

Abstract
Adult T-cell leukemia–lymphoma (ATL), an aggressive neoplasm etiologically associated with HTLV-1, is a chemoresistant malignancy. Heat shock protein 90 (HSP90) is involved in folding and functions as a chaperone for multiple client proteins, many of which are important in tumorigenesis. In this study, we examined NVP-AUY922 (AUY922), a second generation isoxazole-based non-geldanamycin HSP90 inhibitor, and confirmed its effects on survival of ATL-related cell lines. Analysis using FACS revealed that AUY922 induced cell-cycle arrest and apoptosis; it also inhibited the growth of primary ATL cells, but not of normal PBMCs. AUY922 caused strong upregulation of HSP70, a surrogate marker of HSP90 inhibition, and a dose-dependent decrease in HSP90 client proteins associated with cell survival, proliferation, and cell cycle in the G1 phase, including phospho-Akt, Akt, IKKα, IKKβ, IKKγ, Cdk4, Cdk6, and survivin. Interestingly, AUY922 induced downregulation of the proviral integration site for Moloney murine leukemia virus (PIM) in ATL cells. The PIM family (PIM-1, -2, -3) is made up of oncogenes that encode a serine/threonine protein kinase family. As PIM kinases have multiple functions involved in cell proliferation, survival, differentiation, apoptosis, and tumorigenesis, their downregulation could play an important role in AUY922-induced death of ATL cells. In fact, SGI-1776, a pan-PIM kinase inhibitor, successfully inhibited the growth of primary ATL cells as well as ATL-related cell lines. Our findings suggest that AUY922 is an effective therapeutic agent for ATL, and PIM kinases may be a novel therapeutic target.
Keywords: Adult T-cell leukemia–lymphoma, HSP90 inhibitors, NF-κB, NVP-AUY922, PIM kinases
Heat shock protein 90 is involved in folding and functions as a chaperone for multiple client proteins, many of which are important in tumorigenesis. In contrast to normal cells, tumor cells contain an abundance of catalytically active HSP90, which is found in multichaperone complexes. Therefore, HSP90 has emerged as a target of interest in cancer therapy.(1) Inhibition of HSP90 leads to misfolding of client proteins and degradation through the ubiquitin proteasome pathway. Heat shock protein 90 inhibitors target tumor cells on mutated or amplified oncoproteins, such as transmembrane tyrosine kinases (human epidermal growth factor receptor 2, epidermal growth factor receptor, c-Met, insulin-like growth factor 1 receptor), metastable signaling proteins (Akt, Raf-1, IKK), mutated signaling proteins (p53, Kit, Flt-3, v-Src), chimeric signaling proteins (nucleophosmin/anaplastic lymphoma kinase, BCR-ABL), steroid receptors (androgen, estrogen, progesterone receptors), and cell cycle regulators (CDK4, CDK6). The HSP90 inhibitor 17-AAG, derived from geldanamycin, has shown potent antitumor activity against ATL.(2,3) However, geldanamycin derivatives have several limitations, including poor solubility, formulation difficulties, and severe hepatotoxicity in clinical settings,(4–6) which have prompted development of next generation synthetic HSP90 inhibitors including NVP-AUY922 (AUY922), a second generation isoxazole-based non-geldanamycin HSP90 inhibitor that inhibits the ATPase activity of HSP90.(7,8) AUY922 has shown nanomolar efficacy against a wide range of human cancer cells in vitro and also inhibits progression of a variety of tumors in vivo.(7–11) Furthermore, in a phase I clinical trial of AUY922 in patients with advanced solid tumors, the agent showed acceptable tolerability.(12)
Adult T-cell leukemia–lymphoma is a chemoresistant malignancy with a CD4-positive T-lymphocyte origin etiologically associated with HTLV-1.(13) In ATL, activation of NF-κB, AP-1, and PI3K/Akt results in upregulation of expression of a large number of cellular genes involved in cell proliferation and survival.(14–16) Adult T-cell leukemia–lymphoma is generally classified into four clinical subtypes: acute, chronic, smoldering, and lymphoma. Although several approaches have been reported, combination chemotherapy is still the treatment of choice for newly diagnosed aggressive ATL. Patients with aggressive ATL have a median survival time of 13 months, indicating limitations in present treatment strategies.(17) However, agents that interrupt a variety of signal transduction pathways such as HSP90 inhibitors are thought to be potential treatment options for the disease. In this study, we examined the effects of AUY922 on ATL cells in vitro and explored a novel therapeutic target by investigating its molecular mechanisms.
Materials and Methods
Cells and ATL-related cell lines
The ATL-derived cell lines KK1, KOB, SO4, ST1, and LM-Y1, were obtained from ATL patients and established in our laboratory.(18–21) KK1, KOB, SO4, and LM-Y1 were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated FBS and 0.5 U/mL interleukin-2 (kindly provided by Takeda Pharmaceutical Company, Ltd., Osaka, Japan). ST1 and HTLV-1-infected T-cell lines, MT2(22) and HuT102(23), were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated FBS. The KOB, LM-Y1, ST1, MT2, and HuT102 cell lines possess wild-type p53, whereas KK1 and SO4 have mutant-type p53.(24) Primary leukemia cells from patients with ATL were also used. The diagnosis of ATL was based on clinical features, hematological findings, and presence of anti-HTLV-1 antibodies in serum. Monoclonal HTLV-1 provirus integration in the DNA of leukemic cells was confirmed in patients using Southern blot hybridization (data not shown). Peripheral blood mononuclear cells from patients with ATL and a normal healthy donor were isolated by Ficoll–Paque density gradient centrifugation, and washed with PBS. For enrichment of ATL cells, CD4 T cells were negatively enriched using Miltenyi CD4 T-Cell Isolation Kit II (Miltenyi Biotec, Auburn, CA, USA). Each patient sample contained more than 90% leukemia cells at the time of analysis. After receiving approval from the Ethics Committee at Nagasaki University Hospital (Nagasaki, Japan), all patient samples were obtained with informed consent.
Chemicals and cell proliferation assay
AUY922 was kindly provided by Novartis Institutes for Biomedical Research (Basel, Switzerland). 17-AAG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and SGI-1776 (Santa Cruz Biotechnology) were obtained, and dissolved in DMSO. The effect of AUY922 on cell proliferation was examined using the cell viability agent provided in a CellTiter 96 AQueos Cell Proliferation Assay kit (Promega, Madison, WI, USA). Briefly, the cell lines (2–5 × 105/mL) and PBMCs (1 × 106/mL) were separately incubated in 96-well plates in the presence or absence of various concentrations of AUY922. After 72 h, the reagent was added and incubation was continued for 2–4 h, then absorbance at 492 nm was measured using an automated microplate reader. All experiments were carried out in triplicate. Error bars represent the standard error in each experiment. Non-parametric statistical analysis (Mann–Whitney U-test) was carried out using GraphPad Prism version 6.00 software (GraphPad Software, San Diego, CA, USA). P-values <0.05 were regarded as significant.
Flow cytometric analysis (apoptosis assays and cell cycle analysis)
To evaluate apoptotic changes, we used annexin V and a PI Kit (Bender Medsystems, Vienna, Austria). Cell cycle was analyzed using a Cycletest Plus DNA reagent kit (BD Biosciences, San Jose, CA, USA). In brief, 106 cells were washed with a buffer solution containing sodium citrate, sucrose, and dimethyl sulfoxide suspended in a solution containing RNase A, and stained with 125 μg/mL PI for 10 min. All experiments were carried out using a FACSCanto II flow cytometer and FACSDiva software (BD Biosciences).
Western blot analysis and antibodies
Cells were harvested after treatment and washed, then homogenized at 4°C in lysis buffer (0.1% SDS, 1% Igepal CA-630, 0.5% sodium deoxycholate) and a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Cell lysates (20–50 μg) were resolved by electrophoresis on polyacrylamide gels and transferred to PVDF membranes. After blocking the membranes in 5% non-fat dry milk or 5% FBS and 0.1% Tween-20 in Tris-buffered saline for 1 h at room temperature, the blots were hybridized overnight at 4°C with primary antibodies. After hybridization with secondary antibodies conjugated with HRP, immunocomplexes were visualized using an enhanced chemiluminescence kit (GE Healthcare, Chalfont St. Giles, UK). Analyses were carried out with antibodies to HSP90, PIM-1 (Santa Cruz Biotechnology), HSP70, Cdk4, Cdk6, Akt, p-Akt, IκBα, IKKα, IKKβ, IKKγ, Bcl-2, survivin, PIM-2, PIM-3 (Cell Signaling Technology, Beverly, MA, USA), and β-actin (Sigma-Aldrich).
DNA microarray analysis
Gene expression profiling of ATL-related cell lines was examined. KK1, SO4, LM-Y1, and HuT102 cells with or without exposure to 100 nM AUY922 for 24 h were harvested. Total RNA was extracted using ISOGEN (Nippon Gene, Toyama, Japan) and purified with an RNeasy Mini Kit (Qiagen, Germantown, MD, USA), then total purified RNA was amplified with a one-color Low Input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, USA). Cyanine 3–labeled fragmented cRNA was hybridized to a SurePrint G3 Human GE 8 × 60 K Microarray Kit (Agilent Technologies) covering 27 958 Entrez Gene RNAs. The microarrays were washed and scanned with a High-Resolution Microarray Scanner (Agilent Technologies). Data were processed using a quantile normalization method. Significant functions were calculated by Ingenuity Pathways Analysis (Ingenuity Systems, Redwood, CA, USA) with DAVID software (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA; available from http://david.abcc.ncifcrf.gov/) from a list of genes showed a 1.5-fold increase or decrease following treatment with AUY922.
Results
AUY922 inhibits growth of ATL-related cell lines and primary ATL cells
First, we analyzed the effects of AUY922 on proliferation of ATL-related cell lines. Incubation with AUY922 at various concentrations (0–100 nM) for 72 h inhibited cellular proliferation in a dose-dependent manner in a range from 0 to 25 nM, while a plateau was reached at concentrations >25 nM, as assessed by an MTS assay (Fig. 1a). The concentrations of AUY922 required to inhibit cellular proliferation of ATL-related cell lines by 50% (IC50) varied from 12.5 to 25.0 nM. Importantly, AUY922 was effective regardless of the presence of wild-type or mutant p53. We also assessed AUY922-induced cellular inhibition of PBMCs obtained from both normal subjects and patients with ATL. Importantly, primary ATL cells were more susceptible to AUY922 than normal PBMCs, and the difference was statistically significant at 25 nM (Fig. 1b). Also, when compared directly with 17-AAG, AUY922 was between 20- and 50-fold more active at inhibiting growth of ATL-related cell lines (Fig. 2).
Fig. 1.
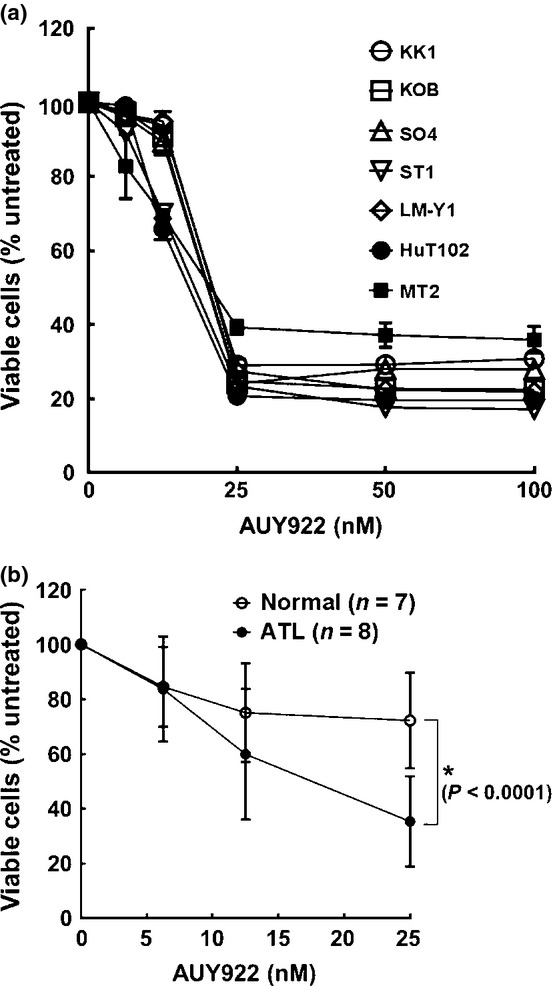
Growth inhibition effects of heat shock protein 90 inhibitor AUY922. Inhibitory effects of AUY922 on cell survival of adult T-cell leukemia–lymphoma-related cell lines (a), and primary adult T-cell leukemia–lymphoma cells (n = 8) and normal PBMCs (n = 7) (b). Cells were incubated in the presence of various concentrations of AUY922 for 72 h and in vitro survival was determined using an MTS assay. A relative viability of 100% was designated as the total number of cells that survived after 72 h in the absence of AUY922. The relative viability of cultured cells was determined from triplicate cultures and is presented as the mean ± SD (bars). *P < 0.0001.
Fig. 2.

Growth inhibition effects of heat shock protein 90 inhibitor 17-AAG. Inhibitory effects of 17-AAG on cell survival of adult T-cell leukemia–lymphoma-related cell lines. Cells were incubated in the presence of various concentrations of 17-AAG for 72 h and in vitro survival was determined using MTS assay. The relative viability of cultured cells is presented as the mean determined from triplicate cultures. A relative viability of 100% was determined based on the total number of cells that survived after 72 h in the absence of 17-AAG. The relative viability of cultured cells was determined from triplicate cultures and is presented as the mean ± SD (bars).
AUY922 induces sub-G1/G1 phase arrest of ATL-related cell lines
Next, we examined the effect of AUY922 on cell cycle progression in the tested cell lines. Cells were incubated with the control, AUY922 at 12.5 nM, or AUY922 at 25.0 nM for 48 h, then cell cycle distribution was analyzed using flow cytometry. Faint increases of G1 and G2–M cell populations were seen in KK1 and KOB, and SO4 cells, at 12.5 nM AUY922, respectively. In all of the tested cell lines, the sub-G1 cell population increased in a dose-dependent manner, indicating apoptotic cell death (Fig. 3).
Fig. 3.

Effects of heat shock protein 90 inhibitor AUY922 on cell cycle. Adult T-cell leukemia–lymphoma-related cell lines were incubated in the absence (−) or presence of AUY922 (12.5 or 25.0 nM) for 48 h and stained with propidium iodide, then DNA content was assayed using flow cytometry. The percentage of cells in various phases of the cell cycle was determined.
AUY922 induces apoptosis of ATL-related cell lines
To examine whether induction of apoptosis accounted for the inhibition of proliferation observed in ATL-related cell lines, cells were treated with the control, 12.5 nM AUY922, or 25.0 nM AUY922 for 48 h, or 100 nM AUY922 for 48–72 h, then examined using the annexin V–PI method. Annexin V binds to cells that express phosphatidylserine on the outer layer of the cell membrane, a characteristic finding in those entering apoptosis. AUY922 increased the proportion of cells positive for annexin V in all cell lines in a dose-dependent manner (Fig. 4a). Moreover, 100 nM AUY922 increased the proportion of cells positive for annexin V in all cell lines in a time-dependent manner (Fig. 4b). We carried out additional apoptosis assays using the non-HTLV-1 related T-cell lines Jurkat and Molt4. Those results were similar to the results obtained with ATL-related cell lines (Fig. S1).
Fig. 4.
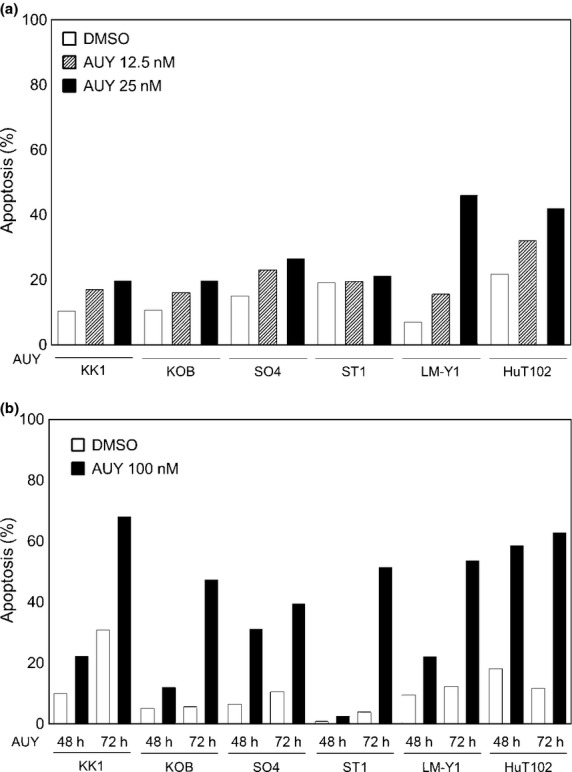
Effects of heat shock protein 90 inhibitor AUY922 on apoptosis. Adult T-cell leukemia–lymphoma-related cell lines were treated with or without AUY922 (12.5 or 25.0 nM [a], or 100 nM [b]) for 48 h, then harvested, stained with annexin V–propidium iodide, and analyzed using flow cytometry. Data shown represent the percentages of apoptotic cells among untreated and AUY922-treated cells.
AUY922 affects induction of HSP70 and depletion of oncogenic proteins through inhibition of HSP90 activity
To verify the molecular mechanisms of the effects of AUY922 on survival and apoptosis of ATL-related cell lines, we examined the expressions of HSP90, HSP70, and several intracellular regulators of cell proliferation, cell cycle, and apoptosis, including p-Akt, Akt, IκBα, IKKα, IKKβ, IKKγ, Cdk4, Cdk6, Bcl-2, and survivin. AUY922 treatment led to induction of HSP70, a surrogate marker for inhibition of HSP90 function, but did not influence the protein level of HSP90 itself. HSP90 and its co-chaperones modulate tumor cell apoptosis, and much of their activity seems to be mediated through effects on the PI3K/Akt pathway and NF-κB function. Suppression of HSP90 function by AUY922 decreases the level of Akt, resulting in a reduction of activated p-Akt. The IKK complex, composed of IKKα, IKKβ, and IKKγ, is a positive regulator of NF-κB. In general, a decrease in the IKK complex inhibits phosphorylation of IκBα, resulting in its increased level. In the present study, AUY922 treatment decreased expression of the IKK complex in all tested cell lines. Among the apoptosis-related proteins examined, we found a decrease in survivin. Overall, we found similar changes in HSP90 client proteins regardless of the presence of wild-type or mutant p53 (Fig. 5).
Fig. 5.
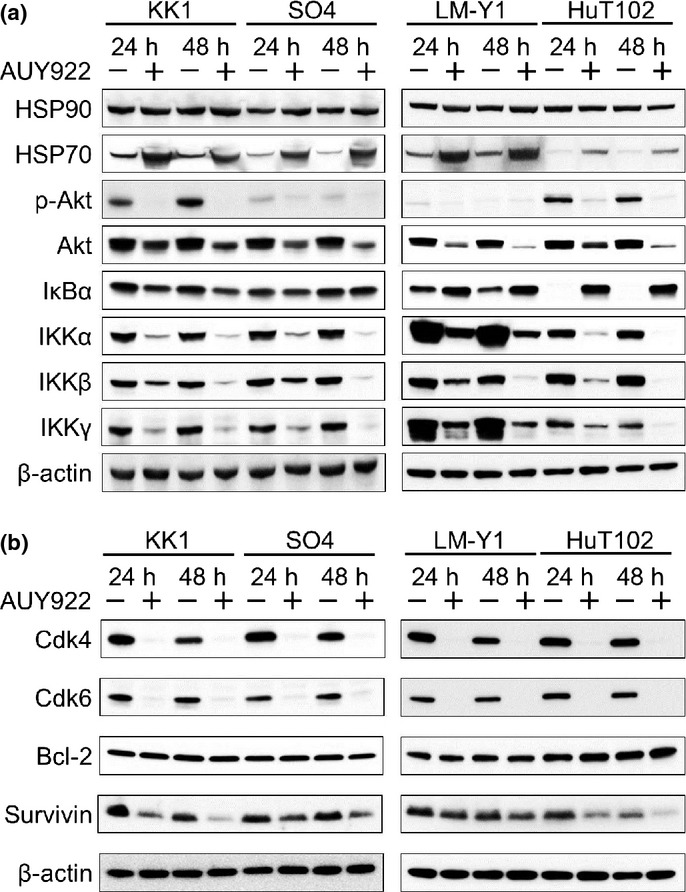
Effects of heat shock protein (HSP) 90 inhibitor AUY922 on HSP90, HSP70, and HSP90 client proteins. Western blot analysis revealed that AUY922 treatment led to strong upregulation of HSP70, a surrogate marker of HSP90 inhibition. In addition, dose-dependent decreases in HSP90 client proteins associated with cell survival, proliferation, and cell cycle, including phospho-Akt (p-Akt), Akt, IκB kinase (IKK)α, IKKβ, IKKγ (a), and Cdk4, Cdk6, and survivin (b), were seen.
Downregulation of PIM kinases in ATL-related cell lines treated by AUY922
To determine which molecules play important roles in AUY922-induced ATL-cell death, gene expression profiling was carried out using DNA microarray analysis. Among genes with changes in average expression of at least 1.5-fold (log2 ratio) in either direction in the four tested cell lines, we selected those with known functions related to apoptosis, cell cycle, and cell proliferation. Our results showed upregulation of HSP70 in those cells, which was consistent with the results of our WB analysis, and we also noted upregulation of HSP90, although the protein level of HSP90 was not changed. Interestingly, decreases in two of the PIM kinases, PIM-1 and -3, were commonly found (Table 1). PIM has multiple cellular functions related to cell survival, proliferation, differentiation, apoptosis, and tumorigenesis, and its expression is also correlated with poor prognosis in most hematopoietic malignancies, although its role in ATL remains unclear. Therefore, to investigate this, we examined the protein expression levels of PIM kinases using WB in ATL-related cell lines treated by AUY922. Although the protein levels of PIM kinases varied in each of the cell lines when untreated, the protein expression levels of PIM-1, -2, and -3 were universally decreased in all treated cell lines (Fig. 6).
Table 1.
Microarray analysis of adult T-cell leukemia–lymphoma-related cell lines treated with heat shock protein 90 (HSP90) inhibitor AUY922
| Fold change (log2 ratio) | ||||||
|---|---|---|---|---|---|---|
| Gene symbol | Gene | KK1 | SO4 | LM-Y1 | HuT102 | Average |
| I. Genes upregulated in AUY922-treated ATL-related cell lines | ||||||
| LGR4 | Leucine-rich repeat containing G protein-coupled receptor 4 | 4.72 | 5.51 | 4.81 | 2.93 | 4.5 |
| CLU | Clusterin | 3.03 | 4.75 | 2.67 | 3.84 | 3.6 |
| HSPA1B | Heat shock 70 kDa protein 1B | 2.58 | 4.05 | 2.31 | 4.14 | 3.3 |
| RGS2 | Regulator of G-protein signaling 2 | 2.41 | 1.97 | 2.78 | 5.86 | 3.3 |
| PDZK1 | PDZ domain containing 1 | 3.34 | 4.70 | 2.06 | 1.22 | 2.8 |
| MXD4 | MAX dimerization protein 4 | 2.91 | 3.20 | 2.06 | 2.91 | 2.8 |
| HSP90AA1 | Heat shock protein 90 kDa alpha, class A member 1 | 2.90 | 3.04 | 1.86 | 2.59 | 2.6 |
| BAG3 | BCL2-associated athanogene 3 | 1.04 | 1.57 | 2.75 | 3.29 | 2.2 |
| NQO1 | NAD(P)H dehydrogenase, quinone 1 | 2.10 | 2.42 | 1.15 | 2.80 | 2.1 |
| CREBBP | CREB binding protein | 2.85 | 2.09 | 1.20 | 1.87 | 2.0 |
| DEDD2 | Death effector domain containing 2 | 1.67 | 1.81 | 1.65 | 2.78 | 2.0 |
| BST2 | Bone marrow stromal cell antigen 2 | 2.09 | 2.28 | 1.64 | 1.76 | 1.9 |
| SPHK2 | Sphingosine kinase 2 | 2.46 | 2.63 | 1.44 | 1.02 | 1.9 |
| HSPD1 | Heat shock 60 kDa protein 1 | 1.98 | 2.08 | 1.30 | 1.56 | 1.7 |
| HDAC4 | Histone deacetylase 4 | 2.00 | 2.13 | 1.19 | 1.48 | 1.7 |
| CEBPA | CCAAT/enhancer binding protein, alpha | 2.38 | 1.74 | 1.47 | 1.09 | 1.7 |
| SAP30BP | SAP30 binding protein | 2.09 | 2.08 | 1.00 | 1.45 | 1.7 |
| SQSTM1 | Sequestosome 1 | 1.59 | 1.21 | 1.56 | 2.10 | 1.6 |
| B9D2 | B9 protein domain 2 | 2.16 | 1.87 | 1.00 | 1.37 | 1.6 |
| TMEM127 | Transmembrane protein 127 | 1.78 | 1.76 | 1.24 | 1.56 | 1.6 |
| CLN3 | Ceroid-lipofuscinosis, neuronal 3 | 1.66 | 1.89 | 1.37 | 1.35 | 1.6 |
| II. Genes downregulated in AUY922-treated ATL-related cell lines | ||||||
| CCL3L3 | Chemokine (C-C motif) ligand 3-like 3 | −4.16 | −4.35 | −7.43 | −4.41 | −5.1 |
| OSM | Oncostatin M | −2.72 | −3.16 | −4.38 | −3.21 | −3.4 |
| PIM1 | Pim-1 oncogene | −3.72 | −4.15 | −1.95 | −1.53 | −2.8 |
| CYP1A1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 | −2.35 | −5.03 | −1.02 | −2.17 | −2.6 |
| IL13 | Interleukin 13 | −2.02 | −3.06 | −2.71 | −2.72 | −2.6 |
| PLAUR | Plasminogen activator, urokinase receptor | −2.73 | −2.06 | −2.18 | −2.95 | −2.5 |
| VEGFA | Vascular endothelial growth factor A | −2.29 | −2.78 | −2.63 | −1.60 | −2.3 |
| CAMK1D | Calcium/calmodulin-dependent protein kinase ID | −2.32 | −2.70 | −2.69 | −1.34 | −2.3 |
| ADAMTSL4 | ADAMTS-like 4 | −2.73 | −2.75 | −2.17 | −1.14 | −2.2 |
| HBEGF | Heparin-binding EGF-like growth factor | −2.00 | −2.95 | −1.98 | −1.84 | −2.2 |
| DMC1 | DMC1 dosage suppressor of mck1 homolog, meiosis-specific homologous recombination (yeast) | −2.09 | −1.22 | −1.81 | −3.54 | −2.2 |
| PTPN6 | Protein tyrosine phosphatase, non-receptor type 6 | −2.65 | −2.89 | −1.06 | −1.96 | −2.1 |
| CEBPB | CCAAT/enhancer binding protein (C/EBP), beta | −2.48 | −3.16 | −1.30 | −1.52 | −2.1 |
| LIF | Homo sapiens leukemia inhibitory factor | −1.39 | −1.22 | −3.94 | −1.75 | −2.1 |
| KLF11 | Kruppel-like factor 11 | −2.12 | −2.21 | −2.23 | −1.60 | −2.0 |
| TERT | Telomerase reverse transcriptase | −1.29 | −2.04 | −2.72 | −2.09 | −2.0 |
| TNFRSF12A | Tumor necrosis factor receptor superfamily, member 12A | −2.38 | −2.41 | −2.30 | −1.01 | −2.0 |
| TRIB3 | Tribbles homolog 3 (Drosophila) | −2.41 | −2.94 | −1.40 | −1.19 | −2.0 |
| BIRC3 | Baculoviral IAP repeat containing 3 | −1.97 | −2.01 | −1.37 | −2.54 | −2.0 |
| BNIP3L | BCL2/adenovirus E1B 19 kDa interacting protein 3-like | −1.82 | −2.65 | −1.20 | −2.07 | −1.9 |
| CAMK2B | Calcium/calmodulin-dependent protein kinase II beta | −1.36 | −1.25 | −3.19 | −1.80 | −1.9 |
| TNFAIP3 | Tumor necrosis factor, α-induced protein 3 | −1.66 | −2.19 | −1.33 | −2.15 | −1.8 |
| TNFSF10 | Tumor necrosis factor (ligand) superfamily, member 10 | −1.65 | −1.53 | −1.73 | −2.34 | −1.8 |
| CDKN2D | Cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) | −2.29 | −1.50 | −1.62 | −1.80 | −1.8 |
| ANG | Angiogenin, ribonuclease, RNase A family, 5 | −2.48 | −2.05 | −1.33 | −1.09 | −1.7 |
| PIM3 | Pim-3 oncogene | −1.98 | −1.71 | −1.66 | −1.58 | −1.7 |
| DTL | Denticleless homolog (Drosophila) | −1.82 | −1.25 | −1.72 | −2.11 | −1.7 |
| IL2RA | Interleukin 2 receptor, alpha | −1.67 | −2.10 | −1.42 | −1.62 | −1.7 |
| MXI1 | MAX interactor 1 | −1.14 | −1.42 | −1.54 | −2.71 | −1.7 |
| NEK6 | NIMA (never in mitosis gene a)-related kinase 6 | −1.44 | −2.19 | −1.39 | −1.56 | −1.6 |
| E2F7 | E2F transcription factor 7 | −2.22 | −1.38 | −1.61 | −1.33 | −1.6 |
| PRIM1 | Primase, DNA, polypeptide 1 | −1.90 | −1.18 | −1.37 | −1.90 | −1.6 |
| SEPT8 | Septin 8 | −1.43 | −1.02 | −1.77 | −2.06 | −1.6 |
| KLF10 | Kruppel-like factor 10 | −1.92 | −1.23 | −1.76 | −1.20 | −1.5 |
To determine which molecules play important roles in AUY922-induced ATL-cell death, gene expression profiling was carried out using DNA microarray analysis. Among genes with changes in average expression of at least 1.5-fold (log2 ratio) in either direction in the four tested cell lines, we selected those with known functions related to apoptosis, cell cycle, and cell proliferation. The results showed upregulation of HSP70 in those, which was consistent with the results of our Western blot analysis. We also noted upregulation of HSP90, although the protein level of HSP90 was not changed. Interestingly, decreases in two of the Moloney murine leukemia virus (PIM) kinases, PIM-1 and PIM-3, were commonly found.
Fig. 6.
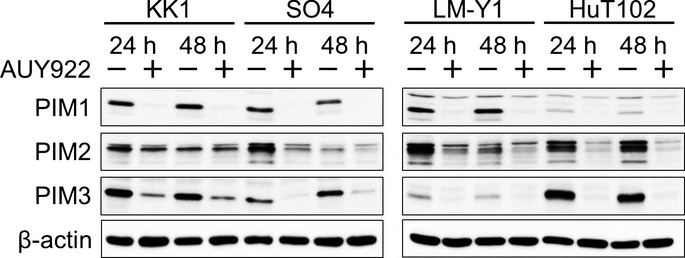
Effects of heat shock protein 90 inhibitor AUY922 on Moloney murine leukemia virus (PIM) kinases in adult T-cell leukemia–lymphoma. Western blot analysis revealed that AUY922 induced downregulation of PIM-1, -2, and -3 in adult T-cell leukemia–lymphoma-related cell lines.
SGI-1776 inhibits cell proliferation by blocking PIM kinases
To confirm the importance of PIM kinases in ATL cells, we evaluated the inhibitory effect of SGI-1776 on those, as well as proliferation of ATL-related cell lines and primary ATL cells. When ATL-related cell lines were cultured with various concentrations (0–10 μM) of SGI-1776 for 72 h, cellular proliferation was inhibited in both dose- and cell-dependent manners (Fig. 7a). In primary ATL cells, SGI-1776 at 10 μM inhibited cellular proliferation (Fig. 7b). Together, these results suggest that PIM kinases may be a novel therapeutic target for treatment of ATL.
Fig. 7.
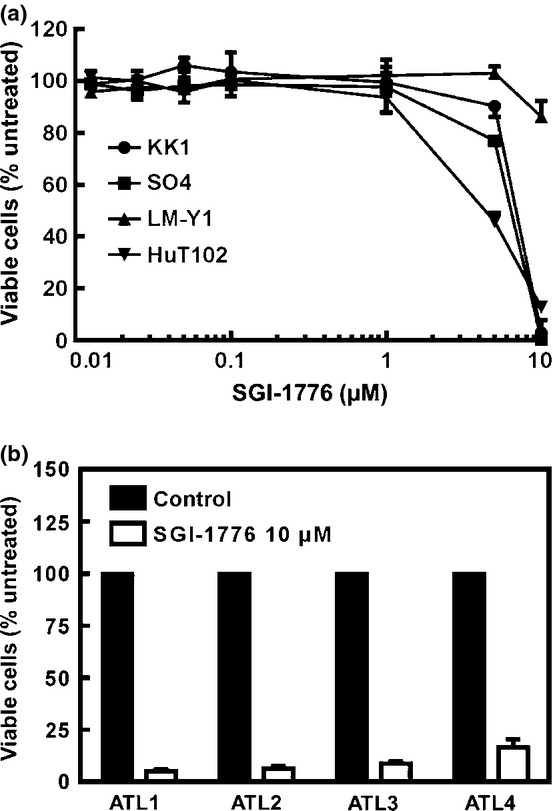
Growth inhibitory effects of SGI-1776 in adult T-cell leukemia–lymphoma. SGI-1776, a pan-PIM kinase inhibitor, inhibited cellular survival suppression in adult T-cell leukemia–lymphoma-related cell lines in both dose- and cell-dependent manners (a). Furthermore, SGI-1776 inhibited cellular survival in primary adult T-cell leukemia–lymphoma cells (b).
Discussion
In cancer cells, HSP90 client proteins play a major role in multiple oncogenic processes, such as cell proliferation and anti-apoptosis. HSP90 inhibitors are promising therapeutic agents for variable cancer, and phase I/II studies of AUY922 with advanced solid tumors and hematological malignancies are underway.(25)
We observed that AUY922 has very high cytotoxicity toward ATL-related cell lines and primary ATL cells. We also found that the inhibitory effect of AUY922 was superior to that of 17-AAG and 17-DMAG,(2,3) and our results confirmed previous reports noting that AUY922 showed potent cell inhibition in a low nanomolar range.(7–9) Moreover, we also showed that ATL-related cell lines and primary ATL cells were more susceptible to inhibition of proliferation by treatment with AUY922 than normal PBMCs. The difference between normal and cancer cells in regard to ATP-binding affinity with HSP90 likely contributed to this selectivity of effect.(26)
We found that the inhibitory effect of AUY922 on ATL cells was due to the induction of cell cycle arrest and apoptosis. Our results showed that AUY922 induced G1 arrest due to decreased protein levels of CDK4 and CDK6, which have been identified as HSP90 client proteins that are important for cell cycle G1 phase progression.(27) Survivin has also been identified as an HSP90 client protein(27) and reported to be overexpressed in ATL cells.(28) Our findings showed that AUY922 induced apoptosis associated with reduction of survivin in ATL-related cell lines. In addition, treatment with AUY922 decreased the IKK complex proteins (IKKα, IKKβ, and IKKγ). HSP90 is a regulator of NF-κB signaling through IKK activation and a reduction in the IKK complex inhibits IκBα phosphorylation followed by a reduction in NF-κB activity.(29) Among apoptosis-related proteins, we found a decrease in survivin and no change in Bcl-2, known as an NF-κB target, following treatment with AUY922. These findings suggest that typical Bcl-2 family members are not involved in AUY922-induced apoptosis. Furthermore, NF-κB activity may contribute to induction of cell cycle arrest and apoptosis of ATL-related cell lines.
AUY922 also induced Akt degradation, which resulted in a reduction of p-Akt. It has been reported that PI3K/Akt plays a role in activation of pro-survival pathways in HTLV-1-infected T-cell lines and primary ATL cells.(16,30–32) In those studies, Akt was shown to be a molecular target in ATL, and it has also been identified as an HSP90 client protein and shown to be sensitive to HSP90 inhibitors.(33,34)
Although the relationship between the p53 mutation and chemosensitivity in ATL remains unknown, Tawara et al. and Nishimura et al.(35,36) noted a tendency for the median survival periods of patients with the p53 mutation and/or loss of heterozygosity of that region to be shorter as compared to patients without a p53 aberration. Importantly, we found that AUY922 had effects on ATL-related cell lines irrespective of their p53 status.
Based on the present DNA microarray results, we focused on the role of PIM kinases in ATL and are the first to present those results. PIM is an oncogene encoding a serine/threonine protein kinase family comprised of PIM-1, -2, and -3; PIM kinases have multiple functions involved in cell proliferation, survival, differentiation, apoptosis, and tumorigenesis.(37,38) Elevated levels of PIM-1 and PIM-2 have been mostly found in hematologic malignancies and prostate cancer, and increased PIM-3 expression has been observed in solid tumors.(39,40) In addition, PIM expression is correlated with poor prognosis in some hematopoietic malignancies.(41–44) Our results indicated an anti-ATL activity of AUY922, which was mediated by degradation of PIM kinases. Those kinases are induced by activation of transcriptional factors downstream of growth factor signaling pathways, such as the Janus kinase and signal transducer and activator of transcription (JAK-STAT) and NF-κB pathways. Therefore, it is possible that the decrease in PIM kinases induced by AUY922 was due to a reduction in NF-κB activity.(45) The present results are the first to show an inhibitory effect of SGI-1776 on ATL-related cell lines and primary ATL cells. We concluded that PIM kinases are partly responsible for cell survival in ATL.
SGI-1776 has been shown to induce apoptosis in cells related to human acute myeloid leukemia and chronic lymphocytic leukemia.(46,47) Although a phase l clinical trial of SGI-1776 in patients with castration-resistant prostate cancer and refractory non-Hodgkin's lymphoma was started, evaluation of this compound was halted due to cardiac toxicity.(48) Our findings suggest that PIM kinases are a novel therapeutic target for treatment of ATL, indicating that a new generation of PIM kinase inhibitors with reduced toxicity in clinical settings is needed.
Heat shock protein 90 mediates protection of PIM kinases from proteosome degradation and PIM-1 was previously reported to be an HSP90 client protein.(49) However, it is not known whether PIM-2 and -3 are also such client proteins. In our WB analysis of SGI1776, even though it was not determined whether PIM-2 and/or -3 directly interact with HSP90, the results suggest that they are HSP90 client proteins in ATL.
In summary, our findings show that AUY922 may be potentially useful as a chemotherapeutic agent and PIM kinases a novel therapeutic target for treatment of ATL.
Acknowledgments
The authors would like to thank Novartis for providing NVP-AUY922. This study was supported in part by a Grant-in-aid for Scientific Research (23590641) from the Japan Society for the Promotion of Science.
Glossary
Abbreviations
- AP-1
activator protein-1
- ATL
adult T-cell leukemia–lymphoma
- HSP90
heat shock protein 90
- IKK
IκB kinase
- NF-κB
nuclear factor-κB
- p-Akt
phospho-Akt
- PI
propidium iodide
- PI3K
phosphatidylinositol 3-kinase
- PIM
proviral integration site for moloney murine leukemia virus
- WB
Western blot
Disclosure Statement
The authors have no conflicts of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Effects of heat shock protein 90 inhibitor AUY922 on apoptosis in non-HTLV-1 related T-cell lines. The non-HTLV-1 related T-cell lines Jurkat and Molt4 were treated with or without 100 nM AUY922 for 48 or 72 h, then harvested, stained, with annexin V–propidium iodide, and analyzed using flow cytometry. Data shown represent the percentages of apoptotic cells among untreated and AUY922-treated cells.
References
- 1.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 2.Kurashina R, Ohyashiki JH, Kobayashi C, et al. Anti-proliferative activity of heat shock protein (Hsp) 90 inhibitors via beta-catenin/TCF7L2 pathway in adult T cell leukemia cells. Cancer Lett. 2009;284:62–70. doi: 10.1016/j.canlet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Ikebe E, Kawaguchi A, Tezuka K, et al. Oral administration of an HSP90 inhibitor, 17-DMAG, intervenes tumor-cell infiltration into multiple organs and improves survival period for ATL model mice. Blood Cancer J. 2013;3:e132. doi: 10.1038/bcj.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goetz MP, Toft D, Reid J, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23:1078–87. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 5.Grem JL, Morrison G, Guo XD, et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23:1885–93. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 6.Banerji U, O'Donnell A, Scurr M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23:4152–61. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 7.Brough PA, Aherne W, Barril X, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51:196–218. doi: 10.1021/jm701018h. [DOI] [PubMed] [Google Scholar]
- 8.Eccles SA, Massey A, Raynaud FI, et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–60. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 9.Jensen MR, Schoepfer J, Radimerski T, et al. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10:R33. doi: 10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stingl L, Stuhmer T, Chatterjee M, Jensen MR, Flentje M, Djuzenova CS. Novel HSP90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cell-cycle impairment, increased DNA damage and repair protraction. Br J Cancer. 2010;102:1578–91. doi: 10.1038/sj.bjc.6605683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garon EB, Finn RS, Hamidi H, et al. The HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growth. Mol Cancer Ther. 2013;12:890–900. doi: 10.1158/1535-7163.MCT-12-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sessa C, Shapiro GI, Bhalla KN, et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19:3671–80. doi: 10.1158/1078-0432.CCR-12-3404. [DOI] [PubMed] [Google Scholar]
- 13.Yamada Y, Tomonaga M. The current status of therapy for adult T-cell leukaemia-lymphoma in Japan. Leuk Lymphoma. 2003;44:611–8. doi: 10.1080/1042819021000055039. [DOI] [PubMed] [Google Scholar]
- 14.Mori N, Fujii M, Ikeda S, et al. Constitutive activation of NF-kappaB in primary adult T-cell leukemia cells. Blood. 1999;93:2360–8. [PubMed] [Google Scholar]
- 15.Mori N, Fujii M, Iwai K, et al. Constitutive activation of transcription factor AP-1 in primary adult T-cell leukemia cells. Blood. 2000;95:3915–21. [PubMed] [Google Scholar]
- 16.Fukuda R, Hayashi A, Utsunomiya A, et al. Alteration of phosphatidylinositol 3-kinase cascade in the multilobulated nuclear formation of adult T cell leukemia/lymphoma (ATLL) Proc Natl Acad Sci U S A. 2005;102:15213–8. doi: 10.1073/pnas.0507184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsukasaki K, Utsunomiya A, Fukuda H, et al. VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J Clin Oncol. 2007;25:5458–64. doi: 10.1200/JCO.2007.11.9958. [DOI] [PubMed] [Google Scholar]
- 18.Yamada Y, Fujita M, Suzuki H, et al. Established IL-2-dependent double-negative (CD4- CD8-) TCR alpha beta/CD3 + ATL cells: induction of CD4 expression. Br J Haematol. 1994;88:234–41. doi: 10.1111/j.1365-2141.1994.tb05012.x. [DOI] [PubMed] [Google Scholar]
- 19.Yamada Y, Ohmoto Y, Hata T, et al. Features of the cytokines secreted by adult T cell leukemia (ATL) cells. Leuk Lymphoma. 1996;21:443–7. doi: 10.3109/10428199609093442. [DOI] [PubMed] [Google Scholar]
- 20.Maeda T, Yamada Y, Moriuchi R, et al. Fas gene mutation in the progression of adult T cell leukemia. J Exp Med. 1999;189:1063–71. doi: 10.1084/jem.189.7.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasegawa H, Yamada Y, Harasawa H, et al. Sensitivity of adult T-cell leukaemia lymphoma cells to tumour necrosis factor-related apoptosis-inducing ligand. Br J Haematol. 2005;128:253–65. doi: 10.1111/j.1365-2141.2004.05289.x. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A. 1982;79:2031–5. doi: 10.1073/pnas.79.6.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posner LE, Robert-Guroff M, Kalyanaraman VS, et al. Natural antibodies to the human T cell lymphoma virus in patients with cutaneous T cell lymphomas. J Exp Med. 1981;154:333–46. doi: 10.1084/jem.154.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasegawa H, Yamada Y, Iha H, et al. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia. 2009;23:2090–101. doi: 10.1038/leu.2009.171. [DOI] [PubMed] [Google Scholar]
- 25.Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat Rev. 2013;39:375–87. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl) 2004;82:488–99. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 28.Che XF, Zheng CL, Owatari S, et al. Overexpression of survivin in primary ATL cells and sodium arsenite induces apoptosis by down-regulating survivin expression in ATL cell lines. Blood. 2006;107:4880–7. doi: 10.1182/blood-2005-08-3423. [DOI] [PubMed] [Google Scholar]
- 29.Broemer M, Krappmann D, Scheidereit C. Requirement of Hsp90 activity for IkappaB kinase (IKK) biosynthesis and for constitutive and inducible IKK and NF-kappaB activation. Oncogene. 2004;23:5378–86. doi: 10.1038/sj.onc.1207705. [DOI] [PubMed] [Google Scholar]
- 30.Peloponese JM, Jr, Jeang KT. Role for Akt/protein kinase B and activator protein-1 in cellular proliferation induced by the human T-cell leukemia virus type 1 tax oncoprotein. J Biol Chem. 2006;281:8927–38. doi: 10.1074/jbc.M510598200. [DOI] [PubMed] [Google Scholar]
- 31.Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene. 2005;24:6719–28. doi: 10.1038/sj.onc.1208825. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Wang Y, Yamakuchi M, et al. Phosphoinositide-3 kinase-PKB/Akt pathway activation is involved in fibroblast Rat-1 transformation by human T-cell leukemia virus type I tax. Oncogene. 2001;20:2514–26. doi: 10.1038/sj.onc.1204364. [DOI] [PubMed] [Google Scholar]
- 33.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–7. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujita N, Sato S, Ishida A, Tsuruo T. Involvement of Hsp90 in signaling and stability of 3-phosphoinositide-dependent kinase-1. J Biol Chem. 2002;277:10346–53. doi: 10.1074/jbc.M106736200. [DOI] [PubMed] [Google Scholar]
- 35.Tawara M, Hogerzeil SJ, Yamada Y, et al. Impact of p53 aberration on the progression of Adult T-cell Leukemia/Lymphoma. Cancer Lett. 2006;234:249–55. doi: 10.1016/j.canlet.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 36.Nishimura S, Asou N, Suzushima H, et al. p53 gene mutation and loss of heterozygosity are associated with increased risk of disease progression in adult T cell leukemia. Leukemia. 1995;9:598–604. [PubMed] [Google Scholar]
- 37.Wang Z, Bhattacharya N, Weaver M, et al. Pim-1: a serine/threonine kinase with a role in cell survival, proliferation, differentiation and tumorigenesis. J Vet Sci. 2001;2:167–79. [PubMed] [Google Scholar]
- 38.Bachmann M, Moroy T. The serine/threonine kinase Pim-1. Int J Biochem Cell Biol. 2005;37:726–30. doi: 10.1016/j.biocel.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 39.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica. 2010;95:1004–15. doi: 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhanasekaran SM, Barrette TR, Ghosh D, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 41.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 42.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9991–6. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poulsen CB, Borup R, Nielsen FC, et al. Microarray-based classification of diffuse large B-cell lymphoma. Eur J Haematol. 2005;74:453–65. doi: 10.1111/j.1600-0609.2005.00429.x. [DOI] [PubMed] [Google Scholar]
- 44.Hsi ED, Jung SH, Lai R, et al. Ki67 and PIM1 expression predict outcome in mantle cell lymphoma treated with high dose therapy, stem cell transplantation and rituximab: a Cancer and Leukemia Group B 59909 correlative science study. Leuk Lymphoma. 2008;49:2081–90. doi: 10.1080/10428190802419640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11:23–34. doi: 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- 46.Chen LS, Redkar S, Taverna P, Cortes JE, Gandhi V. Mechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemia. Blood. 2011;118:693–702. doi: 10.1182/blood-2010-12-323022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen LS, Redkar S, Bearss D, Wierda WG, Gandhi V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2009;114:4150–7. doi: 10.1182/blood-2009-03-212852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batra V, Maris JM, Kang MH, et al. Initial testing (stage 1) of SGI-1776, a PIM1 kinase inhibitor, by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;59:749–52. doi: 10.1002/pbc.23364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shay KP, Wang Z, Xing PX, McKenzie IF, Magnuson NS. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol Cancer Res. 2005;3:170–81. doi: 10.1158/1541-7786.MCR-04-0192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effects of heat shock protein 90 inhibitor AUY922 on apoptosis in non-HTLV-1 related T-cell lines. The non-HTLV-1 related T-cell lines Jurkat and Molt4 were treated with or without 100 nM AUY922 for 48 or 72 h, then harvested, stained, with annexin V–propidium iodide, and analyzed using flow cytometry. Data shown represent the percentages of apoptotic cells among untreated and AUY922-treated cells.