

Abstract
Hepatocellular carcinoma (HCC) is one of the most common human cancers and a major cause of cancer-related death worldwide. The bleak outcomes of HCC patients even after curative treatment have been, at least partially, attributed to its multicentric origin. Therefore, it is necessary to examine not only tumor tissue but also non-tumor liver tissue to investigate the molecular mechanisms operating during hepatocarcinogenesis based on the concept of “field cancerization”. Several studies previously investigated the association of molecular alterations in non-tumor liver tissue with clinical features and prognosis in HCC patients on a genome-wide scale. In particular, specific alterations of DNA methylation profiles have been confirmed in non-tumor liver tissue. This review focuses on the possible clinical value of array-based comprehensive analyses of molecular alterations, especially aberrant DNA methylation, in non-tumor liver tissue to clarify the risk of hepatocarcinogenesis. Carcinogenetic risk estimation based on specific methylation signatures may be advantageous for close follow-up of patients who are at high risk of HCC development. Furthermore, epigenetic therapies for patients with chronic liver diseases may be helpful to reduce the risk of HCC development because epigenetic alterations are potentially reversible, and thus provide promising molecular targets for therapeutic intervention.
Keywords: Carcinogenesis, DNA methylation, epigenetics, hepatocellular carcinoma, microarray
Hepatocellular carcinoma (HCC) is one of the most frequent human cancers, and is not only a leading cause of cancer-related deaths, but is also significantly increasing globally.(1,2) The multistep development of HCC is characterized by the progressive sequential evolution associated with chronic liver damage. Although the precise molecular mechanisms of liver carcinogenesis are still unclear, the increased turnover of hepatocytes and inflammatory cell infiltrate seen in chronic hepatitis and cirrhosis may lead to an accumulation of molecular alterations, which ultimately result in the development of HCC.(3,4)
Many recent studies have shown that specific molecular alterations in tumor tissue can predict early intrahepatic recurrence, possibly due to the intrahepatic metastasis of HCC.(5,6) However, the bleak outcomes of HCC patients even after potentially curative treatment have been, at least partially, attributed to its multicentric origin (late intrahepatic recurrence).(7,8) Therefore, it is necessary to examine not only tumor tissue but also non-tumor liver tissue to investigate the molecular mechanisms during the process of hepatocarcinogenesis based on the concept of “field cancerization”, because multicentric de novo occurrence of HCC is mainly related to underlying chronic liver damage rather than adverse tumor factors.(9–11) Field cancerization is a concept of tumorigenesis, which is defined as a cancer initiating with multiple cumulative epigenetic and genetic alterations that transform a cell or a group of cells in a particular organ.(12) This concept has been described in almost all organ systems,(13,14) including in liver.(15) From a clinical viewpoint, the identification of specific molecular markers to estimate risk of HCC development may provide us with an opportunity to make early diagnoses and effectively intervene with preventative strategies, including salvage liver transplantation and adjuvant interferon therapy.
Currently, several studies have investigated molecular alterations in non-tumor liver tissue on a genome-wide scale, covering different dimensions such as mRNA and microRNA (miRNA) expression, and epigenetic changes. This review focuses on the potential clinical value of array-based comprehensive analysis of molecular alterations in non-tumor liver tissue, particularly aberrant DNA methylation, to clarify hepatocarcinogenesis risk.
Gene Expression Profiling in Non-Tumor Tissue and Hepatocarcinogenesis
There have been at least three reports of DNA microarray analysis that attempted to predict the risk of multicentric recurrence of HCC patients based on the idea of field cancerization.(16–18) One study created a scoring system to estimate the risk of multicentric hepatocarcinogenesis based on the gene-expression profiling of 36 genes commonly associated with both multicentric HCC and multicentric recurrence after hepatectomy. This scoring system successfully predicted the risk of multicentric occurrence of HCC in hepatitis C virus (HCV)-positive patients.(16) Hoshida et al.(17) revealed that the gene expression profiles in non-tumor liver tissue but not primary HCC tissue were highly associated with late recurrence, possibly resulting from the multicentric occurrence of HCC. Both studies used cDNA arrays, and the former evaluated the accuracy of a predictor based only on a training sample set (n = 40), whereas the latter examined it in 82 patients as a training set and in 225 patients as a test sample set. The latter study also confirmed the feasibility of genome-wide expression profiling of formalin-fixed paraffin-embedded tissues, although there was a lack of overlap of predictive genes identified between these two studies.(10) A third study also showed that DNA microarray analysis in non-tumor liver tissue from patients with HCV-associated HCC delivered novel molecular signatures of recurrence-free survival, especially among patients with late recurrence.(18)
These findings suggest that biopsy-based gene expression analysis in cirrhotic patients may be a useful strategy to predict the risk of HCC development and highlight the need for adequate patient screening.(16–18) However, cross-study comparisons for validation of the findings reported on independent cohorts and array platforms may be needed.(18)
MicroRNA Expression Profiling in Non-Tumor Tissue and Hepatocarcinogenesis
MicroRNAs constitute a class of endogenous small regulatory RNA molecules that target mRNAs and trigger either translation repression or mRNA degradation.(19) Many miRNAs regulate genes associated with different biological processes, such as development, cell proliferation, apoptosis, stress response, and tumorigenesis.(20–22) Aberrant expression of several miRNAs are associated with multiple cancer types including HCC.(23–25) By analyzing tumor tissue, many articles have suggested promising results for miRNA-based classifiers in the progression of HCC.(26–31) However, only a few studies have focused on non-tumor liver tissue to identify miRNA-based classifiers for clinical outcomes. Sato et al. (28) examined miRNA expression profiling in paired tumor and non-tumor liver tissue samples from 73 HCC patients who satisfied the Milan Criteria. The expression patterns of tumor-derived miRNAs tended to predict early recurrence better than late recurrence, whereas those of non-tumor-derived miRNAs tended to better predict late recurrence after hepatic resection for HCC. These researchers suggested that miRNA expression profiling in non-tumor liver tissue would reflect the accumulation of genome abnormalities (the “field effect”) in the non-cancerous liver cells. More recently, the miRNA expression patterns in non-tumor liver tissue in HCC patients without multicentric recurrence in more than 3 years and those with multicentric recurrence within 3 years after hepatectomy were compared using a miRNA microarray analysis consisting of 955 probes.(32) Twenty differentially expressed miRNAs associated with multicentric recurrence were identified. The downregulated miRNAs included let-7d, which has been observed in many malignant tumors.(33,34) Expression of let-7d is significantly decreased by treatment with a specific colonic carcinogen at an early stage of carcinogenesis.(33) Downregulation of let-7d promotes pancreatic cancer transformation by post-transcriptional upregulation of crucial related oncogenes, such as K-RAS.(34) Therefore, the reduced expression of let-7d in non-tumor liver tissue might be associated with multicentric recurrence through the upregulation of K-RAS.
These studies suggest that specific miRNA expression signatures in non-tumor liver tissue may help predict the risk of de novo development of HCC.(28,32) However, prospective and external validations are needed before miRNA microarray analysis can be put into practical use.
DNA Methylation Status in Non-Tumor Tissue and Hepatocarcinogenesis
Aberrant DNA methylation is observed in many human cancers, including HCC, in which global hypomethylation and specific promoter hypermethylation have been found as typical epigenetic changes involved in genomic instability and silencing of tumor suppressor genes, respectively.(35,36) Despite numerous examples of aberrant DNA methylation, little is known about the global picture of hyper- or hypomethylated genes in the pathogenesis of HCC. However, several array-based studies indicated that specific DNA methylation signatures in HCC tumor tissues were associated with tumor progression and prognosis in patients with HCC.(37–42)
Aberrant DNA methylation is not only present in HCC tissue, but can also be found in chronically damaged non-tumor liver tissue.(15,43,44) Therefore, a better understanding of methylation alterations in the early stages of hepatocarcinogenesis will provide important molecular insights into the stepwise accumulation of epigenetic changes and may estimate the future risk of developing HCC. Indeed, several researchers have focused on aberrant DNA methylation in non-tumor (precancerous) liver tissue associated with clinicopathological features and prognosis in patients with HCC. Lou et al.(15) revealed that patients with RIZ1 hypermethylation, determined by methylation-specific PCR, in non-tumor liver tissue had a shorter disease-free survival, suggesting the existence of field cancerization in liver. Formeister et al.(43) showed that RIZ1 hypermethylation and LINE-1 hypomethylation, assessed by combined bisulfite restriction analysis, in non-tumor liver tissue were associated with time-to-recurrence. Another study examined the methylation status of nine CpG island loci, including RASSF1A and SOCS1, in dysplastic nodules and early HCC using MethyLight analysis. The frequency of methylated genes increased in a stepwise fashion from cirrhosis to dysplastic nodules, peaked in early HCC, and rather decreased in progressed HCC.(44)
Array-Based Analysis of Aberrant DNA Methylation
As methylation-specific PCR is commonly used in studies examining altered methylation levels in HCC, a single or a limited number of genes have been examined per study.(15,45) New development of DNA methylation analysis using array-based technologies now offers tremendous opportunities to study methylation on a genome-wide scale. This approach can improve our understanding of the epigenetic mechanisms and the effect of DNA methylation on disease-associated molecular networks and pathways, beyond single genes.
Kanai et al.(46) reported that DNA methylation alterations on chromosome 16 were frequently observed even in non-tumor liver tissue. This was one of the earliest reports of DNA methylation alterations at the precancerous stage of HCC. To clarify genome-wide DNA methylation profiles during hepatocarcinogenesis, these researchers later carried out bacterial artificial chromosome (BAC) array-based methylated CpG island amplification. After omitting potentially insignificant BAC clones associated only with inflammation and fibrosis, BAC clones for which DNA methylation status was inherited by HCCs from the precancerous stage were determined. The average numbers of BAC clones showing aberrant DNA methylation increased stepwise from non-tumor liver tissue to HCC tumor tissue, indicating that alterations of DNA methylation during hepatocarcinogenesis occur in a genome-wide manner. Twenty-five BAC clones, whose DNA methylation status was able to discriminate non-tumor liver tissue from normal liver tissue with 100% sensitivity and specificity, were successfully identified.(47) For appropriate surveillance of patients at the precancerous stage of HCC, the criteria for carcinogenetic risk estimation were further explored.(48) The criteria combining DNA methylation status for 30 regions, including the 45 CpG sites in non-tumor liver tissue, could estimate the risk of carcinogenesis in both the learning and validation cohorts. Therefore, it appears that clinicopathologically valid DNA methylation alterations have already accumulated at the precancerous stage.(48) Because even subtle alterations of DNA methylation profiles at the precancerous stage are stably preserved on DNA double strands by covalent bonds, they may be better indicators for risk estimation than mRNA, miRNA, and protein expression profiles that may be easily affected by the microenvironment of precursor cells.(47,49)
To date, at least three studies have used the Illumina (San Diego, CA, USA) GoldenGate Methylation BeadArray Cancer Panel I for simultaneously profiling the methylation state of 1505 CpG sites to identify differentially methylated CpG sites that may be important molecular events involved in liver tumorigenesis.(41,45,50) Hernandez-Vargas et al.(41) revealed the distinct methylation of an independent panel of gene promoters (58 CpG sites) in HCC tumor tissue that was strongly correlated with survival after cancer therapy. However, none of these studies focused on aberrant DNA methylation in non-tumor liver tissues associated with the early events of hepatocarcinogenesis. More comprehensive studies have determined differentially methylated genes between HCC tissues and adjacent non-tumor tissues and/or normal liver tissue using HumanMethylation27 DNA Analysis BeadChips (Illumina), which interrogate 27 578 CpG sites.(51–53) The main purpose of two of these studies was to determine previously unknown regions and genes that are differentially methylated in HCC, and to identify promising candidates of tumor suppressor genes as biomarkers for human HCC.(51,52) A third study revealed that both a hierarchical cluster analysis and the corresponding supervised principal component analysis showed a clear separation of non-tumor tissue from normal liver tissue and HCC tumor tissue.(53) As non-tumor liver tissue is more similar to normal liver tissue than the HCC tissue, the DNA methylation status of the affected loci changed gradually with the transition from normal liver to non-tumor liver tissue and further to a malignant phenotype of HCC. The findings in this study implicated the association of aberrant DNA methylation in non-tumor liver tissue with hepatocarcinogenesis.(53) Furthermore, by using the latest version of the Illumina methylation microarray chip (HumanMethylation450 BeadChip) containing over 485 000 CpG sites, two studies identified aberrant DNA methylation in various known differentially methylated regions, as well as potential new HCC differentially methylated loci.(54,55) Global hypomethylation in HCC tissue was confirmed, especially in the intergenic regions and gene bodies. The main purpose of these studies was also to identify promising biomarkers for human HCC, but they did not focus on the differential methylation in non-tumor liver tissue related to hepatocarcinogenesis.
Aberrant DNA Methylation and Diverse Etiologies
The molecular mechanisms related to aberrant DNA methylation in hepatocarcinogenesis are complicated and multi-factorial, and seem to differ according to the diverse etiologies.(56–58) Nishida et al.(59) quantified hypermethylation of tumor suppressor genes in HCC tissue at 19 CpG loci using combined bisulfite restriction analysis, and found that such hypermethylation was a major mechanism driving human hepatocarcinogenesis, especially in HCV-related HCC rather than hepatitis B virus (HBV)-related HCC. They also revealed that HCV-negative cases may be more likely to develop HCC through DNA hypomethylation based on the results of MethyLight analysis for repetitive DNA sequences, such as LINE-1 and Alu.(60) To further clarify the specific DNA methylation signatures associated with different etiological factors, array-based studies have recently been carried out. Two bead array studies identified the existence of specific methylation profiling in HCC tumor tissue according to diverse etiologies, including alcohol-related, HBV-related, HCV-related, and cryptogenic HCC.(41,52) However, few studies have examined whether specific DNA methylation status associated with etiologies is observed in non-tumor liver tissue.
Hepatitis C virus-related HCC is responsible for the greatest proportion of HCC patients in Japan, however, the proportion of HCC cases negative for hepatitis B surface antigen and hepatitis C antibody, so-called “NBNC-HCC”, is rapidly increasing.(61,62) However, there have been no studies specifically focusing on the implications of the molecular characteristics of non-tumor liver tissue in terms of hepatocarcinogenesis in patients with NBNC-HCC. Therefore, DNA methylation profiling of autosomal CpGs in non-tumor liver tissue of NBNC-HCC (n = 15) was compared with that in normal control (NC) liver tissue (n = 8) using the HumanMethylation450 BeadChip, as described previously.(63) The former (n = 15) included eight hepatitis B core antibody (HBcAb) positive (+) and seven HBcAb negative (−) patients. The latter (n = 7) were non-tumor liver tissue samples from patients who did not undergo any preoperative treatments, such as chemoradiotherapy. The methylation score for each CpG site was represented by a Beta-value calculated according to the normalized probe fluorescence intensity ratios between methylated and unmethylated signals, and Beta-values vary between 0 (fully unmethylated) and 1 (fully methylated). Figure 1 shows volcano plots showing 87 differentially methylated CpG sites in HBcAb (−) liver tissues and 603 CpG sites in HBcAb (+) liver tissue in comparison with NC liver tissue (Beta-value difference >0.2, P < 0.05). It is interesting to note that more differentially methylated CpG sites were identified in HBcAb (+) liver tissue samples compared with those from HBcAb (−) liver tissue, suggesting the possible epigenetic association of occult HBV infection with hepatocarcinogenesis. Thirty CpG sites were commonly hyper- or hypomethylated in both HBcAb (−) and HBcAb (+) liver tissue samples. The overlapping CpG sites were 100% consistent in the direction of methylation changes. The principal component analysis based on these differentially methylated CpG sites showed a clear separation of HBcAb (−) and HBcAb (+) liver tissue samples from NC liver tissue (Fig. 2). Therefore, specific DNA methylation profiling, which possibly contributes to the development of HCC, may exist in the non-tumor liver tissue of patients with NBNC-HCC. A Manhattan plot revealed that such DNA methylation alterations were spread across all chromosomes (data not shown). Table 1 lists the 30 CpG sites corresponding to 14 gene promoters that were commonly hyper- or hypomethylated in both HBcAb (−) and HBcAb (+) liver tissue samples. This genome-wide DNA methylation array system (HumanMethylation450 BeadChip) includes the promoter, gene body, and 3′-UTR.(64) In fact, high percentages of aberrant DNA methylation were observed in gene bodies rather than promoters (Fig. 3). A recent study showed that CpG sites located at promoter regions show a negative correlation whereas CpG sites in gene bodies show a positive correlation between DNA methylation and gene expression levels.(65) It is important to note that correlations between DNA methylation status and gene expression levels may be dependent on the location of CpG sites in the genes. In addition, cautious interpretation of these microarray data with special emphasis on potential signals generated by cross-reactive probes and polymorphic CpGs should also be recommended.(66)
Figure 1.
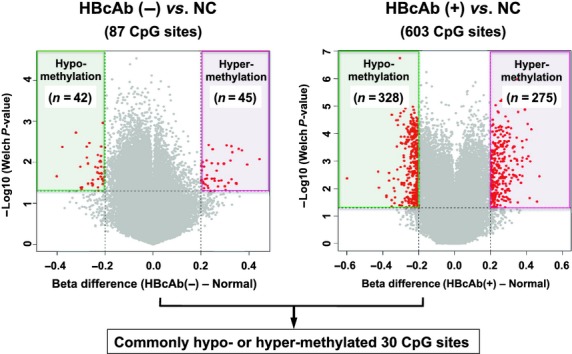
Volcano plots for differential DNA methylation status. The x-axis shows the mean DNA methylation (Beta-value) difference, whereas the y-axis shows the –log10 of the P-value for each CpG site, representing the strength of association. Eighty-seven differentially methylated CpG sites in hepatitis B core antibody (HbcAb) (−) liver tissues and 603 CpG sites in HBcAb (+) liver tissues in comparison with normal control (NC) liver tissues using (Beta-value difference >0.2, P < 0.05) are plotted in red.
Figure 2.
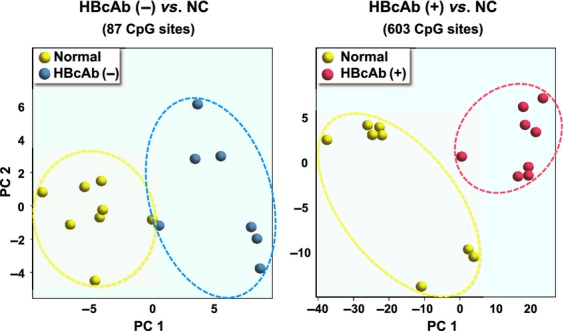
Principal component (PC) analysis for differential DNA methylation status. Normal control (NC), hepatitis B core antibody (HbcAb) (−), and HBcAb (+) liver tissue samples are indicated in yellow, blue, and red, respectively.
Table 1.
Thirty CpG sites corresponding to 14 gene promoters that were commonly hyper- or hypomethylated in both hepatitis B core antibody (HbcAb) (−) and HBcAb (+) liver tissue samples
| Gene name | Target ID | Chromosome | Position† | HBcAb (−) versus NC | HBcAb (+) versus NC | ||
|---|---|---|---|---|---|---|---|
| Mean difference‡ | P-value | Mean difference‡ | P-value | ||||
| Hypermethylated | |||||||
| CD80 | cg21139795 | 3 | 119243933 | 0.224 | 0.025 | 0.270 | 0.005 |
| NPBWR1 | cg26205771 | 8 | 53851156 | 0.272 | 0.039 | 0.300 | 0.002 |
| CD44 | cg13332350 | 11 | 35239907 | 0.392 | 0.011 | 0.471 | 0.004 |
| USP2 | cg10904972 | 11 | 119227328 | 0.249 | 0.005 | 0.267 | 0.001 |
| TTC9 | cg23691406 | 14 | 71112909 | 0.348 | 0.033 | 0.351 | 0.032 |
| CCDC64B | cg27519622 | 16 | 3079877 | 0.213 | 0.043 | 0.324 | 0.001 |
| SOX9 | cg01524174 | 17 | 70119015 | 0.207 | 0.030 | 0.330 | 0.001 |
| SRC | cg25431463 | 20 | 36012946 | 0.203 | 0.027 | 0.219 | 0.003 |
| Hypomethylated | |||||||
| RPS6KA1 | cg24585377 | 1 | 26857774 | −0.216 | −0.216 | −0.203 | 0.002 |
| PTPN14 | cg11188103 | 1 | 214668616 | −0.228 | −0.228 | −0.209 | 0.005 |
| TRIM10 | cg08094206 | 6 | 30122523 | −0.276 | −0.276 | −0.278 | 0.002 |
| PDGFA | cg14496282 | 7 | 544525 | −0.252 | −0.252 | −0.325 | 0.001 |
| ATP11A | cg08464505 | 13 | 113425982 | −0.217 | −0.217 | −0.250 | 0.001 |
| DEGS2 | cg23076361 | 14 | 100622050 | −0.201 | −0.201 | −0.200 | 0.001 |
Positions refer to Genome Research Consortium human genome build 37 (GRCh37)/UCSC human genome 19 (hg19).
Mean difference of Beta-values between HBcAb (−) or HBcAb (+) and normal control liver tissues.
Figure 3.
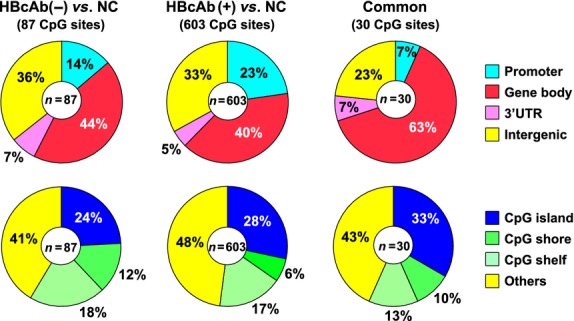
Percentage of CpG sites according to their gene location (upper) and the CpG content in the genes (lower). “Common” indicates the 30 CpG sites, which were commonly hyper- or hypomethylated in both hepatitis B core antibody (HbcAb) (−) and HBcAb (+) liver tissues in comparison with normal control (NC) liver tissues.
Future Directions
Although the potential clinical value of array-based analysis of molecular alterations in non-tumor liver tissue to clarify the risk of hepatocarcinogenesis is still not well characterized, the association of such molecular alterations with HCC patient prognosis has been increasingly investigated. Indeed, the emergence of specific alterations of DNA methylation profiles have already been confirmed in non-tumor liver tissue samples (precancerous lesions) obtained from patients with HCC. Therefore, in addition to the antiviral and anti-inflammatory therapies, epigenetic therapies for patients with chronic liver diseases may be helpful to reduce the risk of HCC development because epigenetic alterations are potentially reversible, and thus provide promising molecular targets for therapeutic intervention, especially for the subset of HCC that develops through the epigenetic pathway. Furthermore, because of continued and rapid progress of mRNA, miRNA, and epigenetic studies, the integrative analyses of all these data will increase our knowledge of the stepwise accumulation of molecular alterations during hepatocarcinogenesis. This will enable us to identify which patients will most benefit from certain therapeutic and preventive strategies, an important step towards personalized medicine in the treatment of early-stage HCC.
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–17. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 2.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 3.Sherlock S. Viruses and hepatocellular carcinoma. Gut. 1994;35:828–32. doi: 10.1136/gut.35.6.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakamoto M. Early HCC: diagnosis and molecular markers. J Gastroenterol. 2009;44:108–11. doi: 10.1007/s00535-008-2245-y. [DOI] [PubMed] [Google Scholar]
- 5.Iizuka N, Oka M, Yamada Okabe H, et al. Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. Lancet. 2003;361:923–9. doi: 10.1016/S0140-6736(03)12775-4. [DOI] [PubMed] [Google Scholar]
- 6.Yoshioka S, Takemasa I, Nagano H, et al. Molecular prediction of early recurrence after resection of hepatocellular carcinoma. Eur J Cancer. 2009;45:881–9. doi: 10.1016/j.ejca.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 7.Poon RT, Fan ST, Ng IO, Lo CM, Liu CL, Wong J. Different risk factors and prognosis for early and late intrahepatic recurrence after resection of hepatocellular carcinoma. Cancer. 2000;89:500–7. [PubMed] [Google Scholar]
- 8.Takenaka K, Adachi E, Nishizaki T, et al. Possible multicentric occurrence of hepatocellular carcinoma: a clinicopathological study. Hepatology. 1994;19:889–94. [PubMed] [Google Scholar]
- 9.Kim JW, Ye Q, Forgues M, et al. Cancer-associated molecular signature in the tissue samples of patients with cirrhosis. Hepatology. 2004;39:518–27. doi: 10.1002/hep.20053. [DOI] [PubMed] [Google Scholar]
- 10.Utsunomiya T, Shimada M, Imura S, et al. Molecular signatures of noncancerous liver tissue can predict the risk for late recurrence of hepatocellular carcinoma. J Gastroenterol. 2010;45:146–52. doi: 10.1007/s00535-009-0164-1. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka S, Mogushi K, Yasen M, et al. Oxidative stress pathways in noncancerous human liver tissue to predict hepatocellular carcinoma recurrence: a prospective, multicenter study. Hepatology. 2011;54:1273–81. doi: 10.1002/hep.24536. [DOI] [PubMed] [Google Scholar]
- 12.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 13.Shen L, Kondo Y, Rosner GL, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330–8. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 14.Chang YL, Wu CT, Lin SC, Hsiao CF, Jou YS, Lee YC. Clonality and prognostic implications of p53 and epidermal growth factor receptor somatic aberrations in multiple primary lung cancers. Clin Cancer Res. 2007;13:52–8. doi: 10.1158/1078-0432.CCR-06-1743. [DOI] [PubMed] [Google Scholar]
- 15.Lou C, Du Z, Yang B, Gao Y, Wang Y, Fang S. Aberrant DNA methylation profile of hepatocellular carcinoma and surgically resected margin. Cancer Sci. 2009;100:996–1004. doi: 10.1111/j.1349-7006.2009.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Utsunomiya T, Okamoto M, Wakiyama S, et al. Specific gene-expression profiles of noncancerous liver tissue predict the risk for multicentric occurrence of hepatocellular carcinoma in hepatitis C virus-positive patients. Ann Surg Oncol. 2006;13:947–54. doi: 10.1245/ASO.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 17.Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuchiya M, Parker JS, Kono H, et al. Gene expression in nontumoral liver tissue and recurrence-free survival in hepatitis C virus-positive hepatocellular carcinoma. Mol Cancer. 2010;9:74. doi: 10.1186/1476-4598-9-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Rooij E, Sutherland LB, Liu N, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–60. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsit CJ, Eddy K, Kelsey KT. MicroRNA responses to cellular stress. Cancer Res. 2006;66:10843–8. doi: 10.1158/0008-5472.CAN-06-1894. [DOI] [PubMed] [Google Scholar]
- 22.Schmittgen TD. Regulation of microRNA processing in development, differentiation and cancer. J Cell Mol Med. 2008;12:1811–9. doi: 10.1111/j.1582-4934.2008.00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 24.Visone R, Croce CM. MiRNAs and cancer. Am J Pathol. 2009;174:1131–8. doi: 10.2353/ajpath.2009.080794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji J, Shi J, Budhu A, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361:1437–47. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang J, Gusev Y, Aderca I, et al. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res. 2008;142:419–27. doi: 10.1158/1078-0432.CCR-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Budhu A, Jia HL, Forgues M, et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;473:897–907. doi: 10.1002/hep.22160. [DOI] [PubMed] [Google Scholar]
- 28.Sato F, Hatano E, Kitamura K, et al. MicroRNA profile predicts recurrence after resection in patients with hepatocellular carcinoma within the Milan criteria. PLoS One. 2011;61:e16435. doi: 10.1371/journal.pone.0016435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viswanathan SR, Powers JT, Einhorn W, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;417:843–8. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong CM, Wong CC, Lee JM, Fan DN, Au SL, Ng IO. Sequential alterations of microRNA expression in hepatocellular carcinoma development and venous metastasis. Hepatology. 2012;55:1453–61. doi: 10.1002/hep.25512. [DOI] [PubMed] [Google Scholar]
- 31.Murakami Y, Tamori A, Itami S, et al. The expression level of miR-18b in hepatocellular carcinoma is associated with the grade of malignancy and prognosis. BMC Cancer. 2013;13:99. doi: 10.1186/1471-2407-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Utsunomiya T, Ishikawa D, Asanoma M, et al. Specific miRNA expression profiles of non-tumor liver tissue predict a risk for recurrence of hepatocellular carcinoma. Hepatol Res. 2013 doi: 10.1111/hepr.12164. doi: 10.1111/hepr.12164 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Davidson LA, Wang N, Shah MS, Lupton JR, Ivanov I, Chapkin RS. n-3 Polyunsaturated fatty acids modulate carcinogen-directed non-coding microRNA signatures in rat colon. Carcinogenesis. 2009;30:2077–84. doi: 10.1093/carcin/bgp245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiao LR, Frampton AE, Jacob J, et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS One. 2012;7:e32068. doi: 10.1371/journal.pone.0032068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–93. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 36.Matsuzaki K, Deng G, Tanaka H, Kakar S, Miura S, Kim YS. The relationship between global methylation level, loss of heterozygosity, and microsatellite instability in sporadic colorectal cancer. Clin Cancer Res. 2005;11:8564–9. doi: 10.1158/1078-0432.CCR-05-0859. [DOI] [PubMed] [Google Scholar]
- 37.Lin CH, Hsieh SY, Sheen IS, et al. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001;61:4238–43. [PubMed] [Google Scholar]
- 38.Shen L, Ahuja N, Shen Y, et al. DNA methylation and environmental exposures in human hepatocellular car- cinoma. J Natl Cancer Inst. 2002;94:755–61. doi: 10.1093/jnci/94.10.755. [DOI] [PubMed] [Google Scholar]
- 39.Calvisi DF, Ladu S, Gorden A, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117:2713–22. doi: 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao W, Kondo Y, Shen L, et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis. 2008;29:1901–10. doi: 10.1093/carcin/bgn170. [DOI] [PubMed] [Google Scholar]
- 41.Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One. 2010;5:e9749. doi: 10.1371/journal.pone.0009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao R, Li J, Xin J, et al. Methylation profile of single hepatocytes derived from hepatitis B virus-related hepatocellular carcinoma. PLoS One. 2011;6:e19862. doi: 10.1371/journal.pone.0019862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Formeister EJ, Tsuchiya M, Fujii H, Shpyleva S, Pogribny IP, Rusyn I. Comparative analysis of promoter methylation and gene expression endpoints between tumorous and non-tumorous tissues from HCV-positive patients with hepatocellular carcinoma. Mutat Res. 2010;692:26–33. doi: 10.1016/j.mrfmmm.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Um TH, Kim H, Oh BK, et al. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J Hepatol. 2011;54:939–47. doi: 10.1016/j.jhep.2010.08.021. [DOI] [PubMed] [Google Scholar]
- 45.Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCVassociated hepatocellular carcinoma. Mol Genet Genomics. 2010;283:341–9. doi: 10.1007/s00438-010-0522-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Sugimura T, Hirohashi S. Aberrant DNA methylation on chromosome 16 is an early event in hepatocarcinogenesis. Jpn J Cancer Res. 1996;87:1210–7. doi: 10.1111/j.1349-7006.1996.tb03135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arai E, Ushijima S, Gotoh M, et al. Genome-wide DNA methylation profiles in liver tissue at the precancerous stage and in hepatocellular carcinoma. Int J Cancer. 2009;125:2854–62. doi: 10.1002/ijc.24708. [DOI] [PubMed] [Google Scholar]
- 48.Nagashio R, Arai E, Ojima H, Kosuge T, Kondo Y, Kanai Y. Carcinogenetic risk estimation based on quantification of DNA methylation levels in liver tissue at the precancerous stage. Int J Cancer. 2011;129:1170–9. doi: 10.1002/ijc.26061. [DOI] [PubMed] [Google Scholar]
- 49.Kanai Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci. 2010;101:36–45. doi: 10.1111/j.1349-7006.2009.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shin SH, Kim BH, Jang JJ, Suh KS, Kang GH. Identification of novel methylation markers in hepatocellular carcinoma using a methylation array. J Korean Med Sci. 2010;25:1152–9. doi: 10.3346/jkms.2010.25.8.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumann O, Kesselmeier M, Geffers R, et al. Methylome analysis and integrative profiling of human HCCs identify novel protumorigenic factors. Hepatology. 2012;56:1817–27. doi: 10.1002/hep.25870. [DOI] [PubMed] [Google Scholar]
- 52.Shen J, Wang S, Zhang YJ, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55:1799–808. doi: 10.1002/hep.25569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ammerpohl O, Pratschke J, Schafmayer C, et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer. 2012;130:1319–28. doi: 10.1002/ijc.26136. [DOI] [PubMed] [Google Scholar]
- 54.Song MA, Tiirikainen M, Kwee S, Okimoto G, Yu H, Wong LL. Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS One. 2013;8:e55761. doi: 10.1371/journal.pone.0055761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen J, Wang S, Zhang YJ, et al. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics. 2013;8:34–43. doi: 10.4161/epi.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiba T, Yokosuka O, Fukai K, et al. Identification and investigation of methylated genes in hepatoma. Eur J Cancer. 2005;41:1185–94. doi: 10.1016/j.ejca.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 57.Feng Q, Stern JE, Hawes SE, Lu H, Jiang M, Kiviat NB. DNA methylation changes in normal liver tissues and hepatocellular carcinoma with different viral infection. Exp Mol Pathol. 2010;88:287–92. doi: 10.1016/j.yexmp.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lambert MP, Paliwal A, Vaissière T, et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. J Hepatol. 2011;54:705–15. doi: 10.1016/j.jhep.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 59.Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908–18. doi: 10.1002/hep.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishida N, Kudo M, Nagasaka T, Ikai I, Goel A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology. 2012;56:994–1003. doi: 10.1002/hep.25706. [DOI] [PubMed] [Google Scholar]
- 61.Utsunomiya T, Shimada M. Molecular characteristics of non-cancerous liver tissue in non-B non-C hepatocellular carcinoma. Hepatol Res. 2011;41:711–21. doi: 10.1111/j.1872-034X.2011.00818.x. [DOI] [PubMed] [Google Scholar]
- 62.Utsunomiya T, Shimada M, Kudo M. Nationwide study of 4741 patients with non-B non-C hepatocellular carcinoma with special reference to the therapeutic impact. Ann Surg. 2014;259:336–45. doi: 10.1097/SLA.0b013e31829291e9. [DOI] [PubMed] [Google Scholar]
- 63.Kinoshita M, Numata S, Tajima A, Shimodera S, Imoto I, Ohmori T. Plasma total homocysteine is associated with DNA methylation in patients with schizophrenia. Epigenetics. 2013;8:584–90. doi: 10.4161/epi.24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 65.Kulis M, Queirós AC, Beekman R, Martín-Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta. 2013;1829:1161–74. doi: 10.1016/j.bbagrm.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 66.Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–9. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]