

Abstract
Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKI) are effective for non-small cell lung cancers (NSCLC) with EGFR-activating mutations. However, most responders develop resistance. Efatutazone, a novel peroxisome proliferator-activated receptor gamma (PPARγ) agonist, is currently under clinical evaluation; it has antiproliferative effects and induces cellular morphological changes and differentiation. The present study investigated the effects of efatutazone in EGFR-TKI-resistant NSCLC cells, while focusing on cell motility. The PC-9-derived NSCLC cell lines PC-9ER and PC-9ZD, resistant to EGFR-TKI due to v-crk avian sarcoma virus CT10 oncogene homolog-like (CRKL) amplification-induced phosphatidylinositol 3-kinase (PI3K)/v-akt murine thymoma viral oncogene homolog (AKT) activation and an EGFR T790M mutation, respectively, were used. These cells exhibit enhanced cell motility due to transforming growth factor β (TGF-β)/Smad2 family member 2 (Smad2) pathway activation. Efatutazone had no growth-inhibitory effect on the tested cells but inhibited the motility of EGFR-TKI-resistant cells in wound closure and transwell assays. Efatutazone plus erlotinib treatment provided greater inhibition of PC-9ER cell migration than efatutazone or erlotinib alone. Efatutazone suppressed increased TGF-β2 secretion from both cell lines (shown by ELISA) and downregulation of TGF-β2 transcription (observed by quantitative RT-PCR). Immunoblot analysis and luciferase assays revealed that efatutazone suppressed Smad2 phosphorylation and its transcriptional activity. These results suggest that efatutazone inhibits cell motility by antagonizing the TGF-β/Smad2 pathway and effectively prevents metastasis in NSCLC patients with acquired resistance to EGFR-TKI regardless of the resistance mechanism.
Keywords: Efatutazone, epidermal growth factor receptor-tyrosine kinase inhibitor resistance, motility, non-small cell lung cancer, peroxisome proliferator-activated receptor gamma
Lung cancer is a leading cause of cancer-related death worldwide,1 and more than 60% of lung cancer patients have inoperable advanced-stage disease at diagnosis.2,3 For years, platinum-based doublet chemotherapy has been the standard therapy for patients with advanced non-small cell lung cancer (NSCLC).4 In recent years, clinical implementation of epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK)-targeted therapies for patients with EGFR-activating mutations and ALK fusion genes, respectively, has led to a paradigm shift in NSCLC treatment.5 However, only a handful of NSCLC patients benefit from these molecular-targeted therapies, and despite an initial dramatic response to the EGFR-tyrosine kinase inhibitors (TKI) gefitinib and erlotinib, the majority of NSCLC patients with EGFR-activating mutations acquire resistance.6 Therefore, the development of novel, efficacious chemotherapeutic agents is urgently needed.
Peroxisome proliferator-activated receptor gamma (PPARγ) agonists have recently been developed as a novel class of anticancer agents. They have been shown to possess antitumor properties in preclinical models of human cancers, including NSCLC.7–9 PPARγ belongs to the ligand-activated nuclear hormone receptor superfamily, and it heterodimerizes with retinoid-X receptors to regulate the transcription of genes associated with energy homeostasis.8,10 PPARγ can be activated by fatty acids, prostaglandins, eicosanoid derivatives and some synthetic thiazolidinediones (TZD), including antidiabetic drugs such as troglitazone, rosiglitazone and pioglitazone.8,10 In addition to their insulin-sensitization effect, in preclinical models of human cancers these TZD have been shown to possess antitumor effects, such as growth inhibition, cell-cycle arrest, apoptosis induction, cell differentiation induction, and migration prevention activities.7–9 Efatutazone (CS-7017, RS5444) is a novel, third-generation, TZD-class PPARγ agonist11 that is an approximate 50-fold and 500-fold more potent PPARγ activator than the second-generation TZD rosiglitazone and troglitazone, respectively.12 Efatutazone inhibits the proliferation of human pancreatic and anaplastic thyroid tumor cells and the growth of tumor colorectal cancer xenografts.11,12 Moreover, efatutazone can induce morphological changes and differentiation.11 The results of a phase I clinical trial demonstrated acceptable tolerability, with evidence of disease control in patients with advanced malignancies.13 A phase II clinical trial of efatutazone in combination with the EGFR-TKI erlotinib in advanced NSCLC patients has been completed, and the data are currently being analyzed.14
We previously reported that EGFR-TKI-resistant NSCLC cells acquire enhanced cell motility via transforming growth factor β2 (TGF-β2)-induced activation of the TGF-β/Smad2 family member 2 (Smad2) pathway.15,16 Interaction between PPARγ and the TGF-β pathway has been reported in numerous studies.17 Therefore, we hypothesized that the PPARγ agonist efatutazone may attenuate the enhanced motility of EGFR-TKI-resistant NSCLC cells. In this study, we investigated the effects of efatutazone on the enhanced cell motility of EGFR-TKI-resistant NSCLC cells and the mechanisms underlying its inhibitory effects.
Materials and Methods
Cell lines, cell culture and reagents
PC-9, a human NSCLC (adenocarcinoma) cell line harboring an EGFR-activating mutation (E746-A750del at exon 19) and gefitinib-resistant PC-9ZD cells were provided by Dr Fumiaki Koizumi (National Cancer Center Hospital, Tokyo, Japan).18 Erlotinib-resistant PC-9 derivative cell lines, PC-9ER1 and PC-9ER4, were also established.15 Resistance to EGFR-TKI in PC-9ER and PC-9ZD cells is conferred by v-crk avian sarcoma virus CT10 oncogene homolog-like (CRKL) amplification-induced constitutive activation of phosphatidylinositol 3-kinase (PI3K)/v-akt murine thymoma viral oncogene homolog (AKT) signaling15,16 and an EGFR T790M mutation,18 respectively. All the cell lines used in the present study were maintained in RPMI1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated FBS (Invitrogen) at 37°C in humidified air containing 5% CO2. Efatutazone was supplied by Daiichi Sankyo (Tokyo, Japan). Erlotinib was purchased from LC Laboratories (Woburn, MA, USA).
Wound closure assay
Cells (5 × 105 cells/well) were seeded into six-well plates (Corning Costar, Cambridge, MA, USA). Once the cells reached confluence, an artificial wound was created with a 200-μL pipette tip (at time point 0). After the cells were washed with FBS-free medium, they were then incubated for 16 h in fresh 1% FBS medium containing either DMSO (0.1%; control), erlotinib (1 μmol/L), efatutazone (10 μmol/L), or a combination of erlotinib and efatutazone. Cell motility was assessed by measuring the movement of the cells by microscopy during closure of the wound.
Transwell assay
Cells (1.5 × 105 cells/well) were seeded in FBS-free medium in the upper chamber of a 5-μm pore filter unit (Corning Costar) and incubated in 1% FBS medium with DMSO (0.1%; control), erlotinib (1 μmol/L), efatutazone (10 μmol/L), or a combination of erlotinib and efatutazone. After 48 h, the cells that had migrated through the pores and reattached to the bottom of the lower chamber were trypsinized, and the number of cells was counted with a Z1 Coulter Particle Counter (Beckman Coulter, Brea, CA, USA).
Quantitative RT-PCR
Cells (7 × 105 cells/dish) were seeded into 60-mm dishes (Corning Costar) and incubated for 30 h. After the cell culture medium was replaced with FBS-free medium, cells were incubated for an additional 12 h in FBS-free medium with DMSO (0.1%; control) or efatutazone (10 μmol/L). Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. To quantify target gene copies, a DNA fragment of each gene was cloned into the pCR2.1-TOPO vector by using the TOPO TA Cloning Kit (Invitrogen). Then, serial dilutions (13 dilutions containing 10–108 copies) of the plasmids were used to generate standard curves. The cDNA template was synthesized from total RNA (1 μg) by using oligo (dT)12–18 primers (Invitrogen) and the Omniscript RT Kit (QIAGEN) according to the manufacturer's instructions. qRT-PCR was performed using the StepOnePlus Real Time PCR system (Applied Biosystems, Foster City, CA, USA), with SYBR Premix Ex Taq II (Tli RNaseH Plus; TAKARA BIO, Shiga, Japan) and Perfect Real Time PCR primers for each gene (TAKARA BIO).
Transforming growth factor β1 and transforming growth factor β2 ELISA
Cells (3 × 105 cells/well) were seeded in six-well plates (Corning Costar), and incubated for 30 h. Then, the cells were incubated in fresh FBS-free medium with DMSO (0.1%; control) or efatutazone (10 μmol/L) for an additional 60 h, and the culture medium was collected and centrifuged. The amount of TGF-β1 or TGF-β2 in the conditioned medium was quantified using the Quantikine ELISA Kit for human TGF-β1 or TGF-β2 (R&D Systems, Minneapolis, MN, USA), respectively. Then, acid activation was performed according to the manufacturer's instructions. The raw data were normalized to cell number.
Immunoblot analysis
Cultured cells were lysed in M-PER mammalian protein extraction reagent with the Halt protease inhibitor cocktail, EDTA-free and the Halt phosphatase inhibitor cocktail (Pierce Chemical, Rockford, IL, USA). Whole cell lysates containing equal amounts of protein (20 μg, as measured by BCA protein assay reagent [Pierce Chemical]) were separated by 8% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked with 5% skim milk or 5% BSA in TBS plus Tween 20 (TBST) for 1 h at room temperature, and then incubated overnight at 4°C with primary antibodies in TBST containing 5% skim milk or 5% BSA. After washing the membranes with TBST, they were incubated with secondary antibodies conjugated to HRP for 1 h at room temperature, and then washed in TBST. Immunoreactive bands were visualized with SuperSignal West Pico Chemiluminescent Substrate (Pierce Chemical). The antibodies used for this analysis are listed in Table S1.
Luciferase assay
Cells (5 × 104 cells/well) were seeded into a 24-well plate (Corning Costar). After a 48-h incubation, the cells were transfected with 1 μg/well of either p3TP-Lux Smad-dependent or pPG2-aP2-TK PPARγ-dependent firefly luciferase reporter plasmid by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfection efficiency was normalized by cotransfecting 16 ng/well of pRL-CMV (Promega, Madison, WI, USA) containing Renilla reniformis luciferase. The next day, the cells were treated with RPMI1640 containing DMSO (0.1%; control), erlotinib (1 μmol/L), efatutazone (10 μmol/L), or a combination of erlotinib and efatutazone. After 24 h, the cells were lysed with reporter lysis buffer (Promega), and luciferase activity was assessed using the Dual-Glo luciferase assay system (Promega) with an ARVO MX luminometer (Perkin Elmer, Norwalk, CT, USA).
Results
Efatutazone attenuated the enhanced motility and migration of PC-9ER and PC-9ZD cells
The EGFR-TKI-resistant cell lines, PC-9ER and PC-9ZD, were approximately 100-fold more resistant to erlotinib than PC-9 cells (Fig. S1). PC-9ER and PC-9ZD cells migrated faster to close a wound than the parental PC-9 cells (Fig. 1a), and this enhanced cell migration was also confirmed by a transwell assay (Fig. 1b). Erlotinib not only effectively abolished the motility of PC-9 cells but also attenuated the enhanced cell motility of PC-9ER cells (Fig. 1a,b). However, erlotinib could not suppress the enhanced cell motility of PC-9ZD cells harboring the T790M resistance mutation (Fig. 1a,b). These results suggest that continuous treatment with erlotinib may have a therapeutic effect by preventing metastasis even after EGFR-TKI failure, except in cases of resistance due to the T790M mutation. In contrast, efatutazone attenuated the motility of not only PC-9 and PC-9ER cells but also PC-9ZD cells in a dose-dependent manner (Fig. 1a and Fig. S2); this was also confirmed by the transwell assay (Fig. 1b). These results imply that efatutazone would be beneficial in preventing metastasis even after EGFR-TKI treatment failure, regardless of the resistance mechanism. Moreover, combined treatment with efatutazone and erlotinib showed a more potent inhibitory effect on the migration of PC-9ER cells than either treatment alone (Fig. 1b), indicating that this combination treatment may be effective for preventing metastasis in patients with EGFR-TKI-resistant NSCLC who do not harbor the EGFR T790M resistance mutation. Efatutazone had no significant antiproliferative effect on any of the tested cell lines (Fig. S1), indicating that cell motility and cell growth are driven by different mechanisms.
Figure 1.
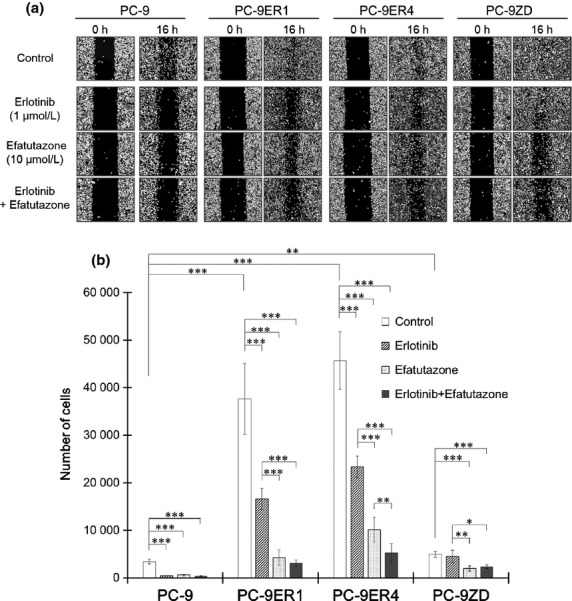
Efatutazone attenuates enhanced cell motility and migration. (a) Cells were seeded and grown to 100% confluence, and then a wound was created by scraping the cells with a 200-μL pipette tip. The wounded cells were then incubated for 16 h in 1% FBS medium with DMSO (0.1%; control), erlotinib (1 μmol/L), efatutazone (10 μmol/L), or a combination of erlotinib and efatutazone. (b) Cells were seeded into transwell chambers and incubated in 1% FBS medium with DMSO (0.1%; control), erlotinib (1 μmol/L), efatutazone (10 μmol/L), or a combination of erlotinib and efatutazone. The number of cells that migrated through the filter and attached to the bottom of the lower chamber was counted 48 h after seeding. *P < 0.05; **P < 0.01; ***P < 0.001 (Student's t-test). The error bars indicate the standard deviations of the mean.
Efatutazone treatment significantly suppressed the transcription and secretion of transforming growth factor β2
We previously reported that PC-9ER cells acquire enhanced motility via TGF-β2-induced activation of the TGF-β/Smad2 pathway, which plays an important role in cell motility and migration.15,19 Therefore, we evaluated the effect of efatutazone on the transcription and secretion of TGF-β ligands. The basal mRNA levels of TGF-β2 in PC-9ER and PC-9ZD cells were higher than those in PC-9 cells. In contrast, no significant differences in the mRNA levels of TGF-β1 were observed among these cell lines (Fig. 2a). Efatutazone treatment suppressed TGF-β2 mRNA expression in all cells, whereas it did not suppress TGF-β1 mRNA expression (Fig. 2a). PC-9ER and PC-9ZD cells secreted significantly higher amounts of TGF-β2 than PC-9 cells did; however, PC-9ER and PC-9ZD cells did not secrete higher levels of TGF-β1 (Fig. 2b). Efatutazone significantly inhibited the secretion of TGF-β2 from all cells, confirming the effect of efatutazone on TGF-β2 transcription (Fig. 2b). These results indicate that efatutazone treatment significantly suppresses TGF-β2 secretion, by suppressing TGF-β2 mRNA expression.
Figure 2.
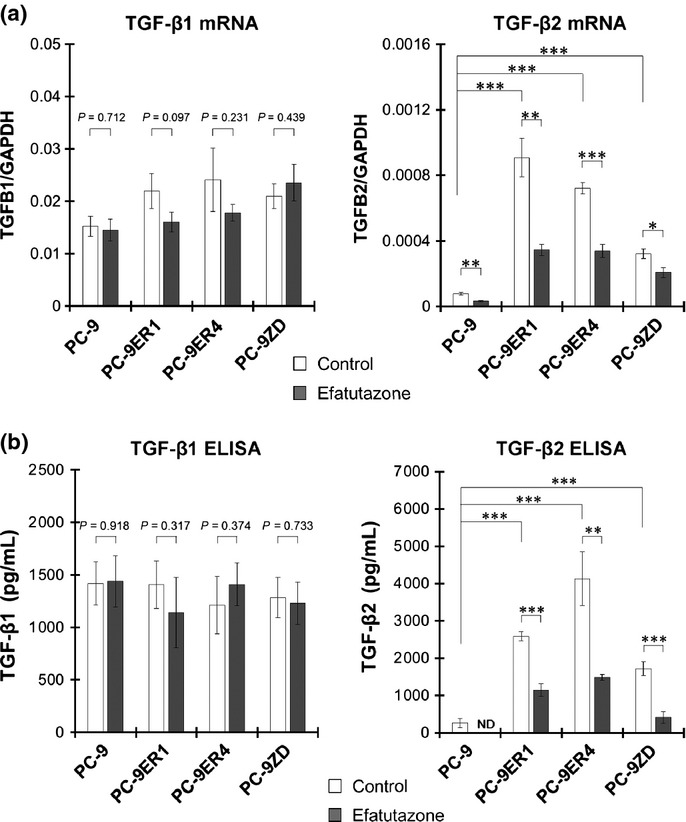
Efatutazone treatment significantly suppresses the mRNA expression and secretion of transforming growth factor β2 (TGF-β2). (a) mRNA levels of TGF-β1 and TGF-β2 after incubation for 12 h in FBS-free medium with DMSO (0.1%; control) or efatutazone (10 μmol/L) were measured by qRT-PCR. *P < 0.05; **P < 0.01; ***P < 0.001 (Student's t-test). The error bars indicate the standard deviations of the mean. (b) Cells were seeded and incubated in six-well plates for 60 h in FBS-free medium with DMSO (0.1%; control) or efatutazone (10 μmol/L). Conditioned medium was collected, and TGF-β1 and TGF-β2 levels were measured by ELISA. **P < 0.01; ***P < 0.001 (Student's t-test). The error bars indicate the standard deviations of the mean. The amount of TGF-β2 in PC-9 cells treated with efatutazone was below the detection limit. ND, not detected.
Efatutazone abrogated activation of the transforming growth factor β/Smad2 pathway in epidermal growth factor receptor-tyrosine kinase inhibitor-resistant cells
We next examined the effect of efatutazone on the activity of Smad2, which is a key downstream effector of the TGF-β pathway. Elevated phosphorylation of Smad2 and enhanced Smad2-mediated transcriptional activity were observed in both PC-9ER and PC-9ZD cells (Figs. 3a,b and Fig. S3). Efatutazone suppressed the elevated phosphorylation of Smad2 in both PC-9ER and PC-9ZD cells (Fig. 3a and Fig. S3). Efatutazone treatment also significantly decreased subsequent Smad-mediated transcriptional regulatory activity in all cells (Fig. 3b), indicating that suppression of TGF-β2 expression by efatutazone abrogates activation of the TGF-β/Smad2 pathway. These observations suggest TGF-β2-mediated cross-talk between PPARγ and the TGF-β pathway.
Figure 3.
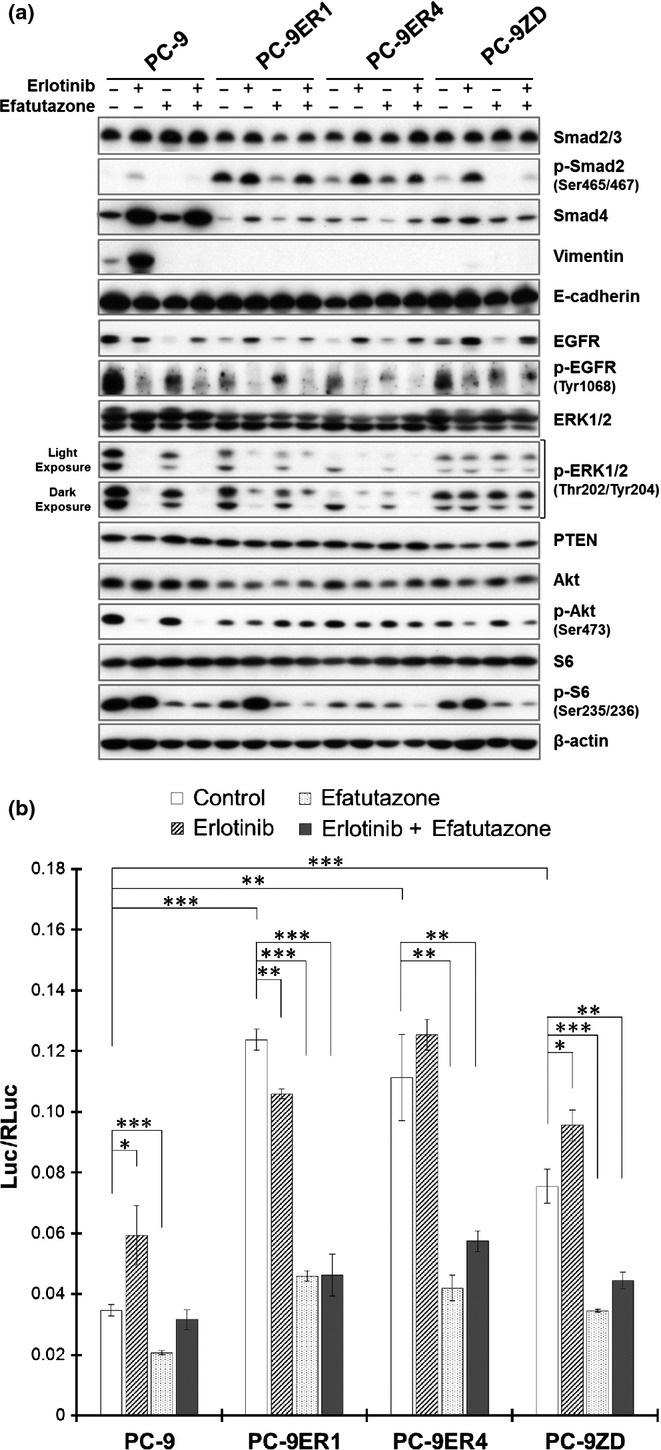
Effect of efatutazone treatment on molecules relevant to the transforming growth factor β (TGF-β), epidermal growth factor receptor (EGFR) and phosphatidylinositol 3-kinase (PI3K)/Akt pathways. (a) The effect of erlotinib and/or efatutazone on relevant molecules was evaluated by immunoblot analysis. Cells were incubated for 48 h in FBS-free medium with DMSO (0.1%; control), erlotinib (1 μmol/L), efatutazone (10 μmol/L) or a combination of erlotinib and efatutazone. The cells were lysed, and the indicated proteins were detected by immunoblotting. (b) The effect of efatutazone on Smad-mediated transcriptional activity was evaluated by using a luciferase reporter assay. After the cells were transfected with the Smad-dependent reporter p3TP-Lux, they were incubated for 24 h in FBS-free medium with DMSO (0.1%; control), erlotinib (1 μmol/L), efatutazone (10 μmol/L), or a combination of erlotinib and efatutazone. The values were normalized relative to the Renilla luciferase activity in cells cotransfected with pRL-CMV. *P < 0.05; **P < 0.01; ***P < 0.001 (Student's t-test). The error bars indicate the standard deviations of the mean.
Erlotinib induced modest phosphorylation of Smad2 and activation of Smad-mediated transcriptional activity in all tested cells (Fig. 3a,b), suggesting a compensatory response to EGFR pathway inhibition and cross-talk between EGFR and the TGF-β pathway.15 When combined with erlotinib, efatutazone effectively suppressed erlotinib-induced activation of the TGF-β pathway (Fig. 3a,b), again suggesting potential cross-talk between PPARγ and the TGF-β pathway and providing a rationale for this combined treatment.
Efatutazone treatment induced PPARγ-mediated transcriptional activity in PC-9 and PC-9ZD cells, but not in PC-9ER cells (Fig. 4), suggesting that the inhibitory effect of efatutazone on the motility of PC-9ER cells may not be due to direct activation of PPARγ signaling, but rather may be due to off-target effects.
Figure 4.
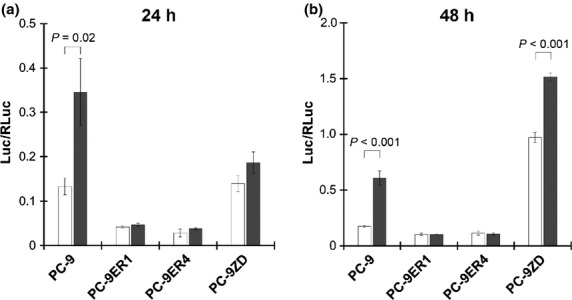
Effect of efatutazone on peroxisome proliferator-activated receptor gamma (PPARγ)-mediated transcriptional activity. After the cells were transfected with the PPARγ-dependent luciferase reporter pPG2-aP2-TK, they were incubated for 24 or 48 h in FBS-free medium with DMSO (0.1%; control) or efatutazone (10 μmol/L). PPARγ-mediated transcriptional activity was measured with a luciferase reporter assay as described in the Materials and Methods. The values were normalized relative to the Renilla luciferase activity in cells cotransfected with pRL-CMV.
Discussion
In this study, we demonstrated that the novel PPARγ agonist efatutazone attenuated the enhanced motility of EGFR-TKI-resistant NSCLC cells regardless of the resistance mechanism. This attenuation was mediated by inhibition of the TGF-β/Smad2 pathway via suppression of TGF-β2 mRNA expression (Fig. 5). These phenomena imply TGF-β2-mediated cross-talk between PPARγ and the TGF-β pathway. Several studies have been performed on the interaction between PPARγ and the TGF-β pathway.17,20–22
Figure 5.
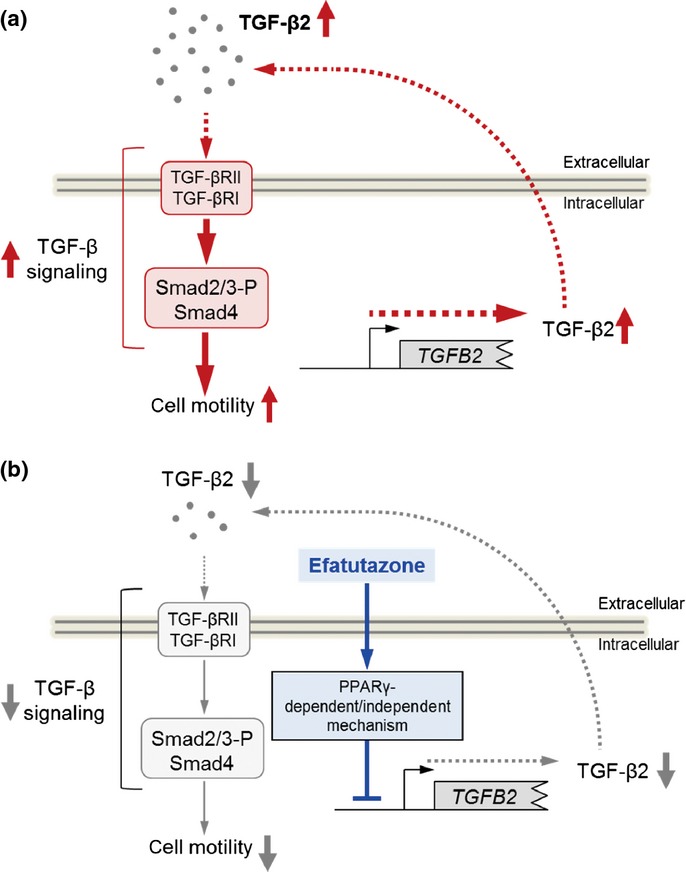
Proposed mechanisms underlying the effect of efatutazone in erlotinib-resistant non-small cell lung cancers (NSCLC) cells. (a) The enhanced motility of PC-9ER and PC-9ZD cells was induced by activation of the transforming growth factor β (TGF-β) pathway due to increased TGF-β2 mRNA expression and subsequent secretion. (b) Enhanced motility of PC-9ER and PC-9ZD cells was suppressed by treatment with efatutazone. Efatutazone treatment significantly antagonized TGF-β2-mediated activation of the TGF-β pathway by suppressing TGF-β2 mRNA expression. The effect of efatutazone on TGF-β2 expression may not be due to direct activation of peroxisome proliferator-activated receptor gamma (PPARγ) signaling. The mechanism underlying efatutazone-mediated TGF-β2 downregulation requires further study.
The TZD troglitazone and rosiglitazone prevent metastasis of NSCLC cells by antagonizing TGF-β stimulation-induced Smad3-mediated epithelial-mesenchymal transition (EMT).20 Troglitazone has also been shown to prevent the TGF-β2 stimulation-induced EMT of retinal pigment epithelium cells by inhibiting Smad2 and Smad3 phosphorylation, which attenuates cell migration.21 In this study, expression of the mesenchymal marker vimentin was only abolished by efatutazone treatment in PC-9 cells with mesenchymal-like morphology,15 suggesting that efatutazone also affects EMT inhibition (Fig. 3a).
Moreover, activated PPARγ can suppress the expression of TGF-β ligand genes, resulting in modulation of TGF-β pathway activity. Activation of the PPARγ pathway by PPARγ agonists represses TGF-β1 mRNA expression via dephosphorylation of zinc finger transcription factor-9 (Zf9), which has been shown to be caused by p70 ribosomal S6 kinase-1 (p70S6K) inhibition due to PPARγ-induced PTEN induction.17,22 Interestingly, in this study, the PPARγ agonist efatutazone attenuated the TGF-β pathway by downregulating TGF-β2 mRNA expression. TZD-induced suppression of TGF-β2 expression has not been previously reported, and further studies are needed to elucidate the mechanism underlying efatutazone-mediated downregulation of TGF-β2 expression.
Efatutazone did not induce PPARγ-mediated transcriptional activity in PC-9ER cells (Figs 4 and 5), suggesting that the inhibitory effect of efatutazone on cell motility may not be due to direct activation of PPARγ signaling. Previous studies have shown that TZD PPARγ agonists exhibit PPARγ-independent antitumor effects via off-target mechanisms.23 For example, rosiglitazone induces AMPK phosphorylation through PPARγ-independent signaling, which inhibits p70S6K phosphorylation in NSCLC cells.24 Troglitazone attenuates EGFR signaling by inducing efficient degradation of endogenous EGFR independent of the PPARγ pathway, which inhibits cell proliferation.25 These PPARγ-independent effects of TZD were also observed in the present study. Efatutazone reduced phopho-S6 levels in all tested cell lines (Fig. 3a and Fig S3), and EGFR expression was abolished by efatutazone treatment in PC-9 and PC-9ZD cells (Fig. 3a and Fig S3). Further studies are required to investigate both the on-target and off-target effects of efatutazone in tumor cells.
Migration of PC-9ER cells was more attenuated by combined treatment with efatutazone and erlotinib than by efatutazone or erlotinib alone (Fig. 1b). Moreover, erlotinib-induced compensatory activation of the TGF-β/Smad2 pathway was also abrogated in all cells treated with this combination (efatutazone and erlotinib) (Fig. 3a,b). These results suggest that this combined treatment is more effective than either treatment alone for preventing metastasis of EGFR-TKI–resistant NSCLC in patients without the EGFR T790M resistance mutation. Combination therapy with efatutazone and erlotinib is currently being evaluated in a phase II study of patients with advanced NSCLC.14
In this study, we also found an additional rationale for continued use of EGFR-TKI after disease progression: resistant tumors, except for those with the T790M resistance mutation, may still remain sensitive to EGFR-TKI in terms of cell motility inhibition. Studies have shown that continuous treatment with EGFR-TKI beyond progressive disease may prolong overall survival in patients with EGFR-activating mutations compared to their survival when chemotherapeutic agents are switched.26–29 Moreover, disease flare, the accelerated progression of disease, has been reported after discontinuation of EGFR-TKI.30 These clinical findings seem to support the hypothesis that tumors with acquired resistance still depend on EGFR signaling and that blocking EGFR signaling with continuous EGFR-TKI treatment may still be beneficial.
Here, we report a rationale for use of the PPARγ agonist efatutazone to prevent the metastasis of EGFR-TKI–resistant NSCLC. In conclusion, these data indicate that efatutazone is a potential therapeutic agent for the prevention of metastasis in NSCLC patients who develop acquired resistance to EGFR-TKI, regardless of the resistance mechanism.
Acknowledgments
This work was supported by a JSPS KAKENHI grant (number 25871225, to MS). The authors thank Junko Suzuki and Akane Naruoka for technical assistance and Daiichi Sankyo Co. Ltd. for kindly providing efatutazone and the pPG2-aP2-TK plasmid.
Disclosure Statement
The authors have no conflict of interest.
Funding information
JSPS KAKENHI grant (25871225)
Supporting Information
Additional supporting information may be found in the online version of this article:
Antibodies used in this study.
Growth-inhibitory effects of erlotinib and efatutazone.
Dose-dependent effect of efatutazone on cell motility.
Effect of efatutazone on the expression and phosphorylation levels of relevant molecules.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Yang P, Allen MS, Aubry MC, et al. Clinical features of 5,628 primary lung cancer patients: experience at Mayo Clinic from 1997 to 2003. Chest. 2005;128:452–62. doi: 10.1378/chest.128.1.452. [DOI] [PubMed] [Google Scholar]
- 3.Koyi H, Hillerdal G, Branden E. A prospective study of a total material of lung cancer from a county in Sweden 1997–1999: gender, symptoms, type, stage, and smoking habits. Lung Cancer. 2002;36:9–14. doi: 10.1016/s0169-5002(01)00451-2. [DOI] [PubMed] [Google Scholar]
- 4.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–8. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 5.Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8:823–59. doi: 10.1097/JTO.0b013e318290868f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007;98:1817–24. doi: 10.1111/j.1349-7006.2007.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanquicett C, Roman J, Hart CM. Thiazolidinediones as anti-cancer agents. Cancer Ther. 2008;6:25–34. [PMC free article] [PubMed] [Google Scholar]
- 8.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004;5:419–29. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 9.Nemenoff RA, Weiser-Evans M, Winn RA. Activation and molecular targets of peroxisome proliferator-activated receptor-gamma ligands in lung cancer. PPAR Res. 2008;2008:156875. doi: 10.1155/2008/156875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–9. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 11.Shimazaki N, Togashi N, Hanai M, et al. Anti-tumour activity of CS-7017, a selective peroxisome proliferator-activated receptor gamma agonist of thiazolidinedione class, in human tumour xenografts and a syngeneic tumour implant model. Eur J Cancer. 2008;44:1734–43. doi: 10.1016/j.ejca.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 12.Copland JA, Marlow LA, Kurakata S, et al. Novel high-affinity PPARgamma agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene. 2006;25:2304–17. doi: 10.1038/sj.onc.1209267. [DOI] [PubMed] [Google Scholar]
- 13.Pishvaian MJ, Marshall JL, Wagner AJ, et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer. 2012;118:5403–13. doi: 10.1002/cncr.27526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Study of erlotinib with or without investigational drug (CS-7017) in subjects with advanced non-small cell lung cancer [ NCT01101334]. [Cited 2014.] Available from URL: http://clinicaltrialsgov/ct2/show/NCT01101334.
- 15.Serizawa M, Takahashi T, Yamamoto N, Koh Y. Combined treatment with erlotinib and a transforming growth factor-beta type I receptor inhibitor effectively suppresses the enhanced motility of erlotinib-resistant non-small-cell lung cancer cells. J Thorac Oncol. 2013a;8:259–69. doi: 10.1097/JTO.0b013e318279e942. [DOI] [PubMed] [Google Scholar]
- 16.Serizawa M, Takahashi T, Yamamoto N, Koh Y. Genomic aberrations associated with erlotinib resistance in non-small cell lung cancer cells. Anticancer Res. 2013b;33:5223–33. [PubMed] [Google Scholar]
- 17.Lee CH, Kim HD, Shin SM, Kim SG. A novel mechanism of PPARgamma regulation of TGFbeta1: implication in cancer biology. PPAR Res. 2008;2008:762398. doi: 10.1155/2008/762398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koizumi F, Shimoyama T, Taguchi F, Saijo N, Nishio K. Establishment of a human non-small cell lung cancer cell line resistant to gefitinib. Int J Cancer. 2005;116:36–44. doi: 10.1002/ijc.20985. [DOI] [PubMed] [Google Scholar]
- 19.Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 20.Reka AK, Kurapati H, Narala VR, et al. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol Cancer Ther. 2010;9:3221–32. doi: 10.1158/1535-7163.MCT-10-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng HC, Ho TC, Chen SL, Lai HY, Hong KF, Tsao YP. Troglitazone suppresses transforming growth factor beta-mediated fibrogenesis in retinal pigment epithelial cells. Molecular Vis. 2008;14:95–104. [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SJ, Yang EK, Kim SG. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: role for Zf9 dephosphorylation. Mol Pharmacol. 2006;70:415–25. doi: 10.1124/mol.106.022954. [DOI] [PubMed] [Google Scholar]
- 23.Wei S, Yang J, Lee SL, Kulp SK, Chen CS. PPARgamma-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009;276:119–24. doi: 10.1016/j.canlet.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han S, Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther. 2006;5:430–7. doi: 10.1158/1535-7163.MCT-05-0347. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Yang X, Xu Y, Jiang X, Nan F, Tang H. Troglitazone inhibits cell proliferation by attenuation of epidermal growth factor receptor signaling independent of peroxisome proliferator-activated receptor gamma. Cell Res. 2009;19:720–32. doi: 10.1038/cr.2009.53. [DOI] [PubMed] [Google Scholar]
- 26.Riely GJ, Kris MG, Zhao B, et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res. 2007;13:5150–5. doi: 10.1158/1078-0432.CCR-07-0560. [DOI] [PubMed] [Google Scholar]
- 27.Maruyama R, Wataya H, Seto T, Ichinose Y. Treatment after the failure of gefitinib in patients with advanced or recurrent non-small cell lung cancer. Anticancer Res. 2009;29:4217–21. [PubMed] [Google Scholar]
- 28.Nishie K, Kawaguchi T, Tamiya A, et al. Epidermal growth factor receptor tyrosine kinase inhibitors beyond progressive disease: a retrospective analysis for Japanese patients with activating EGFR mutations. J Thorac Oncol. 2012;7:1722–7. doi: 10.1097/JTO.0b013e31826913f7. [DOI] [PubMed] [Google Scholar]
- 29.Asami K, Okuma T, Hirashima T, et al. Continued treatment with gefitinib beyond progressive disease benefits patients with activating EGFR mutations. Lung Cancer. 2013;79:276–82. doi: 10.1016/j.lungcan.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 30.Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res. 2011;17:6298–303. doi: 10.1158/1078-0432.CCR-11-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Antibodies used in this study.
Growth-inhibitory effects of erlotinib and efatutazone.
Dose-dependent effect of efatutazone on cell motility.
Effect of efatutazone on the expression and phosphorylation levels of relevant molecules.