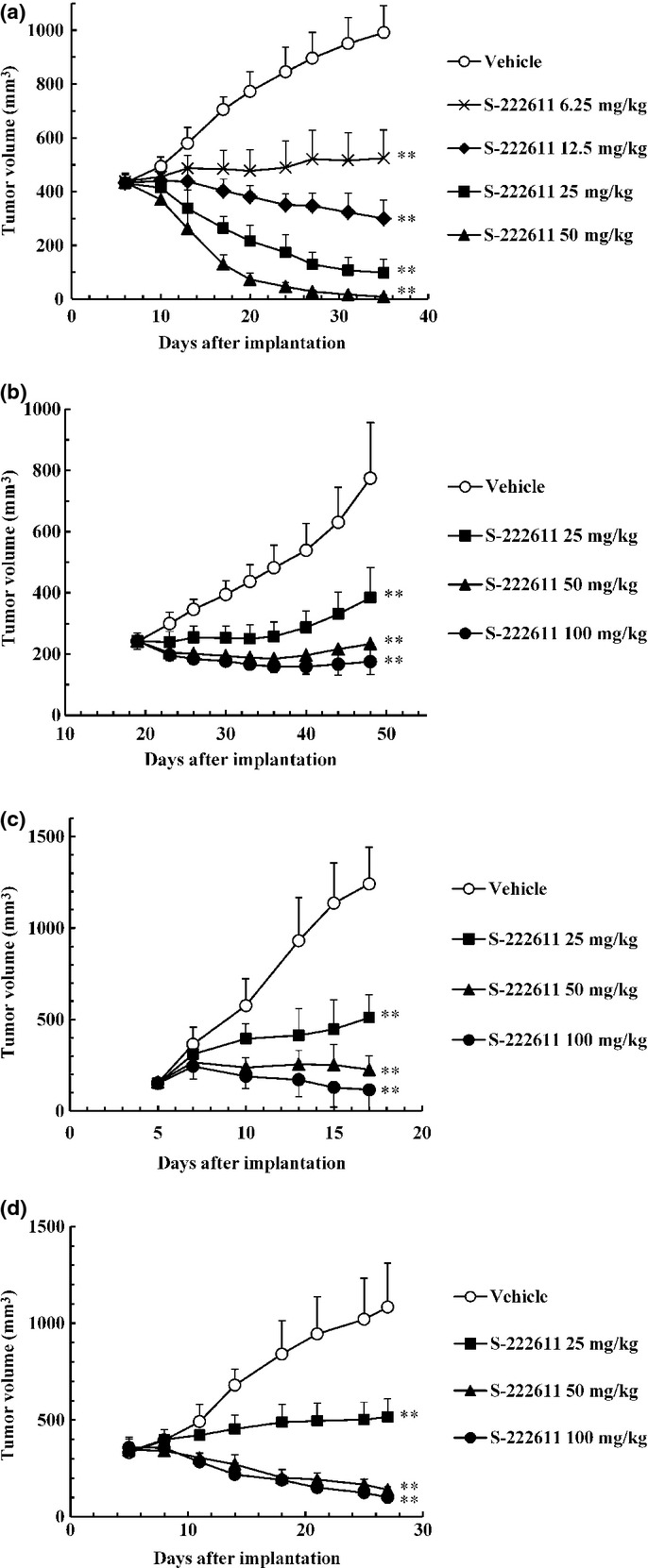
S-222611 inhibits the growth of various types of cancer in animal models. In all animal models, vehicle or indicated doses of S-222611 were orally administered daily. Multiple doses of lapatinib were also administered in the same experiment simultaneously and the ED50 value of both S-222611 and lapatinib were determined (Table S2). Tumor volume was measured twice or thrice weekly and the mean tumor volume with SD is represented by each data point. (a,b) Human breast cancer cells, BT-474 (a) or MDA-MB-361 (b) were implanted orthotopically into the mammary fat pad of mice. Tumor-bearing mice were treated for 28 days. a: n = 7, b: n = 12. (c,d) Human epidermoid carcinoma cells, A431 (c), or human colon cancer cells, HT115 (d), were implanted subcutaneously into the back of mice. Tumor-bearing mice were treated for 11 and 21 days, respectively. c,d: n = 6. **: P < 0.01 (versus vehicle, Dunnett's test).
In vivo activities of S-222611 on the cancer cell growth in bone marrow and brain and on the survival in intracranial implantation model
We evaluated S-222611 in patient-oriented models. Because bone metastasis is observed in more than 80% of advanced breast cancer patients with significant morbidity, we examined S-222611 using a model of luciferase-expressing human breast cancer cells implanted into the femur of nude mice.(30) S-222611 showed approximately four times more potent activity than lapatinib and completely inhibited the growth of cancer cells at 50 mg/kg (Fig. 3a,b).
Fig. 3.

In vivo antitumor activity of S-222611 in patient-oriented preclinical models. (a–d) Luciferase-expressing human breast cancer cell line, BT474-luc, were implanted into the bone marrow of the left femur (a,b) or into the brain (c,d) of mice. (a,c) Tumor growth was monitored by the photons emitted from the tumor as an indicator, which was measured with an IVIS Imaging System 200 (Caliper Life Sciences). (b,d) Bioluminescent images at the last measurement. Tumor-bearing mice were orally administered vehicle or the indicated doses of S-222611 or lapatinib for 21 days (a,b) or 28 days (c,d). The photons were measured once or twice weekly and the mean photons/sec with SD are presented at each data point. a,b: n = 5, c,d: n = 7. In the experiment with intracranially implanted mice (c,d), 1 mouse treated with 200 mg/kg lapatinib died accidentally during the anesthesia for in vivo imaging at 13 days after the initial measurement. The death was not considered to be related to lapatinib treatment. Thus, the data points of the 200 mg/kg lapatinib group at 14 days after initial measurement represent the mean and SD of the data from 6 mice. ns: P ≥ 0.05, **: P < 0.01 (versus vehicle, Dunnett's test). #: P < 0.01 (50 mg/kg S-222611 vs 50 mg/kg lapatinib, Welch's t-test). (e) Human breast cancer cell line, BT474, were implanted into the brain of nude mice. After randomization at 36 days after implantation (n = 10), vehicle or multiple doses of S-222611 or lapatinib were orally administered daily until death or the end of study (106 days after implantation). ns: P ≥ 0.05, **: P < 0.01 (log-rank test with Dunn–Šidák multiple test correction).
Next, we tested S-222611 in the intracranial implantation model of breast cancer cells, because brain metastasis occurs in 10–16% of breast cancer patients.(31) In this model, 25 mg/kg S-222611 significantly inhibited the growth of cancer cells in the brain to the same degree as 200 mg/kg lapatinib (Fig. 3c,d). This strong activity against intracranial tumor was further confirmed in survival assessments. S-222611 significantly prolonged survival at doses over 40 mg/kg, although the activity of lapatinib was not significant even at 200 mg/kg (Fig. 3e). All mice treated with 80 mg/kg S-222611 survived until the last observation (106 days after implantation). These results support the potential clinical value of S-222611 in the treatment of metastatic disease to the brain.
Comparison of S-222611 with irreversible inhibitors
Recently, several irreversible EGFR/HER2 inhibitors have been evaluated in clinical trials and shown promising efficacy.(9)– (15) We compared the antitumor activity of S-222611 with irreversible inhibitors neratinib and afatinib (Fig. 4a). If we simply compared these drugs at the same dose, the efficacy of S-2322611 was weaker than those of both irreversible inhibitors, although statistical significance was observed only between 25 mg/kg S-222611 and 25 mg/kg afatinib. Generally, irreversible inhibitors are more potent in kinase inhibition and antitumor activity than reversible inhibitors, but the clinical trials of these irreversible inhibitors revealed that the frequency and degree of the adverse effect are also significantly increased.(10)– (15,32,33) To assess the toxicity profile, we conducted a histopathological examination with mice used in the antitumor efficacy studies and observed the pivotal toxicity in the colon and eyeball in neratinib-treated and afatinib-treated mice. In the colon, mucosal regeneration was observed in 2/9 mice of the 25 mg/kg neratinib-treated group and 4/9 mice of the 12.5 mg/kg afatinib-treated group (Fig. 4b). These findings indicated prior injuries in the colon intestine. In the eyeball, atrophy in the corneal epithelium was observed in 1/9 mouse of the 6.25 and 25 mg/kg neratinib-treated groups, and 5/9 mice of the 6.25 and 12.5 mg/kg afatinib-treated groups (Fig. 4c). Neither of these toxicological findings was observed with 12.5 and 25 mg/kg S-222611-treated mice. These results indicated that S-222611 should be more effective than irreversible inhibitors when compared at doses that are not associated with toxic changes.
Fig. 4.
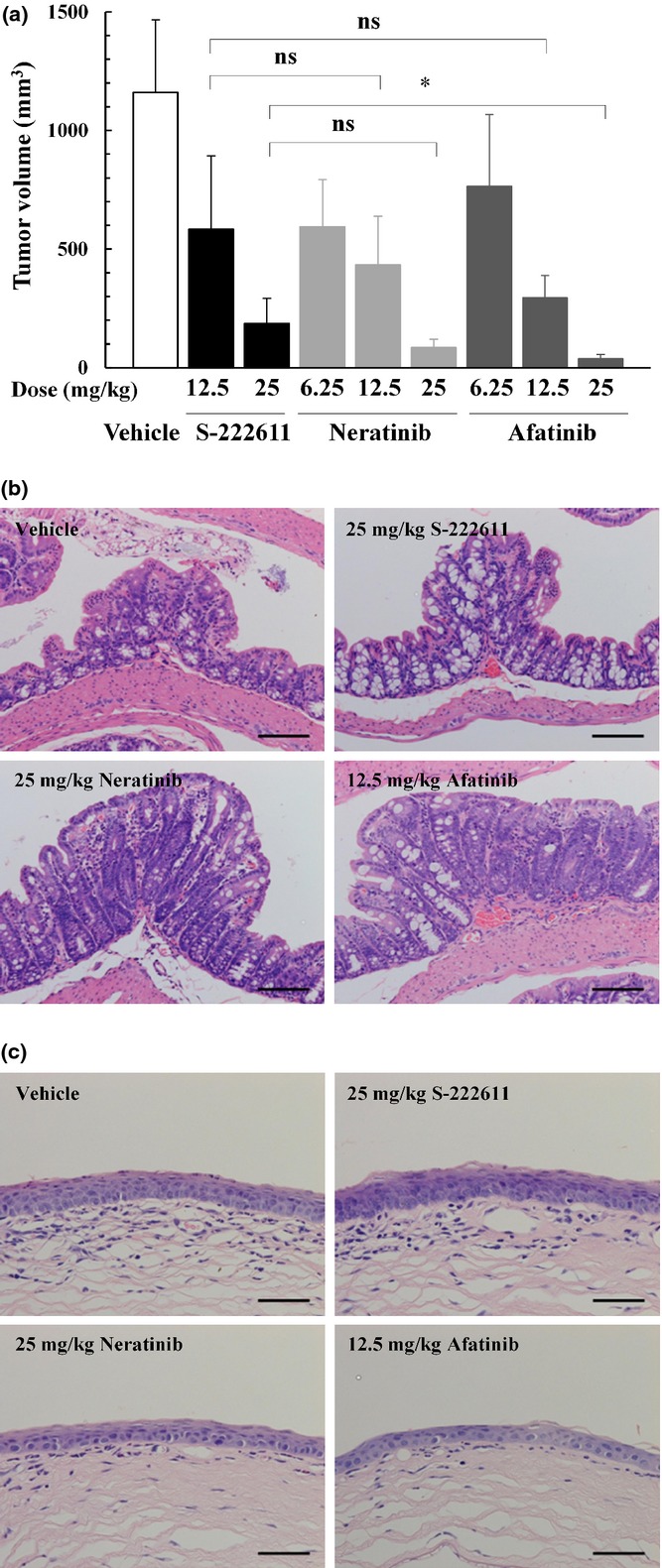
In vivo antitumor activity and toxicity profile of S-222611, neratinib and afatinib in the NCI-N87 model. (a) Human gastric cancer cells, NCI-N87, were implanted subcutaneously into nude mice. After randomization at 3 days after implantation (n = 9), vehicle or multiple doses of S-222611, neratinib or afatinib were orally administered daily for 21 days. Mean tumor volume with SD at the last measurement (the day following the last administration) is represented in each bar graph. ns: P ≥ 0.05, *: P < 0.05 (Welch's t-test with Dunn–Šidák multiple test correction). (b,c) Representative histopatholgical images of the colon (b) and eyeball (c) of mice treated with vehicle, S-222611 (25 mg/kg), neratinib (25 mg/kg) and afatinib (12.5 mg/kg). Regeneration in the colonic epithelium (b) and atrophy in the corneal epithelium were observed in neratinib-treated and afatinib-treated mice. No remarkable changes were observed in vehicle-treated and S-222611-treated mice. Stained with H&E. Bar: 100 μm.
Discussion
Our initial observations documented that S-222611 is a potent kinase inhibitor with an IC50 lower than 10 nmol/L for both EGFR and HER2. Its inhibitory activity was comparable with that of lapatinib by simple comparison of the IC50 value (Table 1a). Subsequently, cell-based evaluation in vitro and antitumor efficacy evaluation in vivo showed S-222611 to be significantly superior to lapatinib (Tables 1a,c,S2 and Figs 1, 2a–d). Extensive characterization of S-222611 revealed relative insensitivity to attenuation by serum protein binding (Table 1d) and sustained kinase-inhibitory activity (Fig. 2c), both of which contribute to the superiority of S-222611 in the in vivo evaluation.
Nonclinical examination and clinical trials to date have established the antitumor efficacy of lapatinib against HER2-expressing cancer while that against EGFR-expressing cancer is limited.(7,8,28,29,34)– (36) Although the ED50 for highly sensitive HER2-dominant cancer, NCI-N87 and BT-474, were smaller than those for other cancers, The ED50 for EGFR-dominant cancer (A431, NCI-H292, SW48, HT115, LoVo and A498) were comparable to those for less-sensitive HER2-dominant cancer (MDA-MB-361 and Calu-3), which indicated that both EGFR inhibition and HER2 inhibition contributed to antitumor activity of S-222611 (Table S2). In addition, we have shown that the in vivo antitumor efficacy of S-222611 was superior to that of lapatinib in all the models tested, including EGFR-dominant cancers (Fig. 2c,d, Table S2).
Human plasma AUC of lapatinib at a clinical dose (1200–1250 mg, once daily) is 14.3–36.2 h μg/mL.(37,38) For S-222611, it is 16.4 h μg/mL at the recommended phase 2 dose, 800 mg.(19,20) With the pharmacokinetic parameters of lapatinib and S-222611 in nude mice (Table S4), the clinically relevant doses in mice are assumed to be approximately 50 and 50–100 mg/kg for lapatinib and S-222611, respectively. Considering clinical relevancy, the ED50 of S-222611 in all tested models was well below the clinically relevant dose (Table S2). For lapatinib, the clinically relevant dose barely reached the ED50 in only three out of 10 models. Thus, the antitumor activity of S-222611 is at the level expected for clinical efficacy against both EGFR-dominant and HER2-dominant cancers.
The potential clinical relevance of antitumor activity of S-222611 was confirmed with two important patient-oriented models: the “bone metastasis” model and “brain metastasis” model (Fig. 3a–e). Excellent antitumor activity of S-222611 was achieved at clinical relevant doses in both models. Lapatinib has been extensively studied as a clinical therapeutic approach in central nervous system metastasis,(39)– (41) but it is of interest that S-222611 was eight times more potent in growth inhibition (Fig. 3b) and resulted in prolonged survival (Fig. 3e) in the “brain metastasis” model. Relatively higher tissue concentrations of S-222611 (Fig. S2b,c) should contribute to these excellent antitumor activities.
Two irreversible EGFR/HER2 inhibitors, neratinib and afatinib, have been developed in clinical trials for breast and non-small lung cancer and the latter has been approved worldwide.(11) The clinical studies have shown efficacy, but the frequency and severity of the adverse effects have been apparently higher than those observed with reversible inhibitors.(32,33) As shown in Figure 4(a), the antitumor activity of S-222611 was slightly weaker than those of neratinib and afatinib at the same dose. However, in histopathological study, mucosal regeneration of the colon and corneal atrophy of the eyeball were observed with neratinib-treated and afatinib-treated mice, but not with S-222611-treated mice (Fig. 4b,c).
The mucosal regeneration pattern is consistent with intestinal injuries and may be related to diarrhea.(42) Diarrhea, a common side effect of EGFR kinase inhibitors, occurred in 85–100% of the patients treated with irreversible inhibitors.(10)– (15,32,33) This frequency was apparently higher than that observed with reversible inhibitors (25–57%).(32,33) Diarrhea was mostly manageable but dose reduction or therapy discontinuation was often needed for the patient experienced higher grade diarrhea.(12)– (15,32)
The corneal abnormality is considered to have resulted from the intervention of EGFR signals in the cornea.(43,44) This adverse effect was less commonly reported with EGFR inhibitiors,(43,45) but rarely led to serious consequences like persistent corneal epithelial defect and perforating corneal ulceration.(46,47) As these histopathological findings have clinical relevance, S-222611 should have a better safety profile than irreversible inhibitors.
Our findings suggest that S-222611 can achieve an excellent balance between efficacy and toxicity. Phase I trials of S-222611 have shown very promising results,(19,20) and phase II trials are in progress.
Acknowledgments
This study was sponsored by Shionogi & Co., Ltd. The authors would like to thank Masaharu Kume, Kazuo Ueda and other chemists at Shionogi & Co., Ltd. who worked together in synthesizing S-222611 and other EGFR/HER2 inhibitors.
Disclosure Statement
HT, MH, MI, SS, TY, KD, KH, YI, MY, KM, TS, KM, SY and MS are employees of Shionogi & Co., Ltd. TW is an employee of Shionogi Techno Advance Research Co., Ltd. EPF consults for Shionogi & Co., Ltd.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1 Dose-dependent antitumor efficacy of lapatinib in the NCI-N87 model.
Fig. S2 Tumor, brain and bone marrow concentrations of S-222611 and lapatinib in tumor-bearing nude mice.
Fig. S3 Slow off-rate dissociation of S-222611 from epidermal growth factor receptor and human epidermal growth factor receptor 2.
Table S1 Kinase inhibition profile of S-222611.
Table S2 Comparison of ED50 values between S-222611 and lapatinib in various tumor models.
Table S3 Pharmacokinetics of S-222611 and lapatinib in nude mice.
Data S1 Legends for supplementary figures and tables.
Data S2 Materials and methods for supplementary figures and tables.
References
- 1.Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12:553–63. doi: 10.1038/nrc3309. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Z, Stiegler AL, Boggon TJ, Kobayashi S, Halmos B. EGFR-mutated lung cancer: a paradigm of molecular oncology. Oncotarget. 2010;1:497–514. doi: 10.18632/oncotarget.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moscatello DK, Holgado-Madruga M, Godwin AK, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995;55:5536–9. [PubMed] [Google Scholar]
- 4.Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer progression. Oncogene. 2007;26:6469–87. doi: 10.1038/sj.onc.1210477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen HX, Cleck JN, Coelho R, Dancey JE. Epidermal growth factor receptor inhibitors: current status and future directions. Curr Probl Cancer. 2009;33:245–94. doi: 10.1016/j.currproblcancer.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee A, Dhadda AS, Shehata M, Chan S. Lapatinib: a tyrosine kinase inhibitor with a clinical role in breast cancer. Expert Opin Pharmacother. 2007;8:2189–204. doi: 10.1517/14656566.8.13.2189. [DOI] [PubMed] [Google Scholar]
- 7.Ravaud A, Hawkins R, Gardner JP, et al. Lapatinib versus hormone therapy in patients with advanced renal cell carcinoma: a randomized phase III clinical trial. J Clin Oncol. 2008;26:2285–91. doi: 10.1200/JCO.2007.14.5029. [DOI] [PubMed] [Google Scholar]
- 8.Iqbal S, Goldman B, Fenoglio-Preiser CM, et al. Southwest Oncology Group study S0413: a phase II trial of lapatinib (GW572016) as first-line therapy in patients with advanced or metastatic gastric cancer. Ann Oncol. 2011;22:2610–5. doi: 10.1093/annonc/mdr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou SHI. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence. Crit Rev Oncol Hematol. 2012;83:407–21. doi: 10.1016/j.critrevonc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 10.López-Tarruella S, Jerez Y, Márquez-Rodas I, Martín M. Neratinib (HKI-272) in the treatment of breast cancer. Future Oncol. 2012;8:671–81. doi: 10.2217/fon.12.66. [DOI] [PubMed] [Google Scholar]
- 11.Keating GM. Afatinib: a review of its use in the treatment of advanced non-small cell lung cancer. Drugs. 2014;74:207–21. doi: 10.1007/s40265-013-0170-8. [DOI] [PubMed] [Google Scholar]
- 12.Burstein HJ, Sun Y, Dirix LY, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28:1301–7. doi: 10.1200/JCO.2009.25.8707. [DOI] [PubMed] [Google Scholar]
- 13.Martin M, Bonneterre J, Geyer CE, Jr, et al. A phase two randomised trial of neratinib monotherapy versus lapatinib plus capecitabine combination therapy in patients with HER2+ advanced breast cancer. Eur J Cancer. 2013;49:3763–72. doi: 10.1016/j.ejca.2013.07.142. [DOI] [PubMed] [Google Scholar]
- 14.Sequist LV, Yang JCH, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3227–34. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 15.Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–22. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 16.Kume M, Matsuo K, Omori N, Takayama M inventors; Shionogi & Co., Ltd., assignee. 2012. Quinazoline derivatives having tyrosine kinase inhibitory activity. United States patent US 8,202,879.; 19 Jun.
- 17.Hirata M, Tanaka H, Dohi K, et al. 2009. S-222611, a potent, orally active small molecule inhibitor of EGFR and HER2: in vitro kinase inhibition and antitumor activity [abstract]. Proceedings of the 100th Annual Meeting of the American Association for Cancer Research; 18−22 Apr 2009, Denver, CO. Philadelphia (PA): AACR,. Abstract number 1761.
- 18.Iguchi M, Tanaka H, Wada T, et al. 2009. S-222611, a potent, orally active small molecule inhibitor of EGFR and HER2: in vivo antitumor effects [abstract]. Proceedings of the 100th Annual Meeting of the American Association for Cancer Research; 18–22 Apr 2009, Denver, CO. Philadelphia, PA: AACR,. Abstract number 1762.
- 19.Baird RD, Papa S, Cresti N, et al. 2012. A phase I study of S-222611 an oral reversible dual inhibitor of EGFR and HER2, in patients with solid tumors. [abstract]. Proceedings of 2012 ASCO Annual Meeting; 1–5 Jun 2012, Chicago, IL. Alexandria, VA: ASCO,. Abstract number 3100.
- 20.Baird RD, Cresti N, Beddowes E, et al. 2013. Phase I trial of S-222611, a dual tyrosine kinase inhibitor of EGFR and HER2, with preliminary evidence of efficacy in patients (pts) with heavily-pretreated HER2-positive metastatic breast cancer. [abstract]. Proceedings of 36th Annual San Antonio Breast Cancer Symposium; 10–14 Dec 2013, San Antonio, TX. San Antonio, TX: SABCS,. Abstract number P4-12-24.
- 21.Lackey K, Spector N, Wood ER, Xia W inventors; Glaxo Group Limited, assignee. 2002. Cancer treatment method. World patent WO 02/056912.; 25 Jul.
- 22.Kimberly GP, Zhang YM, Carter M, et al. Optimization and SAR for dual ErbB-1/ErbB-2 tyrosine kinase inhibition in 6-furanylquinazoline series. Bioorg Med Chem Lett. 2006;16:4686–91. doi: 10.1016/j.bmcl.2006.05.090. [DOI] [PubMed] [Google Scholar]
- 23.Tsou HR, Overbeek-Klumpers EG, Hallett WA, et al. Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J Med Chem. 2005;48:1107–31. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]
- 24.Soyka R, Rall W, Schnaubelt J, Sieger P, Kulinna C inventors; Boehringer Ingelheim International GmbH, assignee. 2005. Process for preparing amino crotonyl compounds. United States patent US 2005/0085495.; 21 Apr.
- 25.Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 26.Polli JW, Humphreys JE, Harmon KA, et al. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36:695–701. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- 27.Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–9. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 28.Konecny GE, Pegram MD, Venkatesan N, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630–9. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 29.Zhang D, Pal A, Bornmann WG, et al. Activity of lapatinib is independent of EGFR expression level in HER2-overexpressing breast cancer cells. Mol Cancer Ther. 2008;7:1846–50. doi: 10.1158/1535-7163.MCT-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozlow W, Guise TA. Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia. 2005;10:169–80. doi: 10.1007/s10911-005-5399-8. [DOI] [PubMed] [Google Scholar]
- 31.Mehta AI, Brufsky AM, Sampson JH. Therapeutic approaches for HER2-positive brain metastases: circumventing the blood–brain barrier. Cancer Treat Rev. 2013;39:261–9. doi: 10.1016/j.ctrv.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang JCH, Reguart N, Barinoff J, et al. Diarrhea associated with afatinib: an oral ErbB family blocker. Expert Rev Anticancer Ther. 2013;13:729–36. doi: 10.1586/era.13.31. [DOI] [PubMed] [Google Scholar]
- 33.Köhler J, Schuler M. Afatinib, erlotinib and gefitinib in the first-line therapy of EGFR mutation-positive lung adenocarcinoma: a review. Onkologie. 2013;36:510–8. doi: 10.1159/000354627. [DOI] [PubMed] [Google Scholar]
- 34.Bekaii-Saab T, Markowitz J, Prescott N, et al. A multi-institutional phase II study of the efficacy and tolerability of lapatinib in patients with advanced hepatocellular carcinomas. Clin Cancer Res. 2009;15:5895–901. doi: 10.1158/1078-0432.CCR-09-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross HJ, Blumenschein GR, Jr, Aisner J, et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin Cancer Res. 2010;16:1938–49. doi: 10.1158/1078-0432.CCR-08-3328. [DOI] [PubMed] [Google Scholar]
- 36.Frank D, Jumonville A, Loconte NK, Schelman WR, Mulkerin D, Lubner S. A phase II study of capecitabine and lapatinib in advanced refractory colorectal adenocarcinoma: a Wisconsin Oncology Network study. J Gastrointest Oncol. 2012;3:90–6. doi: 10.3978/j.issn.2078-6891.2011.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burris HA, 3rd, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23:5305–13. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- 38.Chu QS, Schwartz G, de Bono J, et al. Phase I and pharmacokinetic study of lapatinib in combination with capecitabine in patients with advanced solid malignancies. J Clin Oncol. 2007;25:3753–8. doi: 10.1200/JCO.2007.11.1765. [DOI] [PubMed] [Google Scholar]
- 39.Gril B, Palmieri D, Bronder JL, et al. Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst. 2008;100:1092–103. doi: 10.1093/jnci/djn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin NU, Diéras V, Paul D, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009;15:1452–9. doi: 10.1158/1078-0432.CCR-08-1080. [DOI] [PubMed] [Google Scholar]
- 41.Bachelot T, Romieu G, Campone M, et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol. 2013;14:64–71. doi: 10.1016/S1470-2045(12)70432-1. [DOI] [PubMed] [Google Scholar]
- 42.Araki E, Ishikawa M, Iigo M, Koide T, Itabashi M, Hoshi A. Relationship between development of diarrhea and the concentration of SN-38, an active metabolite of CPT-11, in the intestine and the blood plasma of athymic mice following intraperitoneal administration of CPT-11. Jpn J Cancer Res. 1993;84:697–702. doi: 10.1111/j.1349-7006.1993.tb02031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho WL, Wong H, Yau T. The ophthalmological complications of targeted agents in cancer therapy: what do we need to know as ophthalmologists? Acta Ophthalmol. 2013;91:604–9. doi: 10.1111/j.1755-3768.2012.02518.x. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura Y, Sotozono C, Kinoshita S. The epidermal growth factor receptor (EGFR): role in corneal wound healing and homeostasis. Exp Eye Res. 2001;72:511–7. doi: 10.1006/exer.2000.0979. [DOI] [PubMed] [Google Scholar]
- 45.Tullo AB, Esmaeli B, Murray PI, Bristow E, Forsythe BJ, Faulkner K. Ocular findings in patients with solid tumours treated with the epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (‘Iressa’, ZD1839) in Phase I and II clinical trials. Eye. 2005;19:729–38. doi: 10.1038/sj.eye.6701630. [DOI] [PubMed] [Google Scholar]
- 46.Johnson KS, Levin F, Chu DS. Persistent corneal epithelial defect associated with erlotinib treatment. Cornea. 2009;28:706–7. doi: 10.1097/ICO.0b013e31818fdbc6. [DOI] [PubMed] [Google Scholar]
- 47.Ibrahim E, Dean WH, Price N, et al. Perforating corneal ulceration in a patient with lung metastatic adenocarcinoma treated with gefitinib: a case report. Case Rep Ophthalmol Med. 2012;2012:379132. doi: 10.1155/2012/379132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Dose-dependent antitumor efficacy of lapatinib in the NCI-N87 model.
Fig. S2 Tumor, brain and bone marrow concentrations of S-222611 and lapatinib in tumor-bearing nude mice.
Fig. S3 Slow off-rate dissociation of S-222611 from epidermal growth factor receptor and human epidermal growth factor receptor 2.
Table S1 Kinase inhibition profile of S-222611.
Table S2 Comparison of ED50 values between S-222611 and lapatinib in various tumor models.
Table S3 Pharmacokinetics of S-222611 and lapatinib in nude mice.
Data S1 Legends for supplementary figures and tables.
Data S2 Materials and methods for supplementary figures and tables.