

Abstract
Although the heterogeneities of epithelial and mesenchymal-transitioned cancer cells are often observed within the tumor microenvironment, the biological significance of the interaction between epithelial cancer cells and mesenchymal-transitioned cancer cells is not yet understood. In this study, we show that the mesenchymal-transitioned cancer cells instigate the invasive ability and metastatic potential of the neighboring epithelial cancer cells in vitro and in vivo. We identify WNT3 and WNT5B as critical factors secreted from Transforming growth factor-induced mesenchymal cancer cells for instigating the epithelial cancer cell invasion along with the induction of secondary EMT phenotype. These results shed light on the significance of cancer heterogeneity and the interaction between epithelial and mesenchymal-transitioned cancer cells within the tumor microenvironment in promoting metastatic disease through the WNT-dependent mechanism.
Keywords: Cross-talk, epithelial-to-mesenchymal transition, heterogeneity, metastasis WNT
Tumor tissue consists not of only cancer cells, but also various stromal cells, such as cancer-associated fibroblasts (CAF), immune inflammatory cells, myeloid progenitor cells and vascular endothelial cells.1 The heterogeneity of the complicated environment within tumor tissue, or the tumor microenvironment, plays an important role in cancer malignancy progression.1 Recent evidence suggests that not only the tumor microenvironment but also cancer cells themselves within tumor tissue are heterogeneous by representing numerous subpopulations with both genetic and non-genetic variations.2,3
Cancer metastasis disease, one of the major contributing factors to the high mortality rate in cancer patients, involves multiple biological steps, including intravasation, attachment to vessels, extravasation, angiogenesis and subsequent growth in distal tissues of the primary tumor. Among those steps, the initial acquisition of cellular invasiveness is likely a key stage in metastatic dissemination from the primary tumor site, and the process of epithelial-to-mesenchymal transition (EMT) is known to play an important role during this process.4–7 In accordance with the dynamic yet transient morphological and phenotypic alteration of cancer cells during the EMT process, such mesenchymal-transitioned cancer cells are often seen at the invasive front of tumor tissue beside neighboring epithelial cancer cells.8–13 Even though the importance of transforming growth factor (TGF)-β in the initiation of EMT has been demonstrated,14,15 there are several reports that primary cancer specimens acquire mesenchymal features and develop metastatic disease even in the presence of the deletion of in Smad4, which is a key component of the TGF-β signaling pathway.16,17 These observations might imply that there are alternative pathways to maintain the EMT phenotype other than TGF-β pathway within the tumor microenvironment. Interestingly, the cooperation of mesenchymal-transitioned and surrounding epithelial cancer cells in establishing spontaneous metastasis in a mouse model has been reported.18 It has also been revealed that the heterogeneity of cancer cells and the intra-tumoral cross-talk between distinct types of cancer cells might contribute to the induction of abnormal proliferation and metastasis;19–21 however, the exact mechanism of how those distinct cancer cell types interact with each other is not yet understood.
In the present study, we demonstrate that the coexistence of mesenchymal-transitioned cancer cells with epithelial cancer cells induces the invasive ability and the metastatic potential of epithelial cancer cells in vitro and in vivo. Furthermore, we identified WNT3 and WNT5B as the secretory factors from TGF-β-induced mesenchymal-transitioned cancer cells that induce the invasion of neighboring epithelial cancer cells and secondary EMT phenotype. Collectively, these results strongly implicate secretory WNT ligands as critical soluble factors mediating the invasion instigation of epithelial cancer cells derived from mesenchymal-transitioned cancer cells, and further targeting those secretory WNT proteins could be a new approach to prevent cancer invasion and subsequent metastasis.
Materials and Methods
Cell culture and inhibitors
Human lung adenocarcinoma A549 and human pancreatic ductal adenocarcinoma Panc-1 cells were obtained from the American Type Culture Collection. A549 cells were maintained in RPMI1640, and Panc-1 cells were maintained in DMEM, containing 10% FBS, 1 mM L-glutamine and antibiotics (100 units/mL penicillin and 100 mg/mL streptomycin) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. To establish labeled A549 and Panc-1 cells, A549 or Panc-1 cells were transfected with pGL4.50/Luc2 (Promega, Madison, WI, USA) or pEGFP-C1 (Clontech, Palo Alto, CA, USA), selected, and cloned in growth medium containing 200 μg/mL hygromycin B or 1 mg/mL G418, respectively. The reagents used were: WNT secretion inhibitor, IWP-2 (Sigma-Aldrich, St. Louis, MO, USA) and TGF-β receptor kinase inhibitor (TβRI) (Calbiochem, Dermstadt, Germany).
Preparation of E-cells and M-cells
In this study, non-stimulated epithelial A549 and Panc-1 cells were used as E-cells. To prepare the mesenchymal-transitioned cancer cells (M-cells), A549 or Panc-1 cells were treated with 5 ng/mL recombinant TGF-β (Pepro Tech, Rocky Hill, NJ, USA) for 48 h then washed with fresh culture medium twice and harvested for subsequent experiments.
Direct and separated co-culture experiment
For the direct co-culture experiment, E-cells and M-cells were re-seeded into 60 mm culture dish according to the indicated cell number and co-cultured for 24 h. For the separated co-culture experiment, 1 × 105 E-cells and 3 × 105 cells of M-cells were seeded into lower or upper compartments of the transwell chamber with 1 μm pore diameter (BD Falcon, Bedford, MA, USA) for 24 h.
Generation of conditioned medium
After preparation of E-cells and M-cells, the cells were further cultured in fresh growth medium for an additional 48 h. Finally, these culture supernatants were collected and diluted with fresh growth medium at a ratio of 2:1. The freshly prepared supernatant was used as the conditioned medium (CM) in each experiment.
For preparation of WNT3-depleted and WNT5B-depleted CM by siRNA, parental A549 and Panc-1 cells were transfected with siControl (siGENOME Control Pool Non-targeting siRNA#2), siWNT3 and/or siWNT5B (ON-TARGETplus SMARTpool siRNA) (Thermo Fisher Scientific, Rockford, IL, USA) by Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) 48 h prior to induction of M-cells. For preparation of WNT-depleted CM by IWP-2, E-cells and M-cells were cultured in fresh medium containing IWP-2 for 48 h. The protein level of WNT3 and WNT5B in CM was determined by a specific ELISA (CUSABIO Biotech, Wuhan, China) according to the manufacturer's protocol.
Matrigel invasion assay
Cancer cell invasion through reconstituted basement membrane (Matrigel [BD Biosciences], San Jose, CA, USA) was assayed as previously described.22 After fixing the filter and staining with H&E, the invaded cells were counted manually under a microscope at ×100. For detection of luciferase activity in invaded A549/Luc2 cells, the filters were soaked in passive lysis buffer (Promega) and luciferase activity was determined. For detection of EGFP+ invaded cells, filters were fixed with 4% paraformaldehyde and stained with VECTASHIELD mounting media with DAPI (Vector Laboratories, Burlingame, CA, USA).
Western blotting
Whole cell lysates and nuclear protein extracts were prepared as described previously.23 The primary antibodies used were Epithelial-Mesenchymal transition (EMT) Antibody Sampler Kit (#9782, Cell Signaling Technology, Beverly, MA, USA), antibodies against WNT3 (ab32249) and WNT5B (ab94914) from Abcam, and antibodies against PCNA (PC10), β-actin (C11), Lamin B (C-20) and α-tubulin (D-10) from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Experimental lung metastasis experiment
C.B-17/lcrHsd-Prkdcscid mice were purchased from Japan SLC (Hamamatsu, Japan). All experiments were approved and performed according to the guidelines of the Care and Use of Laboratory Animals of the University of Toyama. Cells were inoculated intravenously (2 × 106 cells/200 μL PBS/mouse) into mice and the lungs were removed 24 h after the tumor inoculation. Mice were intraperitoneally injected with 200 μL of luciferin (1.5 mg/mL [VivoGlo; Promega]) 20 min prior to subject bioluminescent assay by using an in vivo imaging system (IVIS Lumina II, Caliper Life Sciences, Hopkinton, MA, USA). The data was presented as the mean luminescence ± SEM.
Microarray data analysis
The datasets (GSE17708 and GSE23952) were reanalyzed on GenePattern.24 Briefly, the differential expression level of all genes between TGF-β-treated samples and non-treated samples was computed and the top 5% of upregulated genes in TGF-β-treated cells compared with control cells were selected by using the “Comparative Marker Selection” tool from each dataset. Finally, the genes coding the secreted proteins were picked up from the commonly upregulated genes in both datasets.
Gene set enrichment analysis (GSEA) was performed using javaGSEA application v2.0.13 (GSEA, Broad Institute, Boston, MA, USA). These pathway gene sets were provided by the Molecular Signatures Database (MSigDB [http:\\www.broadinstitute.org/gsea/msigdb]).
Statistical analysis
Statistical significance was calculated using Microsoft Excel. More than three means were composed using one-way anova with the Bonferroni correction, and two means were composed using unpaired Student's t-test. P < 0.05 were considered statistically significant.
Results
Epithelial cancer cells acquire metastatic potential by the co-culture with mesenchymal-transitioned cancer cells
To directly evaluate the role of heterogeneity of cancer cells in their invasive potential, we performed Matrigel invasion assays in human epithelial lung adenocarcinoma A549 and pancreatic ductal adenocarcinoma Panc-1 cell co-culture at various ratios of epithelial cancer cells (E-cells) and TGF-β-induced mesenchymal-transitioned cancer cells (M-cells), because TGF-β is known to trigger EMT in both A549 and Panc-1, which is the most potent EMT-inducing cytokine (Fig. S1).25,26 After 24 h co-culture of E-cells and M-cells, the invasive potential of the E-cell and M-cell mixture was more enhanced in both A549 and Panc-1 cell lines, in an M-cell dose-dependent manner, than that in M-cells alone (Fig.1a). To determine the cell population, either E-cells or M-cells, responsible for the enhanced invasive potential after the co-culture, we employed luminescence-labeled E-A549 (E-A549/Luc2) or fluorescent-labeled E-Panc-1 (E-Panc/EGFP) to distinguish E-cells from M-cells within the co-culture. By measuring the invasiveness of labeled E-cells in either A549 or Panc-1, we have found that the invasive potential of E-cells co-cultured with M-cells was higher than that of E-cells alone in both A549 and Panc-1 (Fig.1b) and such induction of invasiveness was observed in an M-cell dose-dependent manner (Fig.1b). We have also found that the invasion potential of E-cells was not altered without the co-culture with M-cells (Fig. S2). Of note, the invasive potential of M-Panc cells was not affected by the co-culture with E-Panc cells as compared with M-Panc cells alone (data not shown). These results clearly indicate that the co-culture of E-cells with M-cells selectively enhances the invasiveness of E-cells in both A549 and Panc-1 cell lines.
Figure 1.
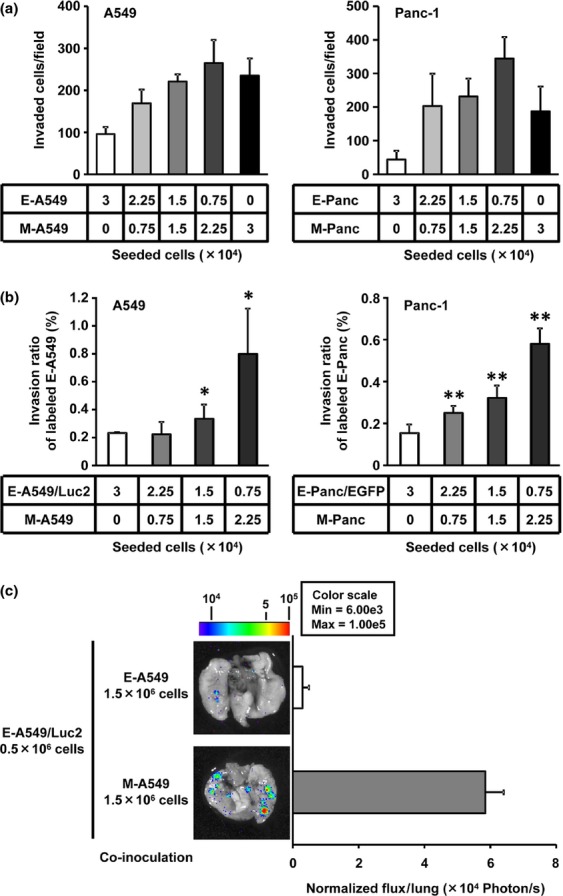
Epithelial cancer cells acquire metastatic potential upon co-culture with mesenchymal-transitioned cancer cells. (a) A549 (left) or Panc-1 (right) cells were subjected to Matrigel invasion assay after 24-h co-culture of epithelial cancer cells (E-cells) with mesenchymal-transitioned cancer cells (M-cells) at the indicated cell numbers. Total invaded cells were counted after H&E staining. Data represented as the mean ± SD of four independent experiments. (b) Labeled E-cells (E-A549/Luc2; left panel or E-Panc/EGFP; right panel) were subjected to Matrigel invasion assay after 24-h-co-culture with M-cells. Invasive abilities were determined by measuring luciferase activity (A549) or counting invaded EGFP+ cells (Panc-1), respectively. Invasion ratio was calculated by the division of invaded E-cells by total E-cells applied. Data represented as the mean ± SD of triplicate experiment. *P < 0.05, **P < 0.01 versus E-cells alone group by Dunnett's test. (c) Epithelial A549 cells overexpressing Luc2 gene (E-A549/Luc2) were co-cultured with either epithelial A549 (E-A549) cells or mesenchymal-transitioned A549 (M-A549) cells and i.v. inoculated into mice. Mice were killed 24 h after the tumor inoculation and lungs were subjected to bioluminescent imaging to determine total flux (photon/s) for lung metastasis quantification. The representative ex vivo images are shown. Data represented as the mean ± SEM (n = 5).
To further investigate the significance of the interaction between E-cells and M-cells in vivo, the metastatic potential of E-A549 cells to the lungs was examined by injecting E-A549/Luc2 cells upon co-culture with either E-A549 or M-A549. The metastatic spread of E-A549/Luc2 in the lungs was much higher after the co-culture with M-A549 cells compared to with E-A549 cells (Fig.1c), therefore indicating the potential of M-cells to promote the metastatic ability of neighboring E-cells in vivo.
Mesenchymal-transitioned cancer cells induce the metastatic potential of the neighboring epithelial cancer cells in a cell–cell contact independent mechanism
To determine whether the direct cell–cell interaction is required for promoting the metastatic potential of E-cells by neighboring with M-cells, we first examined the expression of EMT-related proteins in A549 or Panc-1 cells under the co-culture of E-cells with M-cells in the presence or absence of direct cell–cell contact (Fig.2a,b). Given that the upregulation of the mesenchymal marker (Snail and Vimentin) and the downregulation of the epithelial marker (E-cadherin) were markedly induced by both direct and separated co-culture of E-cells and M-cells, the soluble factor(s) derived from M-cells was at least sufficient for the induction of the secondary EMT phenotype of E-cells upon co-culture with M-cells. We further confirmed that the invasion of E-A549 cells was remarkably enhanced after the culture with M-A549-derived conditioned medium (M-A549-CM) compared with E-A549-CM (Fig.2c and Fig. S3). In concert with the enhanced invasive ability of E-A549 cells after the cultivation with M-A549-CM, the secondary EMT phenotype in E-A549 cells was also induced after the culture with M-A549-CM. Similar results were obtained in the invasion of E-Panc cells after the cultivation with M-Panc-CM (data not shown).
Figure 2.

Cell–cell contact independent induction of secondary epithelial-to-mesenchymal transition (EMT) phenotype and invasiveness in neighboring epithelial cancer cells by mesenchymal-transitioned cancer cells. (a, b) A549 cells were co-cultured directly (a) or separately in transwell cell culture chamber (b) at the indicated cell numbers for 24 h and EMT-related protein expression were determined by western blotting. No cells (−), E-cells (E) or M-cells (M) were seeded in upper compartment of transwell chamber. In the separated co-culture, the total protein from E-cells in lower compartment was examined. (c, d) Epithelial A549 cells were treated with conditioned mediums (CM) from E-/M-A549 cells (c) or E-/M-Panc cells (d) for 48 h and subjected to Matrigel invasion assay or western blotting. Total invaded cells were counted after H&E staining. Data represented as the mean ± SD of triplicate experiment. **P < 0.01 versus E-CM group by two-tailed Student's t test.
Importantly, the CM from M-Panc cells was able to introduce enhanced invasive ability and secondary EMT phenotype in E-A549 cells (Fig.2d and Fig. S3), indicating that the common soluble factor(s) derived from M-A549 cells and M-Panc cells are likely to be involved in this process. Considering that the induction of secondary EMT in E-cells by M-cell-CM was not affected by TGF-β receptor kinase inhibitor (data not shown), the involvement of the TGF-β signaling pathway is less likely. Collectively, these data indicate that mesenchymal-transitioned cancer cell-derived soluble factor(s), which is common in both M-A549 and M-Panc cells, play a significant role in the induction of invasive ability and secondary EMT phenotype in the neighboring epithelial cancer cells.
WNT3 and WNT5B derived from mesenchymal-transitioned cancer cells are the soluble factors that induce metastatic potential in the neighboring epithelial cancer cells
In order to identify the common soluble factor(s) that is secreted from mesenchymal-transitioned A549 and Panc-1 cells, we analyzed the published cDNA microarray datasets (GSE17708 and GSE23952) representing A549 and Panc-1 gene expression following the TGF-β stimulation for 72 or 48 h, respectively. There are 55 candidate genes as the top 5% of encoding secretory proteins that are commonly upregulated in both A549 and Panc-1 cells (Fig.3a and Table S1). By using Gene Set Enrichment Analysis, we further selected candidate pathway gene sets that are significantly enriched in phenotype of TGF-β as shown in Table S2. Among those candidate pathways, WNT pathway was commonly enriched in both M-A549 and M-Panc. Thus, we further focused on WNT3 and WNT5B molecules in the induction of secondary EMT in epithelial cancer cells. WNT3 and WNT5B are known to be a ligand for activating both canonical and non-canonical WNT pathways.27 As shown in Fig.3b, we confirmed the higher expression of WNT3 and WNT5B at protein level in both M-cells compared to E-cells. Consistent with the upregulation of WNT3 and WNT5B, the secretion of these WNT ligands was detected in CM of M-A549 by ELISA (Fig.3c). We also confirmed higher nuclear β-catenin expression and β-catenin transcriptional activity in E-cells with M-cell-CM, indicating that E-cells received the WNT signals from M-cells (data not shown).
Figure 3.

Secretory WNT3 and WNT5B from mesenchymal-transitioned cancer cells induce secondary epithelial-to-mesenchymal transition (EMT) phenotype in epithelial cancer cells. (a) Commonly upregulated genes encoding soluble protein in the top 5% in GSE17708 (Panc-1) and GSE23952 (A549) datasets were shown as Benn diagram. (b) Epithelial or mesenchymal-transitioned A549 or Panc-1 cells were subjected to western blotting to determine the expression of indicated proteins. (c) Conditioned mediums from E-cells or M-cells were subjected to ELISA for detecting WNT3 or WNT5B.
To further examine whether WNT3 and WNT5B are the molecules responsible in M-cell-derived CM for the induction of invasiveness and secondary EMT phenotype in E-cells, we used the siRNA of WNT3 and WNT5B or the chemical WNT ligand secretion inhibitor, IWP-2, during the preparation of CM. The knockdown efficiencies or the inhibition of secretion were confirmed by qRT-PCR, western blotting and ELISA (Fig. S4). While the M-cell-CM derived from single knockdown of either WNT3 or WNT5B did not completely diminish the induction of invasive potential and Vimentin/Snail expression of E-A549 and E-Panc cells, the knockdown of both WNT3 and WNT5B completely abrogated the activity of M-cell-CM in the induction of invasive potential and secondary EMT phenotype of E-A549 and E-Panc cells (Fig.4a). Importantly, the M-cell-CM prepared in the presence of IWP-2 also completely diminished its activity to induce invasion and secondary EMT phenotype in both E-A549 and E-Panc cells (Fig.4b,c). These results strongly indicate that WNT3 and WNT5B are likely to be key soluble factors produced by mesenchymal-transitioned cancer cells to instigate the metastatic potential of neighboring epithelial cancer cells by enhancing their invasiveness and inducing secondary EMT phenotype.
Figure 4.
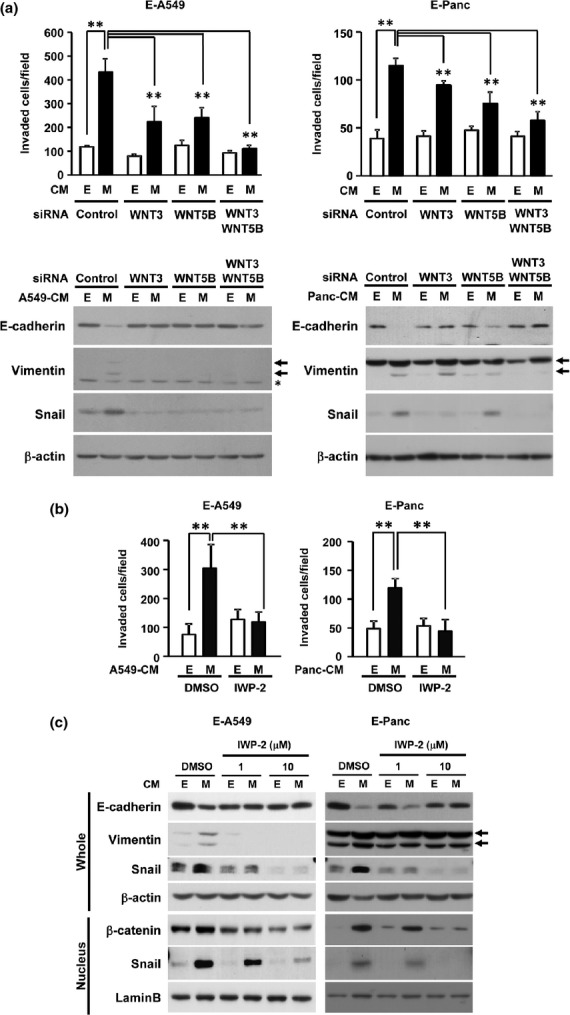
Critical requirement of secretory WNT3 and WNT5B from mesenchymal-transitioned cancer cells for inducing secondary epithelial-to-mesenchymal transition (EMT) phenotype of epithelical cancer cells. (a, b) E-A549 (left) and E-Panc (right) cells were treated with WNT-depleted CM derived from E-cells or M-cells either by siRNAs against WNT3/WNT5B (a) or by 10 μM WNT secretion inhibitor (IWP-2) (b) for 48 h. The cells were subjected to Matrigel invasion assay and western blotting. **P < 0.01 by one-way anova with the Bonferroni correction. (c) E-A549 or E-Panc cells were treated with WNT-depleted CM derived from E-cells or M-cells by the indicated dose of IWP-2 for 48 h and the expression of proteins were determined by western blotting.
Finally, we have tested whether the mesenchymal-transitioned cancer cells can instigate metastatic spread of neighboring epithelial cancer cells through providing secretory WNT3 and WNT5B ligands. Consistent with the induction of invasiveness and secondary EMT phenotype in vitro, the lung colonization of E-A549/Luc2 cells co-cultured with M-A549 cells was largely enhanced compared to that of E-A549/Luc2 cells co-cultured with E-A549 cells (Fig.5). Such increased lung colonization was fully abrogated when E-A549/Luc2 cells co-cultured with M-A549 cells in the presence of IWP-2. Collectively, these results strongly indicate that the secretory WNT3 and WNT5B derived from mesenchymal-transitioned cancer cells are able to enhance the metastatic potential of neighboring epithelial cancer cells in vivo.
Figure 5.
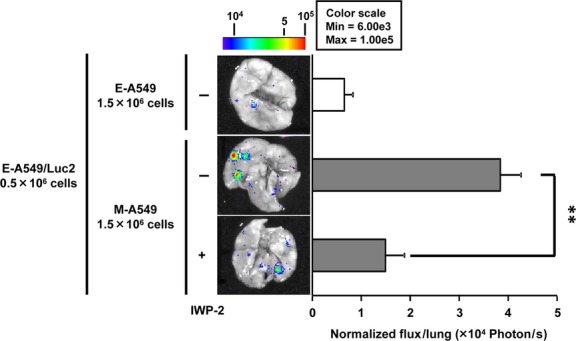
Secretory WNT-dependent metastasis instigation of epithelial cancer cells by mesenchymal-transitioned cancer cells. E-A549 cells were co-cultured with either control (E-A549) cells or mesenchymal-transitioned A549 (M-A549) cells in the presence or absence of IWP-2 for 48 h and i.v. inoculated into mice. Mice were killed 24 h after the tumor inoculation and lungs were subjected to bioluminescent imaging to determine total flux (photon/s) for lung metastasis quantification. The representative ex vivo images are shown. Data represented as the mean ± SEM (n = 3–5).
Discussion
Considering the significance of the in vivo metastatic ability of epithelial cancer cells upon co-culture with mesenchymal-transitioned cancer cells (Figs1 and 5), there are additional effects on epithelial cancer cells other than inducing invasive ability and secondary EMT phenotype through the secretion of WNT ligands by neighboring mesenchymal-transitioned cancer cells. First, it is reported that mesenchymal-transitioned cancer cells play a unique role in escorting epithelial cancer cells to metastatic organ in vivo.18 Second, the interaction with other host stromal cells, such as platelets or fibroblasts, might be involved in cancer metastasis by preventing cancer cells from cellular stresses, inducing EMT within the blood stream,28 or leading to collective cell invasion through gap junctions or integrins.29,30 Third, WNT ligands are known to not only enhance the invasiveness or secondary EMT of epithelial cancer cells but also affect the multiple steps in cancer metastasis and progression. Along with the paracrine WNT signaling as seen in this study, the autocrine WNT signaling of mesenchymal-transitioned cancer cells has been reported to contribute to the maintenance of mesenchymal phenotypes and stem cell-like states in breast cancer.31 It has also been reported that WNT signaling is involved in the expression of matrix metalloproteinases for digesting extracellular matrix during intra-vasation or extra-vasation of the metastasis process,32 niche formation,33 and enhancement of anchorage-independent sphere formation to increase the metastatic ability of pancreatic cancer cells.34 Collectively, this evidence might indicate that not only the cell-contact independent interaction but also the involvement of direct cell–cell interaction between epithelial cancer cells and mesenchymal-transitioned cancer cells play important roles in the regulation of the metastasis process.
Although WNT3 and WNT5B secreted from mesenchymal-transitioned cancer cells are indispensable in inducing invasiveness and secondary EMT in neighboring epithelial cancer cells (Fig.4), we have also identified other gene candidates of secretory proteins, which include other known-inducers of cancer cell invasiveness and EMT, such as CXCL12,35 LOX/LOXL236 and HB-EGF37. Those secretory proteins from mesenchymal-transitioned cancer cells might also contribute to induce invasiveness and secondary EMT in epithelial cancer cells by cooperating with WNT ligands; therefore, further studies are required in this context.
Transforming growth factor-β is one of the most potent inducers of EMT and metastasis; however, TGF-β pathway could be often genetically abrogated in relatively late-stage tumors because of the deletion or mutation of Smad4 (DPC4) or TGF-β receptors.16,17,38 In addition, TGF-β -induced EMT is reversible unless there is long-term exposure to TGF-β as reported previously,39,40. We also clarified that M-cell-CM-dependent EMT was also a reversible process, at least in vitro (data not shown). Thus, the heterogeneity of epithelial and mesenchymal-transitioned cancer cells might be maintained within the tumor microenvironment through such dynamic cellular transition between E-cell and M-cell states. Considering TGF-β receptor kinase inhibitor did not affect the induction of invasive ability and secondary EMT phenotype in this study (data not shown), the paracrine WNT stimulation can be an alternative inducer of EMT and metastasis to TGF-β in the cross-talk between mesenchymal-transitioned cancer cells and epithelial cancer cells. In this study, we focused on the WNT ligands secreted from mesenchymal-transitioned cancer cells; however, other stromal cells in the cancer microenvironment might also produce WNT ligands and, therefore, be involved in the cancer metastasis process. Although we did not observe the induction of WNT3 and WNT5B by TGF-β stimulation in mouse NIH3T3 fibroblast or primary human lung fibroblasts (data not shown), it has been reported that upregulation of WNT3A in CAF could result in the aggressive progression of prostate tumor.41 Besides secretion of WNT ligands, the hyperactivation of WNT signaling pathway has been observed in highly metastatic lung adenocarcinoma, colon cancer42,43 and pancreatic cancer.34 In the context of the clinical significance, WNT3 was reported to be associated with poor prognosis of non-small cell lung cancer,44 and to promote EMT in HER2-overexpressing breast cancer cells.45 Although we cannot exclude the possibility that WNT5B need to be coordinated with WNT3 to induce cellular invasion, we believe WNT5B could be solely responsible for impaired instigation considering that the non-canonical WNT pathway through WNT5B is reported to be involved in inducing tumor invasion.46,47 Furthermore, WNT5A, a paralog of WNT5B, and its receptor (FZD3) are known to be involved in the promotion of cell motility through the activation of paracrine non-canonical WNT signaling in skin cancer.48 Even though IWP-2 is a pan Wnt ligand secretion inhibitor, our presented data by knockdown both WNT3 or 5B with siRNA almost completely diminished the activity of M-cell CM to induce invasion of E-cells; therefore, these results strongly suggest that both WNT3 and WNT5B from M-cells are important for the induction of E-cell invasion. Given that E-cell CM in the presence of IWP-2 downregulated Snail or nuclear β-catenin expression in E-A549 cells (Fig.4c and Fig S5), we speculate that even E-cells may produce substantial levels of WNT ligand, by which Snail or β-catenin expression of E-cells is maintained in an autocrine manner. Collectively, WNT ligands derived from cancer stromal cells as well as mesenchymal-transitioned cancer cells and subsequent activation of WNT-signaling pathway may play a significant role in the malignant behavior of cancer cells, including metastatic spread to distant organs.
In conclusion, the intra-tumoral heterogeneity has been considered to be one of hallmarks in cancer malignancy and we have newly identified that secretory WNT ligands from mesenchymal-transitioned cancer cells instigate the invasion of neighboring epithelial cancer cells. This novel function of WNT signaling in the cancer microenvironment could be an attractive target not only for the new therapeutic opportunity but also for the new biomarker candidate in metastatic disease.
Acknowledgments
This work was supported in part by Grants-in-aid for Challenging Exploratory Research 24659348 (IS), by Grant-in-Aid 24700971 for Young Scientists (B) (SY) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and by Grant-in-Aid for JSPS Fellows (DC2) 2510159 from Japan Society for the Promotion of Science (SK). We thank members of the Saiki laboratory for discussions and suggestions.
Disclosure Statement
The authors have no conflict of interest.
Funding information
Grants-in-aid for Challenging Exploratory Research (24659348) and Grant-in-aid for Young Scientists (B) (24700971) from Ministry of Education, Culture, Sports, Science, and Technology, Japan. Grant-in-Aid for JSPS Fellows (DC2) (2510159) from Japan Society for the Promotion of Science, Japan.
Supporting Information
Additional supporting information may be found in the online version of this article.
Expression of epithelial-to-mesenchymal transition (EMT)-related proteins following transforming growth factor (TGF-β-induced EMT.
Effect of seeded cell number on invasion potential in E-cells.
Quantification of western blotting bands related to Fig. 2 by densitometry.
mRNA expression of WNT3 and WNT5B upon siRNA and transforming growth factor (TGF-β treatment.
Quantification of western blotting bands related to Fig. 4a and c by densitometry.
Common upregulated genes (top 5%) in both mesenchymal-transitioned A549 and Panc-1 cells by the stimulation of transforming growth factor (TGF-β for 7 and 48 h, respectively.
Enriched pathways in transforming growth factor (TGF- -treated cells.
Materials and Methods.
References
- 1.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 3.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–34. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 4.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 5.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–36. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ledford H. Cancer theory faces doubts. Nature. 2011;472:273. doi: 10.1038/472273a. [DOI] [PubMed] [Google Scholar]
- 7.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2012;331:1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 8.Brabletz T, Jung A, Reu S, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA. 2001;98:10356–61. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11:1287–96. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnomet A, Syne L, Brysse A, et al. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene. 2012;31:3741–53. doi: 10.1038/onc.2011.540. [DOI] [PubMed] [Google Scholar]
- 11.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franci C, Takkunen M, Dave N, et al. Expression of Snail protein in tumor–stroma interface. Oncogene. 2006;25:5134–44. doi: 10.1038/sj.onc.1209519. [DOI] [PubMed] [Google Scholar]
- 13.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells – An integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–9. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 14.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris J, Wang SC, Hebrok M. KRAS, Hedgehog Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683–95. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–49. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 17.Rasheed ZA, Yang J, Wang Q, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102:340–51. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuji T, Ibaragi S, Shima K, et al. Epithelial–mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008;68:10377–86. doi: 10.1158/0008-5472.CAN-08-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc Natl Acad Sci USA. 2006;103:13474–9. doi: 10.1073/pnas.0606053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunewald TG, Herbst SM, Heinze J, Burdach S. Understanding tumor heterogeneity as functional compartments–superorganisms revisited. J Transl Med. 2011;9:79. doi: 10.1186/1479-5876-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calbo J, van Montfort E, Proost N, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 2011;19:244–56. doi: 10.1016/j.ccr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 22.Saiki I, Murata J, Watanabe K, Fujii H, Abe F, Azuma I. Inhibition of tumor cell invasion by ubenimex (bestatin) in vitro. Jpn J Cancer Res. 1989;80:873–8. doi: 10.1111/j.1349-7006.1989.tb01729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakurai H, Suzuki S, Kawasaki N, et al. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–23. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- 24.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–1. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 25.Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J Biol Chem. 2009;284:245–53. doi: 10.1074/jbc.M804777200. [DOI] [PubMed] [Google Scholar]
- 26.Kim JH, Jang YS, Eom KS, et al. Transforming growth factor beta1 induces epithelial-to-mesenchymal transition of A549 cells. J Korean Med Sci. 2007;22:898–904. doi: 10.3346/jkms.2007.22.5.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benhaj K, Akcali KC, Ozturk M. Redundant expression of canonical Wnt ligands in human breast cancer cell lines. Oncol Rep. 2006;15:701–7. [PubMed] [Google Scholar]
- 28.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial–mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–90. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14:777–83. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 30.Gaggioli C, Hooper S, Hidalgo-Carcedo C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 31.Scheel C, Eaton EN, Li SH, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–40. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 33.Malanchi I, Santamaria-Martinez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85–9. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 34.Yu M, Ting DT, Stott SL, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487:510–3. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Ma Q, Xu Q, et al. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012;322:169–76. doi: 10.1016/j.canlet.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peinado H, Portillo F, Cano A. Switching on-off Snail: LOXL2 versus GSK3beta. Cell Cycle. 2005;4:1749–52. doi: 10.4161/cc.4.12.2224. [DOI] [PubMed] [Google Scholar]
- 37.Yagi H, Yotsumoto F, Miyamoto S. Heparin-binding epidermal growth factor-like growth factor promotes transcoelomic metastasis in ovarian cancer through epithelial-mesenchymal transition. Mol Cancer Ther. 2008;7:3441–51. doi: 10.1158/1535-7163.MCT-08-0417. [DOI] [PubMed] [Google Scholar]
- 38.Yanagisawa K, Uchida K, Nagatake M, et al. Heterogeneities in the biological and biochemical functions of Smad2 and Smad4 mutants naturally occurring in human lung cancers. Oncogene. 2000;19:2305–11. doi: 10.1038/sj.onc.1203591. [DOI] [PubMed] [Google Scholar]
- 39.Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–98. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong C, Wu Y, Yao J, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122:1469–86. doi: 10.1172/JCI57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Placencio V, Iturregui JM, et al. Prostate tumor progression is mediated by a paracrine TGF-beta/Wnt3a signaling axis. Oncogene. 2008;27:7118–30. doi: 10.1038/onc.2008.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen DX, Chiang AC, Zhang XH, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62. doi: 10.1016/j.cell.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh A, Sweeney MF, Yu M, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148:639–50. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakashima N, Liu D, Huang CL, Ueno M, Zhang X, Yokomise H. Wnt3 gene expression promotes tumor progression in non-small cell lung cancer. Lung Cancer. 2012;76:228–34. doi: 10.1016/j.lungcan.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Wu Y, Ginther C, Kim J, et al. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res. 2012;10:1597–606. doi: 10.1158/1541-7786.MCR-12-0155-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deraz EM, Kudo Y, Yoshida M, et al. MMP-10/stromelysin-2 promotes invasion of head and neck cancer. PLoS ONE. 2011;6:e25438. doi: 10.1371/journal.pone.0025438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klemm F, Bleckmann A, Siam L, et al. beta-catenin-independent WNT signaling in basal-like breast cancer and brain metastasis. Carcinogenesis. 2011;32:434–42. doi: 10.1093/carcin/bgq269. [DOI] [PubMed] [Google Scholar]
- 48.Pourreyron C, Reilly L, Proby C, et al. Wnt5a is strongly expressed at the leading edge in non-melanoma skin cancer, forming active gradients, while canonical Wnt signalling is repressed. PLoS ONE. 2012;7:e31827. doi: 10.1371/journal.pone.0031827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of epithelial-to-mesenchymal transition (EMT)-related proteins following transforming growth factor (TGF-β-induced EMT.
Effect of seeded cell number on invasion potential in E-cells.
Quantification of western blotting bands related to Fig. 2 by densitometry.
mRNA expression of WNT3 and WNT5B upon siRNA and transforming growth factor (TGF-β treatment.
Quantification of western blotting bands related to Fig. 4a and c by densitometry.
Common upregulated genes (top 5%) in both mesenchymal-transitioned A549 and Panc-1 cells by the stimulation of transforming growth factor (TGF-β for 7 and 48 h, respectively.
Enriched pathways in transforming growth factor (TGF- -treated cells.
Materials and Methods.