

Abstract
Clonal heterogeneity in lymphoid malignancies has been recently reported in adult T-cell lymphoma/leukemia, peripheral T-cell lymphoma, not otherwise specified, and mantle cell lymphoma. Our analysis was extended to other types of lymphoma including marginal zone lymphoma, follicular lymphoma and diffuse large B-cell lymphoma. To determine the presence of clonal heterogeneity, 332 cases were examined using array comparative genomic hybridization analysis. Results showed that incidence of clonal heterogeneity varied from 25% to 69% among different types of lymphoma. Survival analysis revealed that mantle cell lymphoma and diffuse large B-cell lymphoma with clonal heterogeneity showed significantly poorer prognosis, and that clonal heterogeneity was confirmed as an independent predictor of poor prognosis for both types of lymphoma. Interestingly, 8q24.1 (MYC) gain, 9p21.3 (CDKN2A/2B) loss and 17p13 (TP53, ATP1B2, SAT2, SHBG) loss were recurrent genomic lesions among various types of lymphoma with clonal heterogeneity, suggesting at least in part that alterations of these genes may play a role in clonal heterogeneity.
Keywords: Array comparative genomic hybridization, heterogeneity, malignant lymphoma, patient outcome assessment, tumor cell population
Clonal heterogeneity is defined as the presence of several subclones possessing different genomic alterations within a tumor.(1,2) Clonal heterogeneity has been associated with tumor development,(3) invasion and metastasis,(4) and poor clinical outcome.(5) In regards to malignant lymphoma, there are some previous studies on clonal heterogeneity,(6,7) including our recent reports.(8–10) We analyzed adult T-cell leukemia/lymphoma and found that clonal heterogeneity existed in approximately 70% of investigated cases.(11) It was also found that cases of peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) with clonal heterogeneity had a poorer prognosis compared with cases without heterogeneity.(9,10)
Genomic alterations in cases with clonal heterogeneity are believed to contribute to clonal heterogeneity and several methods can be used to identify these alterations. Genomic alterations found in malignancies are somatic mutations, chromosomal translocations and copy number alterations (CNA). Tumors with many somatic mutations in general tend to lack CNA, while tumors with CNA have fewer somatic mutations.(12) Therefore, it is important to choose an appropriate method to analyze genomic alterations for each tumor. Tumors with many somatic mutations require next-generation sequencing analysis, while tumors with CNA such as lymphoma are suitable for analysis by metaphase cytogenetics and/or array comparative genomic hybridization (CGH).(5)
Array CGH analysis is a powerful tool that can explore CNA in detail and evaluate tumor cell populations in biopsy samples, which comprise tumor cells with genomic alterations and normal cells without alterations. In this study, we developed a method of evaluating clonal heterogeneity by taking into account tumor cell populations, and extended our study to other types of lymphoma including Burkitt lymphoma, follicular lymphoma and diffuse large B-cell lymphoma (DLBCL) to investigate the relationship between clonal heterogeneity and clinicopathological features.
Materials and Methods
Patients for array comparative genomic hybridization analysis
We studied previously untreated 332 cases of lymphoma, comprising 29 cases of mantle cell lymphoma, 117 cases of DLBCL, 79 cases of Follicular lymphoma, 25 cases of Burkitt lymphoma, 31 cases of mucosa-associated lymphoid tissue (MALT) lymphoma and 51 peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS). The patients were selected from those hospitalized between 1983 and 2004 at the Aichi Cancer Center hospital and hospitals related to Nagoya University Hospital. Lymphoma was diagnosed in accordance with World Health Organization classification criteria.(13) The clinical features, hematologic characteristics, histology and immunophenotypes were reviewed by an expert hematopathologist (SN). Patients had received standard chemotherapy (mainly CHOP-like regimen). Clinical information is summarized in Table S1. This study was approved by the ethics committee of the Aichi Cancer Center Research Institute.
Analysis procedure of array comparative genomic hybridization data
Array comparative genomic hybridization data
We analyzed data obtained from previous results of BAC array CGH for these 332 lymphoma samples.(14–18) The array CGH glasses contained from 1778 to 2163 BAC clones (1.4–1.7 Mb resolutions). These clones were aligned with each of the chromosomes based on data from the National Center for Biotechnology Information (GRCh37) or from the University of California Santa Cruz (hg 19). Data can be accessed at http://www.ncbi.nlm.nih.gov/gds: Accession Number GSE54303.
Background signal removal
In an effort to remove background signals from the array CGH data, the data obtained from 332 samples were treated individually. One objective sample was selected and Pearson's correlation coefficients between the objective sample and the other 331 samples were calculated, where the correlation was computed based on log2 ratios after manually removing obvious CNA. A subset of samples were then selected where the correlation coefficients to the object sample were >0.5 (P = 4E-113). The median value of those selected samples for each probe was then taken as the background signal, and this value was subtracted from the log2 ratio of the objective sample. This procedure was repeated 332 times.
Identification of copy number alterations and log2 ratio
We defined CNA on array CGH data by interactively updating and labeling a maximum likelihood segmentation model using SegAnnDB software.(19) On each continuous CNA, an average of the log2 ratios was calculated.
Calculation of tumor cell population from log2 ratio
The tumor cell population was calculated from the log2 ratio based on the functional relationship between the tumor cell population to the total cells in the sample (X) and the log2 ratio of CNA from array CGH data (Y):
for gain, while
where n is the gained or deleted chromosomal copy number on the CNA in tumor cells.
Definition of clonal heterogeneity
We defined a case indicating the same log2 ratio for all CNA as a sample without clonal heterogeneity (Fig. 1a) because the single tumor cell population was calculated from the log2 ratio(s) of all CNA in this sample. We defined a case having different log2 ratios in one sample as a sample with clonal heterogeneity (Fig. 1a). Differences in log2 ratios were evaluated using Student's t-test (P < 0.001) as previously described.(9) In a few cases, the same tumor cell population was applied when the log2 ratios were proportional at variable copy numbers (one copy gain versus two copy gain) in the same sample. For example, a log2 ratio of 0.685 indicates a tumor cell population of 61% in a case of one copy gain and 0.39 is 63% in two copy gain (Fig. S1d). We defined such a case with different log2 ratios showing one tumor cell population as a sample without clonal heterogeneity.
Figure 1.
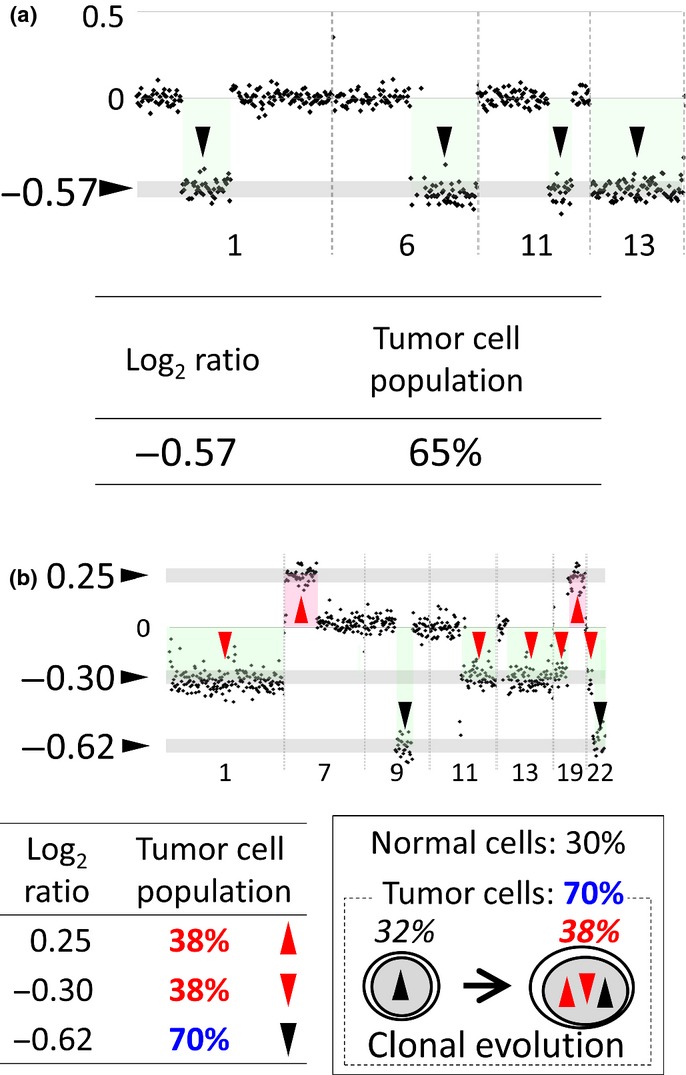
Results of array comparative genomic hybridization (CGH) analysis for cases with and without clonal heterogeneity. (a) A case without clonal heterogeneity showing a constant log2 ratio for all copy number alterations (CNA). (Top) The result of array CGH analysis with chromosomal regions on the x-axis and log2 ratios on the y-axis. There are four CNA (arrowheads) whose log2 ratios are −0.57. (Bottom) Log2 ratio and tumor cell population. The population of tumor cells with the CNA whose log2 ratio is −0.57 is calculated as 65% (Fig. S1a–c). This case is assessed as a sample without clonal heterogeneity since populations of tumor cells with each and every CNA in this sample are the same. (b) A case with clonal heterogeneity showing different log2 ratios. (Top) The result of array CGH analysis. There are two CNA (red up arrowheads) whose log2 ratio is 0.25, five CNA (red down arrowheads) whose log2 ratios are −0.30, and two CNA (black down arrowheads) whose log2 ratios are −0.62. (Bottom left) Log2 ratios and assessed tumor cell populations. This case is assessed as a sample with clonal heterogeneity because of the different tumor cell populations. (Bottom right) The simplest model of a tumor biopsy sample with different log2 ratios explained by clonal evolution. The solid box shows a tumor biopsy sample from a patient. It consists of 30% of normal cells (30%) and 70% of tumor cells (70%, dotted box). The tumor cells consist of a subclone (32%, left) and the other subclone (38%, right), which can have evolved from the subclone in the left.
Statistics
Statistical analysis included Student's t-test, Fisher's exact test, the Cox proportional hazard model and Kaplan–Meier analysis, which were performed using the free software R (http://www.r-project.org/).
Analysis of published data
We also studied the existence of clonal heterogeneity in MALT lymphoma samples using independently published data.(20) Data was obtained from http://www.ncbi.nlm.nih.gov/gds; Accession Number GSE35278.
Results
Assessment of clonal heterogeneity using array comparative genomic hybridization data
Identification of copy number alterations
We identified CNA for 332 malignant lymphoma cases using array CGH data. Of these, 51 cases showed no CNA and were removed from further analysis. The removed cases were 13 of the 79 cases of follicular lymphoma, five of the 25 cases of Burkitt lymphoma, 21 of the 31 cases of MALT lymphoma, and 12 of the 51 cases of PTCL-NOS. In the remaining 281 samples, one to 38 (8.5 ± 6.5, mean ± SD) CNA per sample were identified. When the log2 ratio of each CNA was examined, some cases showed different log2 ratios in a sample (Fig. 1a). Detailed analysis suggested that several subclones with different CNA existed in such cases.
Assessment of clonal heterogeneity
The average log2 ratios of identified CNA were used to calculate tumor cell populations for the examination of clonal heterogeneity. When a sample does not contain any normal tissue components (e.g. cell lines), the log2 ratio is calculated as shown in Figure S1(a). However, a tumor biopsy sample consists of tumor cells with CNA and normal cells without any CNA (Fig. S1b). We can evaluate an average chromosomal copy number for a sample containing a mixture of tumor cells and a normal tissue component using the formula shown in Figure S1(c). Therefore, the percentage of tumor cells can be assessed by the log2 ratio of such CNA. When all CNA in a sample show the same log2 ratio, the case is assessed as a sample without clonal heterogeneity because the population of tumor cells with each CNA is the same in that case (Fig. 1a). A case with different log2 ratios is assessed as a sample with clonal heterogeneity because there were different populations of tumor cells with different CNA in most cases. Clonal evolution is suggested to have taken place in such cases (Fig. 1a).
Assessment by G-banding of method for the examination of clonal heterogeneity
Assessment of the method employed for the examination of clonal heterogeneity based on array CGH analysis was performed using G-banding analysis. Sometimes the same tumor cell population was determined from different log2 ratios of array CGH analysis in a sample, indicating that there are CNA with different chromosomal copy numbers in a tumor clone. G-banding revealed that CNA were present with different copy numbers in such cases (Fig. S1d), indicating that the method employed for the examination of clonal heterogeneity accurately reflected the karyotype.
Incidence of clonal heterogeneity
The 281 cases of six types of lymphoma were assessed for clonal heterogeneity based on the method described above. Incidence of clonal heterogeneity varied significantly among lymphoma types, from 25% (Burkitt lymphoma) to 69% (mantle cell lymphoma; Table 1, P = 0.003 by Fisher's exact test). Incidence of clonal heterogeneity also varied among lymphoma subtypes. It differed between GCB-DLBCL and ABC-DLBCL (Table S2, 28% and 64%, P = 0.007). It was correlated with stages in follicular lymphoma (Table S3, 17%, 29%, 33% and 44% in stage I, II, IIIa and IIIb, respectively). In cases of MALT lymphoma, any CNA did not exist in cases with t(11:18) and clonal heterogeneity was only found in cases without t(11:18). This result was confirmed using independent published data (Table S4).(20)
Table 1.
Sample number for array CGH analysis and incidence of clonal heterogeneity for each types of lymphoma
| Type of lymphoma | Total sample number | Number of samples without CNA | Samples with CNA | |
|---|---|---|---|---|
| Number of samples with CNA (number of samples with clonal heterogeneity) | Clonal heterogeneity (%) | |||
| MCL | 29 | 0 | 29 (20) | 69% |
| DLBCL | 117 | 0 | 117 (55) | 47% |
| FL | 79 | 13 | 66 (19) | 29% |
| BL | 25 | 5 | 20 (5) | 25% |
| MALT | 31 | 21 | 10 (3) | 30% |
| PTCL-NOS | 51 | 12 | 39 (16) | 41% |
| Total | 332 | 51 | 281 (118) | 42% |
CGH, comparative genomic hybridization; CNA, copy number alterations; BL, Burkitt lymphoma; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MALT, mucosa-associated lymphoid tissue lymphoma; MCL, mantle cell lymphoma; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified.
Numbers of copy number alterations for samples with and without clonal heterogeneity
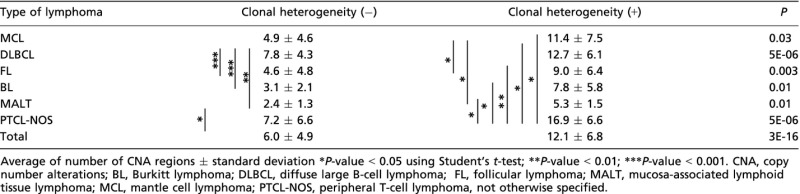
Numbers of CNA were compared between cases with and without clonal heterogeneity (Table 2). The average numbers of CNA were significantly higher for the cases with clonal heterogeneity than those without clonal heterogeneity for any types of lymphoma (Table 2). The averages varied significantly among each type of lymphoma, from 2.4 (MALT) to 7.8 (DLBCL) in cases without clonal heterogeneity and from 5.3 (MALT) to 16.9 (PTCL-NOS) in cases with clonal heterogeneity.
Table 2.
Number of CNA regions for each type of lymphoma
 |
Differences of chromosomal regions and frequencies of copy number alterations between cases with and without clonal heterogeneity
Chromosomal regions and frequencies of CNA were compared between cases with and without clonal heterogeneity (Fig. 2, Fig. S2–7; Table 3). Recurrent CNA among various types of lymphoma with clonal heterogeneity were found to be 8q24.1 (minimal overlapping lesion: MYC) gain, 9p21.3 (CDKN2A/2B) loss and 17p13 (TP53, ATP1B2, SAT2, SHBG) loss. The 8p24.1 (MYC) gain was found in significantly more cases with clonal heterogeneity than in those without clonal heterogeneity in follicular lymphoma and PTCL-NOS. The 9p21.3 loss was found in mantle cell lymphoma, DLBCL and PTCL-NOS. The 17p13 loss was found in mantle cell lymphoma, DLBCL, Burkitt lymphoma and PTCL-NOS (Fig. 3).
Figure 2.
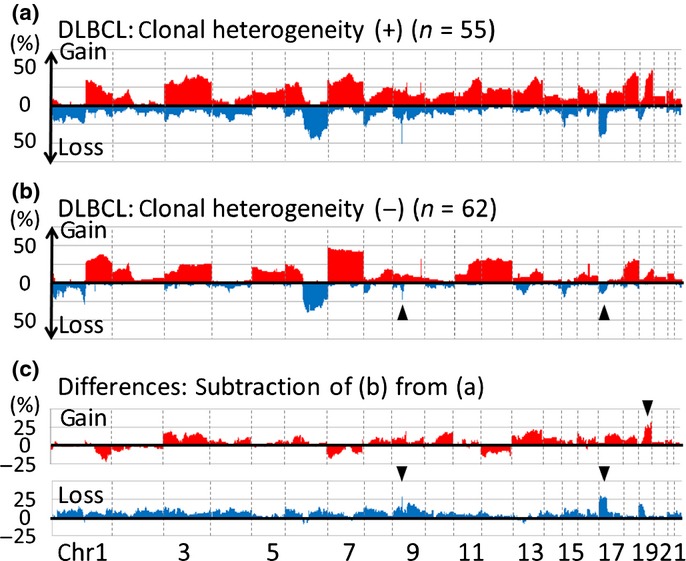
Chromosomal regions and frequencies of copy number alterations (CNA) for DLBCL with and without clonal heterogeneity. (a) CNA for 55 diffuse large B-cell lymphoma (DLBCL) cases with clonal heterogeneity. The x-axis represents chromosomal regions and the y-axis represents frequencies of gain (above 0) or loss (below 0). (b) CNA for 62 DLBCL cases without clonal heterogeneity. The profile of cases without clonal heterogeneity resembles those of cases with clonal heterogeneity (a) while loss of 9p and 17p are less common in cases without heterogeneity than in those with (arrowheads). (c) Differences in CNA between DLBCL with and without clonal heterogeneity. (Top) Subtraction of frequencies of gain in cases without clonal heterogeneity (b, red area) from those with (a, red area). The y-axis represents differences in frequency (>0: higher frequencies in cases with clonal heterogeneity; <0: higher frequencies in those without clonal heterogeneity). The characteristic gain in DLBCL cases with clonal heterogeneity is 19p (arrowhead) with more than 25% differences between cases with and without clonal heterogeneity. (Bottom) Subtraction of frequencies of loss in cases without clonal heterogeneity (b, blue area) from those with clonal heterogeneity (a, blue area). The y-axis represents differences (>0: higher frequencies in cases with clonal heterogeneity; <0: higher frequencies in those without clonal heterogeneity). The characteristic loss in DLBCL cases with clonal heterogeneity are 9p and 17p (arrow heads). All gain and loss with differences >25% are summarized in Table 3.
Table 3.
Differences in CNA between samples with and without clonal heterogeneity (more than 25% differences)
| Types of lymphoma | Clonal heterogeneity (−) | Clonal heterogeneity (+) | Detailed in | ||
|---|---|---|---|---|---|
| Gain | Loss | Gain | Loss | ||
| MCL | — | — | 6q21, 8q13.3–q24.23† | 1p22.1–q12, 5q23.1,§ 7q36.3, 8p23.3–11.21, 9p24.3–q31.1,¶ 17p13.3–11.2†† | Figure S2 |
| DLBCL | — | — | 19q13.12–.43 | 9p21.3,¶ 17p13. 2–11. 2†† | Figure2 |
| FL | — | — | 2p16.1–14, 7p11.1, 7q31.1–.2, ‡ 8q22.1–24.23,† 9q12–34.3,‡ 10p15.3–13, 12p13.33–q21.1, 18p11.21–q23 | — | Figure S3 |
| BL | 1q44, 12p13.33–q24.33, 15q22.32–15q25.1 | — | 1q25.2, 3q26.33–29, 13q31.3–32.3, 17q23.1–24.1 | 6p25.3–22.3, q13–21, 10q23.33–24.1, 11q23.3–25, 14q11.1–32.33,‡‡ 16q24.3, 17p13.3–q12†† | Figure S4 |
| MALT | 1p36.33–.32, 2q14.3, 3p26.3–q29, 5q31.1, 10p12.31–.1, 18q11.2 -q23 | — | 13q34, 20p11.22–q12, 22q11.21 | 7q31.31–q34, 20p12.1 | Figure S5 |
| PTCL-NOS | 5p15.1 | — | 7q21.11–q35,‡ 8q24.13–.21,† 9q33.3–34,‡ 16p13.3–11.1, 17q21.2–25.3 | 2p23.3, q21.3–37.3, 4p16.1–15.2, 5q14.3–34,§ 9p21.3–q31.1,¶ 10q11.1–q23.31, 12p13.1–.13, 13q21.31–34, 14q13.1–32.12,‡‡ 15q23, 17p13.3–11.2†† | Figure S6 |
| All lymphomas | — | — | — | 9p21.3,¶ 17p13.3–11.2†† | Figure S7 |
Common aberrations more than two lymphomas are indicated in bold with underline.
Aberration is found in MCL, FL and PTCL-NOS;
aberration found in FL and PTCL-NOS;
aberration found in MCL and PTCL-NOS;
aberration found in MCL, DLBCL and PTCL-NOS;
aberration found in MCL, DLBCL, BL and PTCL-NOS;
aberration found in BL and PTCL-NOS. —, no region is identified; BL, Burkitt lymphoma; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MALT, mucosa-associated lymphoid tissue lymphoma; MCL, mantle cell lymphoma; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified.
Figure 3.
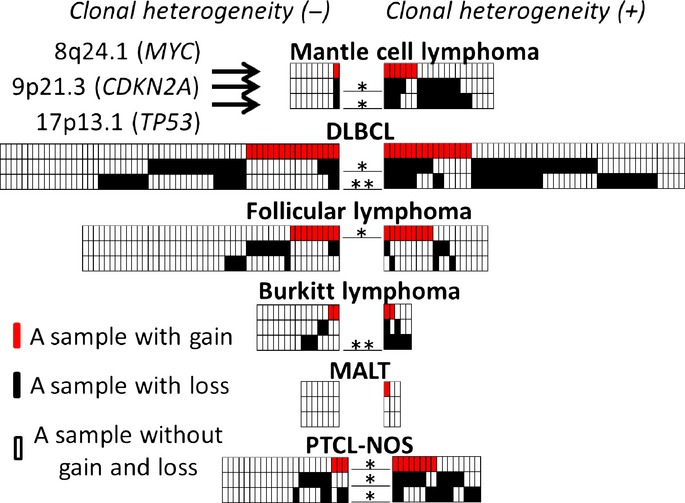
Various types of lymphoma cases with or without 8q24.1 gain, 9p21.3 loss and 17p13.1 loss. Cases without clonal heterogeneity (left) are compared with those with clonal heterogeneity (right). Lymphoma types are shown over each box. Red, black and white boxes indicate cases with gain, with loss, and without loss and gain, respectively; the upper, middle and lower lanes of the boxes represent 8p24.1 (MYC locus), 9p21.3 (CDKN2A) and 17p13.1 (TP53), respectively. *P < 0.05 and **P < 0.01 by Fisher's exact test.
Relationship between clonal heterogeneity and clinical outcomes
Survival analysis revealed that clonal heterogeneity was correlated with poor prognosis in several types of lymphoma. Significantly poor prognosis was found in mantle cell lymphoma (P = 0.005) and DLBCL (P = 6e-5; Fig. 4). Burkitt lymphoma and PTCL-NOS also showed a poorer prognosis in cases with clonal heterogeneity than those without clonal heterogeneity, although the differences were not statistically significant (P = 0.17 and P = 0.15, Fig. S10b,c). There was no difference between cases with clonal heterogeneity and those without for cases involving follicular lymphoma (P = 0.7, Fig. S10a). Because all patients with MALT lymphoma were alive, statistical analysis was not performed.
Figure 4.
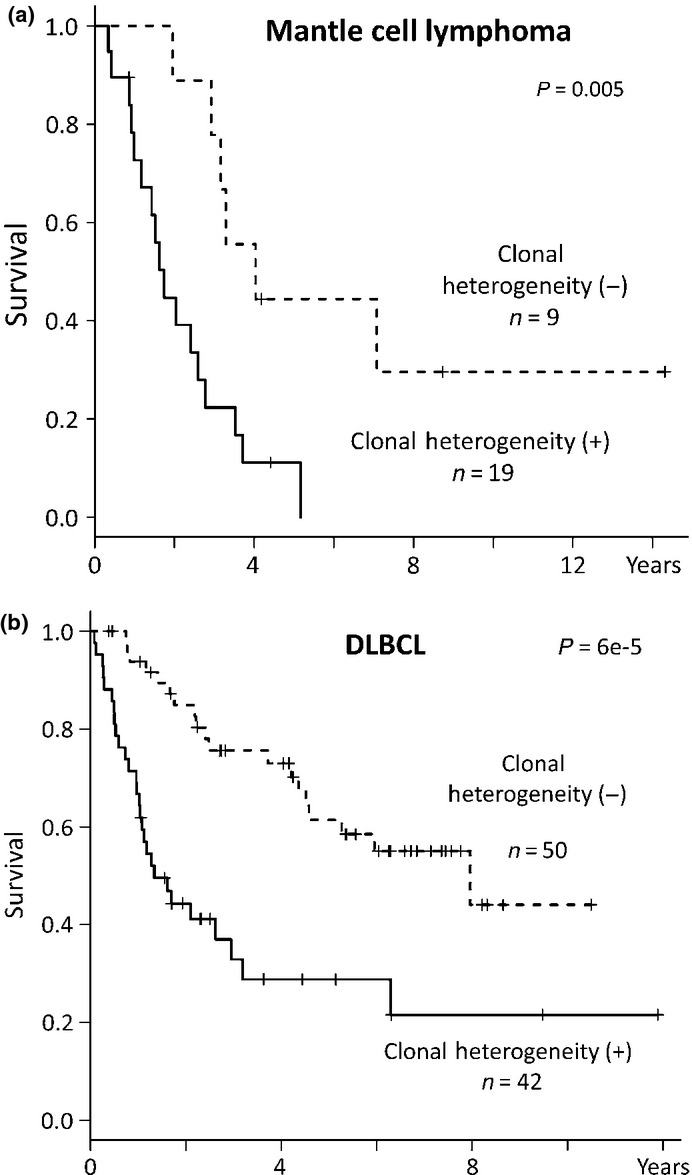
Clonal heterogeneity correlated with poor prognosis. (a) Kaplan–Meier analysis to compare survival in mantle cell lymphoma patients with and without clonal heterogeneity. Patients with clonal heterogeneity demonstrated a poorer prognosis than those without. (b) Kaplan-Meier analysis in diffuse large B-cell lymphoma (DLBCL) patients. Patients with clonal heterogeneity demonstrated poorer prognosis than those without clonal heterogeneity.
Monovariate analysis was conducted on the factors affecting prognosis in mantle cell lymphoma and DLBCL. The results revealed that IPI (international prognostic Index; P = 0.03 for mantle cell lymphoma, P = 0.002 for DLBCL) and clonal heterogeneity (P = 0.02 and P = 0.00005, respectively) were correlated with prognosis in both of mantle cell lymphoma and DLBCL (Table 4). 9p21.3 (CDKN2A) loss was correlated with poor prognosis in DLBCL (P = 0.001) and 17p13.1 (TP53) loss was correlated in mantle cell lymphoma (P = 0.02). Multivariate analysis compared IPI, number of CNA, clonal heterogeneity, 8q24.1 (MYC) gain, 9p21.3 loss and 17p13.1 loss. It demonstrated that IPI (P = 0.0009), clonal heterogeneity (P = 0.006) and 8q24.1 gain (P = 0.005) were associated with a poor prognosis for mantle cell lymphoma while clonal heterogeneity (P = 0.0003) and 9p21.3 loss (P = 0.003) were associated with a poor prognosis for DLBCL (Table 4). Clonal heterogeneity was confirmed by multivariate analysis as an independent predictor of poor prognosis for both types of lymphoma.
Table 4.
Monovariate and multivariate analysis of prognostic factors affecting overall survival
| Mantle cell lymphoma (n = 26) | DLBCL (n = 84) | |||||
|---|---|---|---|---|---|---|
| Variable | Unfavorable factors | Odds ratio (95% CI) | P | Hazard ratio (95% CI) | P | |
| Monovariate analysis | IPI | High intermediate/High | 2.67 (1.12–6.37) | 0.03 | 2.66 (1.45–4.88) | 0.002 |
| Number of CNA(s) | More than 7 | 4.25 (1.56–11.55) | 0.005 | 1.89 (0.97–3.68) | 0.06 | |
| Clonal heterogeneity | Heterogeneity (+) | 3.81 (1.25–11.63) | 0.02 | 3.70 (1.97–6.96) | 0.00005 | |
| 8q24.2 (MYC locus) | Gain | 2.16 (0.9–5.22) | 0.09 | 1.06 (0.55–2.02) | 0.9 | |
| 9p21.3 (CDKN2A locus) | Loss | 2.32 (0.94–5.76) | 0.07 | 2.78 (1.51–5.13) | 0.001 | |
| 17p13.1 (TP53 locus) | Loss | 3.14 (1.18–8.38) | 0.02 | 1.26 (0.69–2.29) | 0.4 | |
| Multivariate analysis | IPI | High intermediate/High | 7.15 (2.25–22.77) | 0.0009 | 1.66 (0.87–3.18) | 0.1 |
| Number of CNA(s) | More than 7 | 3.59 (0.96–13.36) | 0.06 | 1.72 (0.8–3.72) | 0.2 | |
| Clonal heterogeneity | Heterogeneity (+) | 8.06 (1.8–36.15) | 0.006 | 4.16 (1.93–8.98) | 0.0003 | |
| 8q24.2 (MYC locus) | Gain | 8.11 (1.86–35.42) | 0.005 | 1.4 (0.67–2.92) | 0.4 | |
| 9p21.3 (CDKN2A locus) | Loss | 4.94 (1.02–24.03) | 0.05 | 2.69 (1.4–5.14) | 0.003 | |
| 17p13.1 (TP53 locus) | Loss | 1.54 (0.3–7.75) | 0.6 | 2.03 (0.9–4.61) | 0.09 | |
CI, confidence interval; DLBCL, diffuse large B-cell lymphoma; IPI, International Prognostic Index.
Discussion
We have developed a method for the assessment of clonal heterogeneity by taking into account the normal tissue components in a tumor sample. Based on such an analysis, we investigated the presence of clonal heterogeneity in 332 cases comprising six types of lymphoma, and found relationships between clonal heterogeneity and poor prognosis and recurrent CNA.
Clonal heterogeneity was associated with poor prognosis in several types of lymphoma. Cases with clonal heterogeneity showed significantly poorer prognosis in mantle cell lymphoma and DLBCL (Fig. 4). Cases with clonal heterogeneity also showed poorer prognosis, although not significantly, in Burkitt lymphoma and PTCL-NOS (Fig. S10b,c), and showed no difference in prognosis in follicular lymphoma (Fig. S10a). One reason for the poor prognosis may relate to clonal evolution of the tumor cells, which could have taken place in cases with clonal heterogeneity (Fig. 1b). Recently, prognostic biomarkers in several types of lymphoma have been identified by analysis based on next-generation sequencing or SNP array,(21–27) such as somatic mutations of notch 1 and 2(26,28) and TP53.(29) In addition to somatic mutations, CNA,(30,31) aberrant mRNA expression,(32–34) and cell surface markers(35) have also been identified as prognostic biomarkers. Our current analysis examined the presence of clonal heterogeneity in lymphoma cases and found a correlation between clonal heterogeneity and poor clinical outcome. Therefore, the evaluation of clonal heterogeneity could potentially be adopted as a new biological marker and the combination of this marker and previously established prognostic biomarkers could contribute toward the more accurate prediction of clinical outcome for B and T-cell lymphoma.
Cases with clonal heterogeneity irrespective of lymphoma type indicated recurrent CNA: gain of MYC, loss of CDKN2A/2B and loss of TP53 (Table 3; Fig. 3). Messenger RNA expression of TP53 was lower in DLBCL cases (P = 0.04, data not shown). Aberrations of these oncogene and tumor suppressor genes could contribute toward clonal heterogeneity by promoting and/or progressing lymphoma in various ways. For example, overexpression of MYC stimulates tumor cell proliferation, TP53 loss inhibits apoptosis, including apoptosis induced by overexpression of MYC, and CDKN2A/2B loss damages cell cycle checkpoint responses. Although it is not known whether these CNA were generated during clonal evolution or as an early event during lymphoma development, it is important to note that these oncogene and tumor suppressor genes reported in a variety of tumors(36–38) were also involved in cases with clonal heterogeneity.
Although the relationships between clonal heterogeneity and recurrent CNA and poor prognosis irrespective of lymphoma type were shown, distinctive features of each lymphoma types were also shown in our analysis. The incidence of clonal heterogeneity varied among lymphoma types (Table 1) and subtypes (Table S2–4). In addition to the incidence of clonal heterogeneity, the numbers of CNA also varied among lymphoma types (Table 2). Moreover, examination of chromosomal regions and frequency of CNA indicated distinct differences among lymphoma types (Fig. 2; Fig. S2–S7) and subtypes (Fig. S8–9). These distinctive differences might reflect physiological and pathological differences among lymphoma types. Beyond these differences, clonal heterogeneity was present in all types of lymphoma.
In summary, we have developed a method of evaluating clonal heterogeneity based on array CGH analysis by taking into account tumor cell populations and found in the present study that clonal heterogeneity was present in every type of lymphoma. The relationships between clonal heterogeneity and poor prognosis and recurrent CNA were identified. These findings suggest that the evaluation of clonal heterogeneity could potentially be adopted as a new biological marker, and that the combination of this marker and previously established prognostic biomarkers could contribute toward the more accurate prediction of clinical outcome for B and T-cell lymphoma.
Acknowledgments
The outstanding technical assistance of Yumiko Kasugai and Kyoko Hirano is very much appreciated. The authors thank Drs Tatsuo Kakiuchi and Taishi Takahara for their critical discussions and constructive suggestions. This work was supported in part by a grant-in-Aid from the Ministry of Health, Labour and Welfare of Japan, the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Japan Society for the Promotion of Science, and from the Takeda Science Foundation (M. Seto).
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Method for calculating a tumor cell population using the result of array comparative genomic hybridization (CGH) analysis.
Fig. S2. Chromosomal regions and frequencies of copy number alterations (CNA) for mantle cell lymphoma with or without clonal heterogeneity.
Fig. S3. Chromosomal regions and frequencies of copy number alterations (CNA) for follicular lymphoma with and without clonal heterogeneity.
Fig. S4. Chromosomal regions and frequencies of copy number alterations (CNA) for Burkitt lymphoma with or without clonal heterogeneity.
Fig. S5. Chromosomal regions and frequencies of copy number alterations (CNA) for mucosa-associated lymphoid tissue (MALT) lymphoma with and without clonal heterogeneity.
Fig. S6. Chromosomal regions and frequencies of copy number alterations (CNA) for peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS) with and without clonal heterogeneity.
Fig. S7. Chromosomal regions and frequencies of copy number alterations (CNA) for all lymphoma with and without clonal heterogeneity.
Fig. S8. Chromosomal regions and frequencies of copy number alterations (CNA) for ABC-diffuse large B-cell lymphoma (DLBCL) with and without clonal heterogeneity.
Fig. S9. Chromosomal regions and frequencies of copy number alterations (CNA) for GCB-diffuse large B-cell lymphoma (DLBCL) with and without clonal heterogeneity.
Fig. S10. Survival differences between cases with and without clonal heterogeneity in each type of lymphoma.
Table S1. Clinical information and summary of analysis for 332 malignant lymphoma cases.
Table S2. Incidence of clonal heterogeneity for diffuse large B-cell lymphoma (DLBCL) subgroups.
Table S3. Incidence of clonal heterogeneity for follicular lymphoma.
Table S4. Incidence of clonal heterogeneity of mucosa-associated lymphoid tissue (MALT) lymphoma with or without MALT1 translocation.
References
- 1.Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842–51. doi: 10.1056/NEJMra1204892. [DOI] [PubMed] [Google Scholar]
- 2.Yang T, Rycaj K, Liu Z, et al. Cancer stem cells: constantly evolving and functionally heterogeneous therapeutic targets. Cancer Res. 2014 doi: 10.1158/0008-5472.CAN-14-0266. doi: 10.1158/0008-5472.CAN-14-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–13. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bochtler T, Stolzel F, Heilig CE, et al. Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J Clin Oncol. 2013;31:3898–905. doi: 10.1200/JCO.2013.50.7921. [DOI] [PubMed] [Google Scholar]
- 6.Villamor N, Lopez-Guillermo A, Lopez-Otin C, et al. Next-generation sequencing in chronic lymphocytic leukemia. Semin Hematol. 2013;50:286–95. doi: 10.1053/j.seminhematol.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Brioli A, Melchor L, Cavo M, et al. The impact of intra-clonal heterogeneity on the treatment of multiple myeloma. Br J Haematol. 2014;165:441–54. doi: 10.1111/bjh.12805. [DOI] [PubMed] [Google Scholar]
- 8.Liu F, Yoshida N, Suguro M, et al. Clonal heterogeneity of mantle cell lymphoma revealed by array comparative genomic hybridization. Eur J Haematol. 2013;90:51–8. doi: 10.1111/ejh.12030. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida N, Umino A, Liu F, et al. Identification of multiple subclones in peripheral T-cell lymphoma, not otherwise specified with genomic aberrations. Cancer Med. 2012;1:289–94. doi: 10.1002/cam4.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seto M. XV. Malignant lymphoma as a consequence of clonal evolution. Hematol Oncol. 2013;31(Suppl 1):84–8. doi: 10.1002/hon.2073. [DOI] [PubMed] [Google Scholar]
- 11.Umino A, Nakagawa M, Utsunomiya A, et al. Clonal evolution of adult T-cell leukemia/lymphoma takes place in the lymph nodes. Blood. 2011;117:5473–8. doi: 10.1182/blood-2010-12-327791. [DOI] [PubMed] [Google Scholar]
- 12.Ciriello G, Miller ML, Aksoy BA, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008. [Google Scholar]
- 14.Fukuhara N, Nakamura T, Nakagawa M, et al. Chromosomal imbalances are associated with outcome of Helicobacter pylori eradication in t(11;18)(q21;q21) negative gastric mucosa-associated lymphoid tissue lymphomas. Genes Chromosom Cancer. 2007;46:784–90. doi: 10.1002/gcc.20464. [DOI] [PubMed] [Google Scholar]
- 15.Nakagawa M, Nakagawa-Oshiro A, Karnan S, et al. Array comparative genomic hybridization analysis of PTCL-U reveals a distinct subgroup with genetic alterations similar to lymphoma-type adult T-cell leukemia/lymphoma. Clin Cancer Res. 2009;15:30–8. doi: 10.1158/1078-0432.CCR-08-1808. [DOI] [PubMed] [Google Scholar]
- 16.Tagawa H, Karnan S, Suzuki R, et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene. 2005;24:1348–58. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- 17.Tagawa H, Karube K, Guo Y, et al. Trisomy 3 is a specific genomic aberration of t(14;18) negative follicular lymphoma. Leukemia. 2007;21:2549–51. doi: 10.1038/sj.leu.2404817. [DOI] [PubMed] [Google Scholar]
- 18.Tagawa H, Suguro M, Tsuzuki S, et al. Comparison of genome profiles for identification of distinct subgroups of diffuse large B-cell lymphoma. Blood. 2005;106:1770–7. doi: 10.1182/blood-2005-02-0542. [DOI] [PubMed] [Google Scholar]
- 19.Hocking TD, Boeva V, Rigaill G, et al. SegAnnDB: interactive web-based genomic segmentation. Bioinformatics. 2014;30:1539–46. doi: 10.1093/bioinformatics/btu072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braggio E, Dogan A, Keats JJ, et al. Genomic analysis of marginal zone and lymphoplasmacytic lymphomas identified common and disease-specific abnormalities. Mod Pathol. 2012;25:651–60. doi: 10.1038/modpathol.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389–401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–7. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohr JG, Stojanov P, Lawrence MS, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–84. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bea S, Colomo L, Lopez-Guillermo A, et al. Clinicopathologic significance and prognostic value of chromosomal imbalances in diffuse large B-cell lymphomas. J Clin Oncol. 2004;22:3498–506. doi: 10.1200/JCO.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 26.Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:18250–5. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villamor N, Conde L, Martinez-Trillos A, et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia. 2013;27:1100–6. doi: 10.1038/leu.2012.357. [DOI] [PubMed] [Google Scholar]
- 29.Li C, Kim SW, Rai D, et al. Copy number abnormalities, MYC activity, and the genetic fingerprint of normal B cells mechanistically define the microRNA profile of diffuse large B-cell lymphoma. Blood. 2009;113:6681–90. doi: 10.1182/blood-2009-01-202028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oscier D, Owen R, Johnson S. Splenic marginal zone lymphoma. Blood Rev. 2005;19:39–51. doi: 10.1016/j.blre.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Young KH, Leroy K, Moller MB, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood. 2008;112:3088–98. doi: 10.1182/blood-2008-01-129783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3460–7. doi: 10.1200/JCO.2011.41.4342. [DOI] [PubMed] [Google Scholar]
- 33.Johnson NA, Slack GW, Savage KJ, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3452–9. doi: 10.1200/JCO.2011.41.0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navarro A, Clot G, Royo C, et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012;72:5307–16. doi: 10.1158/0008-5472.CAN-12-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyazaki K, Yamaguchi M, Suzuki R, et al. CD5-positive diffuse large B-cell lymphoma: a retrospective study in 337 patients treated by chemotherapy with or without rituximab. Ann Oncol. 2011;22:1601–7. doi: 10.1093/annonc/mdq627. [DOI] [PubMed] [Google Scholar]
- 36.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 37.Velimezi G, Liontos M, Vougas K, et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat Cell Biol. 2013;15:967–77. doi: 10.1038/ncb2795. [DOI] [PubMed] [Google Scholar]
- 38.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Method for calculating a tumor cell population using the result of array comparative genomic hybridization (CGH) analysis.
Fig. S2. Chromosomal regions and frequencies of copy number alterations (CNA) for mantle cell lymphoma with or without clonal heterogeneity.
Fig. S3. Chromosomal regions and frequencies of copy number alterations (CNA) for follicular lymphoma with and without clonal heterogeneity.
Fig. S4. Chromosomal regions and frequencies of copy number alterations (CNA) for Burkitt lymphoma with or without clonal heterogeneity.
Fig. S5. Chromosomal regions and frequencies of copy number alterations (CNA) for mucosa-associated lymphoid tissue (MALT) lymphoma with and without clonal heterogeneity.
Fig. S6. Chromosomal regions and frequencies of copy number alterations (CNA) for peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS) with and without clonal heterogeneity.
Fig. S7. Chromosomal regions and frequencies of copy number alterations (CNA) for all lymphoma with and without clonal heterogeneity.
Fig. S8. Chromosomal regions and frequencies of copy number alterations (CNA) for ABC-diffuse large B-cell lymphoma (DLBCL) with and without clonal heterogeneity.
Fig. S9. Chromosomal regions and frequencies of copy number alterations (CNA) for GCB-diffuse large B-cell lymphoma (DLBCL) with and without clonal heterogeneity.
Fig. S10. Survival differences between cases with and without clonal heterogeneity in each type of lymphoma.
Table S1. Clinical information and summary of analysis for 332 malignant lymphoma cases.
Table S2. Incidence of clonal heterogeneity for diffuse large B-cell lymphoma (DLBCL) subgroups.
Table S3. Incidence of clonal heterogeneity for follicular lymphoma.
Table S4. Incidence of clonal heterogeneity of mucosa-associated lymphoid tissue (MALT) lymphoma with or without MALT1 translocation.