

Abstract
Double-strand breaks (DSBs) are one of the severest types of DNA damage. Unrepaired DSBs easily induce cell death and chromosome aberrations. To maintain genomic stability, cells have checkpoint and DSB repair systems to respond to DNA damage throughout most of the cell cycle. The failure of this process often results in apoptosis or genomic instability, such as aneuploidy, deletion, or translocation. Therefore, DSB repair is essential for maintenance of genomic stability. During mitosis, however, cells seem to suppress the DNA damage response and proceed to the next G1 phase, even if there are unrepaired DSBs. The biological significance of this suppression is not known. In this review, we summarize recent studies of mitotic DSB repair and discuss the mechanisms of suppression of DSB repair during mitosis. DSB repair, which maintains genomic integrity in other phases of the cell cycle, is rather toxic to cells during mitosis, often resulting in chromosome missegregation and aberration. Cells have multiple safeguards to prevent genomic instability during mitosis: inhibition of 53BP1 or BRCA1 localization to DSB sites, which is important to promote non-homologous end joining or homologous recombination, respectively, and also modulation of the non-homologous end joining core complex to inhibit DSB repair. We discuss how DSBs during mitosis are toxic and the multiple safeguard systems that suppress genomic instability.
Keywords: Chromosome segregation, DSB repair, homologous recombination, mitosis, NHEJ
The vast majority of studies for DSB repair have been carried out using cells during G1 or S/G2 phase. Cells have two main DSB repair pathways: C-NHEJ, or HR.(1) DSBs are often repaired by one of the two pathways in a cell-cycle dependent manner: C-NHEJ in G1 phase, or HR in S/G2 phase. A-NHEJ, a third, less-characterized repair pathway, also plays a critical role in DSB repair.(2–6) During mitosis, however, regulation of these repair pathways is not well understood.
In G1 or S/G2 phase, DSBs initiate a massive signaling cascade, called the DDR. The DDR sensor protein complex, MRN, recognizes damaged DNA and results in recruitment of PIKKs such as: ATM through the interaction with Nbs1; ATR through the interaction of the ATR partner, ATRIP, with replication protein A, a single-stranded DNA binding protein, on single-stranded DNAs; and a third PIKK member, DNA-PKcs, through the binding with the Ku complex, to facilitate DDR and also C-NHEJ at the DSB sites.(7) The ATM phosphorylates histone H2AX on S139, generating γH2AX (Fig. 1a).(8) The MDC1 protein recognizes γH2AX. ATM also phosphorylates MDC1 on TQXF motifs,(9) and then the RNF-containing E3 ubiquitin ligase RNF8, which mediates a protein ubiquitination cascade, is recruited to the damage site through binding to the phosphorylated TQXF motif on MDC1 (Fig. 1b, left). RNF168, a second E3 ubiquitin ligase, is accumulated at the DSB sites by recognizing products ubiquitinated by RNF8 (Fig. 1c, left). RNF8/RNF168-mediated ubiquitination of histone H2A at lysine 13/15 is believed to be important for remodeling of the chromatin flanking DSBs,(10,11) followed by RNF8/RNF168 mediated lysine 63-linked and lysine 48-linked ubiquitin chain synthesis(10,12) and multi-ubiquitination.(13) The ubiquitination cascade results in the recruitment of BRCA1 or 53BP1 to the DSB sites(14–18) (Fig. 1d, left, middle).
Fig. 1.
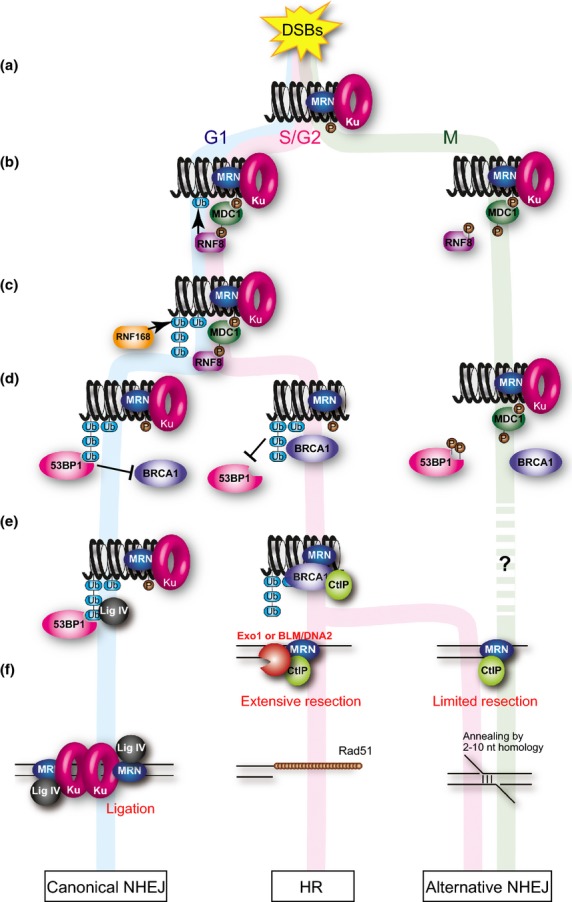
Role of ubiquitin in the response of double-strand breaks (DSBs) in interphases and mitosis. (a) DSB induces ATM-dependent phosphorylation of histone H2AX. (b) MDC1 recognizes the phosphorylation in both interphase and mitosis. ATM also phosphorylates MDC1 to promote RNF8 recruitment to the DSB sites in interphase. CDK1 phosphorylates RNF8 to inhibit the recruitment during mitosis. (c) RNF8 works with RNF168 to ubiquitinate histone H2A and other molecules to amplify the ubiquitin-mediated DSB signaling. In mitotic cells, the ubiquitination is suppressed. (d) Ubiquitination leads to recruitment of multiple effector proteins such as 53BP1 and BRCA1 in interphases. Both 53BP1 and BRCA1 fail to localize to the DSB sites during mitosis. (e) 53BP1 promotes non-homologous end joining (NHEJ) in G1 phase, whereas BRCA1 promotes homologous recombination (HR) by interacting with CtIP in S/G2 phase. (f) In G1 phase, DSB is ligated by DNA ligase IV, an NHEJ-specific DNA ligase. In S/G2 phase, DSBs are resected by functions of CtIP and the MRN complex to promote Rad51-ssDNA filament formation to execute HR. Alternative NHEJ (A-NHEJ) dependent on CtIP function, which induces limited resection at the DSB site. Cell-cycle regulation of A-NHEJ is not clear.
The proportion of 53BP1 or BRCA1 at DSB sites determines whether the repair pathway is NHEJ or HR. The 53BP1 and BRCA1 proteins have opposite functions in DSB end resection during DSB repair. 53BP1 binds to the DSB site by recognizing ubiquitinated histone H2A using its ubiquitin-dependent recruitment motif(19) and inhibits DSB end resection.(20,21) Unresected DSB ends prefer DNA ligase IV-dependent C-NHEJ rather than HR or A-NHEJ (Fig. 1d–f, left). However, BRCA1 somehow associates with initiation of DSB end resection.(22,23) It overcomes the inhibitory function of 53BP1 on DSB end resection and promotes HR (20) (Fig. 1d,e, middle). The recruitment of BRCA1 to DSB sites largely depends on RNF8- and RNF168-dependent ubiquitination.(24) However, the molecular mechanism of the recruitment of BRCA1 and its function at DSB sites are still unclear. There are at least three different protein complexes containing BRCA1: BRCA1-A (RAP80, Abraxas, BRCC36), -B (BACH1), and -C (CtIP).(22) The BRCA1-C complex promotes DSB end resection. The DSB end is processed to produce a 3′-overhanging single-stranded DNA region by the collaboration of CtIP and Mre11 nuclease of the MRN, followed by an extension step mediated by exonuclease I or BLM helicase/DNA2 nuclease.(25) Then, replication protein A is recruited to the single-stranded DNA region. Rad51 recombinase (Fig. 1f, middle), a eukaryotic RecA, forms nucleoprotein filaments on single-stranded DNAs in the aid of Rad51 mediators such as BRCA2, and then Rad51 paralogs including XRCC2, XRCC3, Rad51B, Rad51C, and Rad51D to facilitate HR.(26–28)
A-NHEJ is defined as an NHEJ activity when core NHEJ factors (DNA ligase IV, Ku70, and Ku80) are inactivated.(5) The A-NHEJ process, initiated by MRN- and CtIP-dependent DSB end resection, needs the ERCC1–XPF nuclease and DNA ligase I/III–XRCC1 complex, but is independent of the DNA-PKcs, DNA ligase IV, or Ku complex.(29,30) A-NHEJ in vertebrates is often used as a synonym of microhomology-mediated end joining in yeast.(31) Generally, A-NHEJ is classified as a part of microhomology-mediated end joining, which is defined as any end joining involving microhomology at the joined end.(29) Precise molecular mechanisms of the pathways and their biological significance are still unclear.
Effect of DSB Introduction During Mitosis
Human cells show a different DDR during mitosis than in other cell cycle phases. During most of the cell cycle, cells induce checkpoints in response to DNA damage. Until late prophase, the entry into mitosis can be suppressed by the damage. After cells reach the point of no return, however, DSBs on mitotic chromosomes do not trigger cell-cycle delay or arrest; and the cells rather proceed through mitosis even if they contain unrepaired DSBs or fragmented chromosomes.(32) Indeed, the sensitivity to ionizing radiation is higher in mitotic cells than in interphase cells.(33,34) Moreover, DSB introduction into mitotic cells by etoposide treatment induces massive chromosome aberrations in the next cell cycle in a cancer cell line (Fig. 2). Thus, DSBs during mitosis are very toxic to cells because of the induction of severe genomic instability. However, the biological significance of suppression of the DDR remains unclear.
Fig. 2.
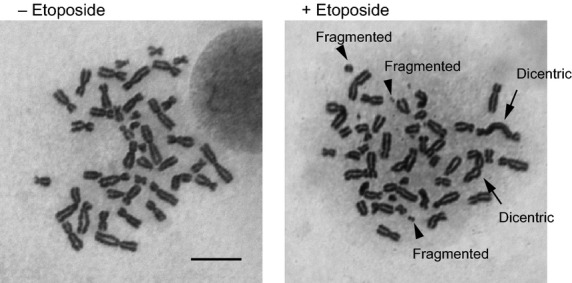
Effect of etoposide-induced double-strand breaks (DSBs) during mitosis on genomic instability of mitotic chromosomes. Representative images of chromosome spreads from etoposide-treated (+Etoposide) and non-treated (−Etoposide) in HCT116 human colon cancer cells arrested in mitosis. Arrows indicate dicentric chromosomes. Arrowheads show fragmented chromosomes. Scale bar = 10 μm.
Double-Strand Break Repair During Mitosis is Inactivated by Mitotic Kinases
During mitosis, DSBs activate PIKKs to induce phosphorylation of H2AX, and MDC1 as well as the MRN complex are recruited to the DSB sites, as seen during interphases (Fig. 1a). However, the recruitment of RNF8, RNF168, 53BP1, and BRCA1 to DSB is largely suppressed during mitosis (Fig. 1b,d, right).(30,33) The mechanism of the suppression and its purpose were not well understood. Two groups recently showed that localization of RNF8 and 53BP1 to DSBs is inhibited during mitosis.(35,36) The first work done by Orthwein et al. (2014) showed that, in human cells, RNF8 and 53BP1 are phosphorylated during mitosis by CDK1 and that inhibition of the phosphorylation restores their localization to DSB sites (Fig. 1b,d, right). Orthwein et al. also identified T198 on RNF8 as a CDK1 phosphorylation site and showed that RNF8-T198A, a phosphorylation-defective protein, can localize to mitotic chromatin after DSB introduction. RNF8 prepared from mitotic extracts cannot bind to MDC1 in vitro, but RNF8 prepared from CDK1 activity-inhibited cells interacts with MDC1 in vitro. The RNF8-T198A protein restores localization of BRCA1 but not 53BP1 to the DSB site. They also found that T1609 and S1618 on 53BP1 are phosphorylated during mitosis (Fig. 3a) and that 53BP1-T1609A/S1618A double mutant protein can localize to mitotic chromatin and restores DSB repair in cells expressing RNF8-T198A. Their study confirmed that T1609 is phosphorylated by CDK1, whereas S1618 is a target of PLK1 in vitro. These show that there are at least two distinct mechanisms to regulate the recruitment of these DDR effector proteins.(35)
Fig. 3.
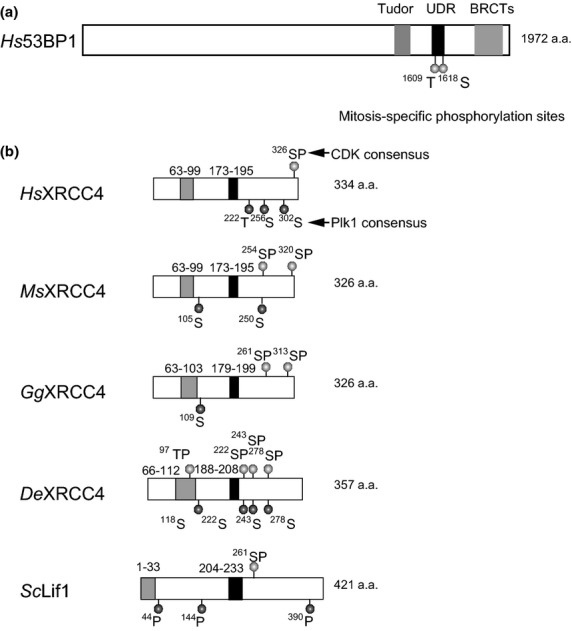
Domain structure of 53BP1 and comparison of CDK1 and PLK1 sites among XRCC4 orthologs. (a) Domain structure and mitosis-specific phosphorylation sites of 53BP1. (b) Domain structure of XRCC4 and conservation of phosphorylation sites among various species. S326 in the C-terminus of human XRCC4 (Hs) is a potential CDK1 phosphorylation site. T222, S256, and S303 are putative PLK1 phosphorylation sites. The gray and black boxes show XLF and DNA ligase IV binding sites, respectively. Phosphorylation of CDK1 or PLK1 sites are shown in mouse (Ms), chicken (Gg), zebrafish (De), and budding yeast (Sc). DNA ligase IV and XLF binding sites in mouse, chicken, and zebrafish were predicted by aligning with the DNA ligase IV and XLF binding sites of human XRCC4 using T-Coffee software.
The second work by Lee et al. (2014) indicated that T1609 and S1618 of 53BP1 are hyperphosphorylated in the absence of PP4c phosphatase in human cell lines.(36,37) They analyzed the function of PP4c phosphatase during DDR and found that these sites are phosphorylated specifically during mitosis.(36) These mitosis-specific phosphorylation sites are located in the ubiquitin-dependent recruitment motif of 53BP1, a recognition site of ubiquitinated H2A, which is required for the localization of the protein to DSB sites.(15–17) Both of the reports(35,36) showed that DNA damage during mitosis in cells expressing phosphorylation-defective 53BP1-T1609A/S1618A mutant, which restores its localization to DSB sites, leads to increased micronuclei formation. This suggests that restoration of DSB repair during mitosis causes defects in proper chromosome segregation and that inhibition of DSB repair during mitosis serves to maintain genomic stability. In addition to micronuclei formation, Orthwein et al. showed that restoration of accumulation of RNF8 and 53BP1 to damaged mitotic chromosomes increases telomere fusions, which seems to be mediated by DSB repair such as C-NHEJ.(35) Therefore, suppression of DSB repair by phosphorylation of RNF8 or 53BP1 by CDK1 or PLK1 may protect the genome during mitosis. In particular, suppression of RNF8- and 53BP1-dependent DSB repair during mitosis may prevent telomere fusions.
Involvement of C-NHEJ Core Complex in Segregation Defects
Two groups showed that inhibition of the DNA-PKcs suppresses micronuclei formation induced by mitotic DSBs in 53BP1-T1609A/S1618A-expressing or 53BP1-T1609A/S1618A RNF8-T198A-expressing cells, suggesting and that the core C-NHEJ complex plays a role in the segregation defects.(35,36) Recently, we showed another DSB repair suppression mechanism by modulating the C-NHEJ core complex during mitosis.(38) DSB repair is largely suppressed during mitosis, but a substantial level of repair still occurs.(38) C-NHEJ causes formation of anaphase bridges, which are bridge-like DNA structures that span daughter chromosomes and frequently induce genomic instability through inappropriate chromosome segregation.(39,40) In our study, severe chromosome aberrations such as dicentric and fragmented chromosomes are induced by mitosis-specific DSB introduction by transient treatment with etoposide (Fig. 2).(38) This observation raises a possibility that anaphase bridges are caused by dicentric chromosome formation by interchromosomal connections between telomeres or other chromosomal loci. These results suggest that there is a process to connect sister chromatids or individual chromosomes in DDR during mitosis. DNA ligase IV-dependent C-NHEJ contributes to dicentric chromosome formation through telomere fusion in cells with dysfunctional telomeres,(41) which act as DSB ends.(42) Therefore, we hypothesized that C-NHEJ is involved in the formation of anaphase bridges. Both XRCC4 and XLF are regulatory subunits of the DNA ligase IV complex and are essential for its activity.(43–45) Anaphase bridge formation is reduced after XRCC4 knockdown, suggesting that C-NHEJ contributes to the formation of some of these bridges during mitosis.(38) Knockdown of the HR-specific factor XRCC3 has almost no effect on anaphase bridge formation, indicating that C-NHEJ is more critical than HR for the formation of anaphase bridges in mitotic cells. Our studies strongly suggest that inappropriate use of the C-NHEJ pathway causes chromosome bridges, which lead to chromosome aberrations from mitotic DSBs.
Mitosis-Specific Phosphorylation of XRCC4
A phosphoproteomics study revealed that many sites are phosphorylated during mitosis, including those on XRCC4,(46) so we evaluated cell-cycle regulation of XRCC4 phosphorylation.(38) This mitosis-specific phosphorylation of XRCC4 contributes to the suppression of anaphase bridge formation after induction of DSBs during mitosis.(38) The mitosis-specific phosphorylation of XRCC4 depends on both CDK1 and PLK1 activities.(38) S326 is a putative CDK phosphorylation site, and S326A substitution substantially reduces mitosis-specific phosphorylation of XRCC4 (Fig. 3b).(38) Interestingly, the S326 phosphorylation residue overlaps with the polo box recognition motif.(47) Both S256 and S326 are phosphorylated during mitosis.(46) Because S256 resides in a putative PLK1 phosphorylation site (Fig. 3b), CDK-dependent phosphorylation of S326 might prime the mitosis-specific phosphorylation of XRCC4 by PLK1.
Although the ortholog of XRCC4 has not yet been identified in nematode or fission yeast, most of the DNA ligase IV subunits have been identified in various species (Table 1). We previously analyzed cell-cycle regulation of Lif1p, which is the S. cerevisiae ortholog of XRCC4.(48–50) Lif1p is phosphorylated by CDKs from S phase through mitosis and this phosphorylation plays a role in suppressing C-NHEJ in S to M phases through the pathway dependent on Sae2, the S. cerevisiae ortholog of CtIP.(49) These findings prompted us to assess the conservation of the phosphorylation sites among XRCC4 orthologs. Although the locations of the phosphorylation sites are different, both S. cerevisiae Lif1p and human XRCC4 have one putative target site for CDK1 and multiple sites for PLK1 (Fig. 3b). We found conservation of the phosphorylation sites among various species. Both the CDK1 and PLK1 sites near the C-terminus were conserved between human and mouse (Fig. 3b). In chicken, there are two CDK1 sites near the C-terminus and one PLK1 site. However, in zebrafish, although the CDK1 site near the C-terminus was not conserved, there are four CDK1 sites in other locations. Three of them are located downstream of the DNA ligase IV binding site and overlap with PLK1 sites (Fig. 3b). All of the XRCC4 orthologs have CDK1 or PLK1 phosphorylation sites downstream region of the DNA ligase IV interaction domain. Thus, the function of mitosis-specific phosphorylation of XRCC4 is likely to be conserved among many species. We showed that XRCC4 phosphorylation-defective mutant (XRCC4-S326A) restores rapid repair of mitotic DSBs associated with more anaphase bridge formation.(38) This suggests that mitotic XRCC4 phosphorylation is involved in suppressing C-NHEJ to prevent chromosome instability in human cells. CtIP promotes DSB end resection in HR and A-NHEJ.(51) Because anaphase bridge formation is increased in CtIP-depleted cells,(38) CtIP may have a function to suppress anaphase bridges, possibly through the A-NHEJ pathway. BRCA1, which is important for recruitment of CtIP to DSB sites, does not localize to mitotic DSB sites.(33) However, only the function of CtIP in HR, but not in A-NHEJ, is dependent on interaction with BRCA1.(52) Moreover, in Xenopus M-phase extract, CtIP can bind mitotic chromatin.(53) Thus, CtIP may function as an A-NHEJ factor in suppression of genomic instability during mitosis. Alternatively, CtIP-dependent end resection may have functions to suppress anaphase bridge formation.
Table 1.
Conservation of DNA ligase IV complex subunits among species
| DNA ligase IV | XRCC4 | XLF | |
|---|---|---|---|
| Human | DNA ligase IV | XRCC4 | XLF |
| Mouse | DNA ligase IV | XRCC4 | XLF |
| Chicken | DNA ligase IV | XRCC4 | XLF |
| Flog | DNA ligase IV | XRCC4 | XLF |
| Zebrafish | DNA ligase IV | XRCC4 | Nhej1 |
| Fruit fly | Lig4 | XRCC4 | XLF |
| Nematode | LIG-4 | ? | ? |
| Arabidopsis | LIG4 | XRCC4 | ? |
| Fission yeast | Lig4 | ? | Xlf1 |
| Budding yeast | Dnl4 | Lif1 | Nej1 |
?, Unknown.
Although DNA ligase IV (catalytic subunit) localizes to mitotic chromosomes, XRCC4 does not,(54) suggesting that the DNA ligase IV complex function during mitosis is different from those in other cell cycles. Because complex formation of the DNA ligase IV with XRCC4 is believed to be essential for its activity, an activity of the DNA ligase IV complex may be modified during mitosis. We confirmed the difference in the localization of DNA ligase IV and XRCC4 during mitosis.(38) Like wild-type protein, XRCC4 phosphorylation-defective mutant protein also fails to localize to mitotic chromosomes, showing that the phosphorylation of XRCC4 is not responsible for inhibiting the localization to chromosomes during mitosis. On the basis of these findings, we propose that there may be two mechanisms to suppress C-NHEJ: the reduced recruitment of C-NHEJ-specific factors to mitotic chromosomes, and mitosis-specific phosphorylation of XRCC4 by modulating the function of the DNA ligase IV complex. Taken together, these results suggest that XRCC4, a key regulatory subunit of the DNA ligase IV complex, is required not only for C-NHEJ in interphase but also for suppression of C-NHEJ during mitosis to prevent genomic instability in human cells.
Multiple Layers of Safeguards for Suppression of DSB Repair Prevent Genomic Instability During Mitosis
Cells prevent carryover of DNA lesions at the G2/M checkpoint because DNA damage as well as the DNA repair process are toxic during mitosis. As described above, there are several types of DSB repair suppression systems that occur during mitosis. The DDR pathway consists of two steps (Fig. 4a). However, mitotic cells do not undergo the second level of DDR: neither 53BP1 nor BRCA1 localizes to DSB sites. Mitosis-specific phosphorylation of 53BP1 and RNF8 prevents their localization to DSB sites.(35,36) Because 53BP1 and BRCA1 localization to DSB sites is important for NHEJ and HR, respectively, DSB repair pathways should be largely suppressed on mitotic chromosomes (Fig. 4b). We found that the third level of DDR is also suppressed or modified by mitosis-specific phosphorylation of XRCC4, a component of the core C-NHEJ complex.(38) Even if DSB repair pathways are largely suppressed by mitosis-specific phosphorylation of 53BP1 and RNF8, considerable levels of DSB repair still occur and cause genomic instability during mitosis. Phosphorylation of XRCC4 during mitosis slows DSB repair.(38) Thus, XRCC4 phosphorylation has some functions to modulate DNA ligase IV complex activity (Fig. 4c). In addition, XRCC4 fails to localize to mitotic chromatin.(54) The failure of XRCC4 to localize to mitotic chromosomes also may modify the composition of the DNA ligase IV complex. CtIP, but not Rad51, is recruited to mitotic chromatin.(53) This also suggests that there is the third level of suppression mechanisms in HR and that CtIP has some functions to prevent genomic instability without the requirement of BRCA1 localization.
Fig. 4.
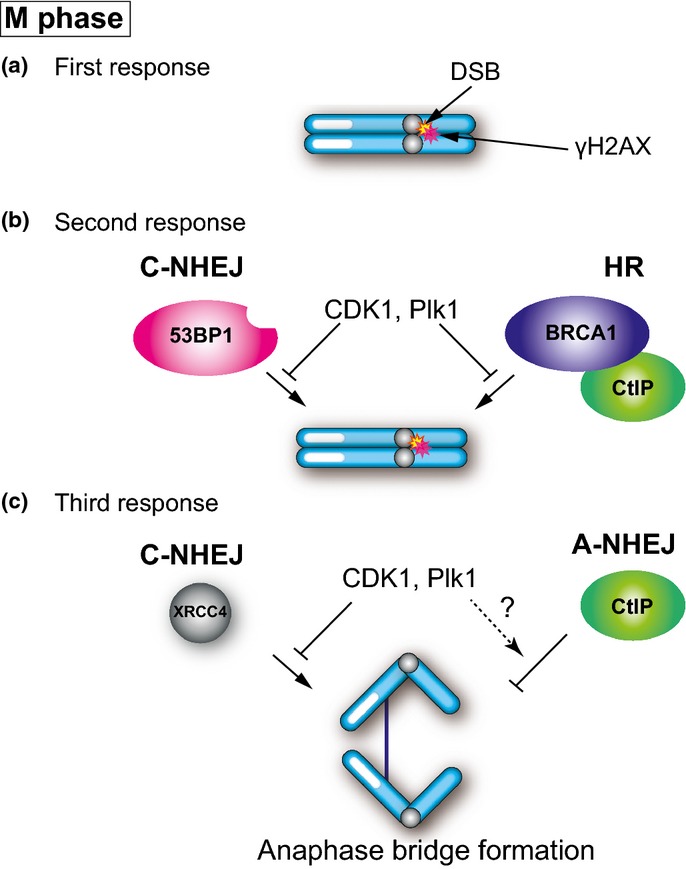
DNA damage response in mitosis. (a) Double-strand break (DSB) induces histone H2AX phosphorylation (γH2AX) by ATM. (b) CDK1 and PLK1 phosphorylate RNF8 and 53BP1 to inhibit 53BP1 and BRCA1 localization of DSB sites. (c) CDK1 and PLK1 phosphorylate XRCC4, a regulatory subunit of the DNA ligase IV complex, to suppress canonical non-homologous end joining (C-NHEJ) activity. CtIP-dependent alternative non-homologous end joining (A-NHEJ) may prevent anaphase bridge formation.
Conclusion and Future Directions
Cells use multiple safeguards to prevent genomic instability by suppression or modification of DSB repair activities during mitosis, through the activation of mitotic kinases, CDK1 and PLK1. The molecular mechanisms of mitosis-specific phosphorylation and the change in localization of the DNA ligase IV component XRCC4 are still unknown. Analyzing how DNA ligase IV activity in mitotic cells differs at the molecular level from during other cell-cycle phases is critical to understand the mechanisms of these controls. Another C-NHEJ component, DNA-PKcs also phosphorylated by PLK1, is dephosphorylated by PP6 and is required for accurate chromosome segregation.(55) It is also interesting to analyze how those C-NHEJ factors are involved in accurate mitosis.(56) Active DSB repair during mitosis affects chromosome segregation, which often results in apoptosis, aneuploidy, or other chromosome aberrations. Thus, studies of DSB repair control during mitosis are important to understand the origin of genomic instability, which causes cell tumorigenesis. In addition, activation of C-NHEJ during mitosis causes severe damage to growing cells like cancer cells, but the activation of NHEJ by itself would not be detrimental to interphase cells. This property would be useful for the development of anticancer drugs in the future.
Acknowledgments
We thank Ms. Chie Watanabe for technical assistance with chromosome aberration analysis. This work was supported by Grants-in-Aid (Kakenhi) to M.T. and A.S. from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by the NEXT program to M.S. from Japan Society of Promotion of Science.
Glossary
Abbreviations
- 53BP1
p53-binding protein 1
- A-NHEJ
alternative non-homologous end joining
- ATM
ataxia–telangiectasia mutated protein
- ATR
ataxia–telangiectasia mutated related protein
- BRCA1
breast cancer 1, early onset
- C-NHEJ
canonical non-homologous end joining
- DDR
DNA damage response
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- DSB
double-strand break
- HR
homologous recombination
- MDC1
mediator of DNA damage checkpoint 1
- MRN
Mre11–Rad50–Nbs1 complex
- NHEJ
non-homologous end joining
- PIKK
phosphatidylinositol-3-kinase-like kinase
- PLK1
polo-like kinase 1
- RNF
RING finger
- XRCC
X-ray repair cross-complementing group
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 2.Bogue MA, Wang C, Zhu C, Roth DB. V(D)J recombination in Ku86-deficient mice: distinct effects on coding, signal, and hybrid joint formation. Immunity. 1997;7:37–47. doi: 10.1016/s1074-7613(00)80508-7. [DOI] [PubMed] [Google Scholar]
- 3.Kabotyanski EB, Gomelsky L, Han JO, Stamato TD, Roth DB. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 1998;26:5333–42. doi: 10.1093/nar/26.23.5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang F, Romanienko PJ, Weaver DT, Jeggo PA, Jasin M. Chromosomal double-strand break repair in Ku80-deficient cells. Proc Natl Acad Sci USA. 1996;93:8929–33. doi: 10.1073/pnas.93.17.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson LH. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res. 2012;751:158–246. doi: 10.1016/j.mrrev.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 8.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 10.Mallette FA, Mattiroli F, Cui G, et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–78. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatti M, Pinato S, Maspero E, Soffientini P, Polo S, Penengo L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle. 2012;11:2538–44. doi: 10.4161/cc.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meerang M, Ritz D, Paliwal S, et al. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat Cell Biol. 2011;13:1376–82. doi: 10.1038/ncb2367. [DOI] [PubMed] [Google Scholar]
- 13.Kato K, Nakajima K, Ui A, Muto-Terao Y, Ogiwara H, Nakada S. Fine-tuning of DNA damage-dependent ubiquitination by OTUB2 supports the DNA repair pathway choice. Mol Cell. 2014;53:617–30. doi: 10.1016/j.molcel.2014.01.030. [DOI] [PubMed] [Google Scholar]
- 14.Doil C, Mailand N, Bekker-Jensen S, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–46. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 15.Huen MS, Grant R, Manke I, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolas NK, Chapman JR, Nakada S, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–40. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mailand N, Bekker-Jensen S, Faustrup H, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 18.Stewart GS, Panier S, Townsend K, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–34. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 19.Fradet-Turcotte A, Canny MD, Escribano-Diaz C, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–4. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li ML, Greenberg RA. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem Sci. 2012;37:418–24. doi: 10.1016/j.tibs.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49:795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 25.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17:1260–2. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bishop DK, Ear U, Bhattacharyya A, et al. Xrcc3 is required for assembly of Rad51 complexes in vivo. J Biol Chem. 1998;273:21482–8. doi: 10.1074/jbc.273.34.21482. [DOI] [PubMed] [Google Scholar]
- 28.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–70. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 29.Goodarzi AA, Jeggo PA. The repair and signaling responses to DNA double-strand breaks. Adv Genet. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Peng G, Lin SY, Zhang P. DNA damage response is suppressed by the high cyclin-dependent kinase 1 activity in mitotic mammalian cells. J Biol Chem. 2011;286:35899–905. doi: 10.1074/jbc.M111.267690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daley JM, Palmbos PL, Wu D, Wilson TE. Nonhomologous end joining in yeast. Annu Rev Genet. 2005;39:431–51. doi: 10.1146/annurev.genet.39.073003.113340. [DOI] [PubMed] [Google Scholar]
- 32.Rieder CL, Cole RW. Entry into mitosis in vertebrate somatic cells is guarded by a chromosome damage checkpoint that reverses the cell cycle when triggered during early but not late prophase. J Cell Biol. 1998;142:1013–22. doi: 10.1083/jcb.142.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol. 2010;190:197–207. doi: 10.1083/jcb.200911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stobbe CC, Park SJ, Chapman JD. The radiation hypersensitivity of cells at mitosis. Int J Radiat Biol. 2002;78:1149–57. doi: 10.1080/09553000210166570. [DOI] [PubMed] [Google Scholar]
- 35.Orthwein A, Fradet-Turcotte A, Noordermeer SM, et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344:189–93. doi: 10.1126/science.1248024. [DOI] [PubMed] [Google Scholar]
- 36.Lee DH, Acharya SS, Kwon M, et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA Breaks. Mol Cell. 2014;54:512–25. doi: 10.1016/j.molcel.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee DH, Goodarzi AA, Adelmant GO, et al. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J. 2012;31:2403–15. doi: 10.1038/emboj.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terasawa M, Shinohara A, Shinohara M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 2014;10:e1004563. doi: 10.1371/journal.pgen.1004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montgomery E, Wilentz RE, Argani P, et al. Analysis of anaphase figures in routine histologic sections distinguishes chromosomally unstable from chromosomally stable malignancies. Cancer Biol Ther. 2003;2:248–52. doi: 10.4161/cbt.2.3.362. [DOI] [PubMed] [Google Scholar]
- 40.Artandi SE, Chang S, Lee SL, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–5. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 41.Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol. 2002;12:1635–44. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- 42.Longhese MP. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008;22:125–40. doi: 10.1101/gad.1626908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valencia M, Bentele M, Vaze MB, et al. NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature. 2001;414:666–9. doi: 10.1038/414666a. [DOI] [PubMed] [Google Scholar]
- 44.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–13. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 45.Grawunder U, Wilm M, Wu X, et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–5. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- 46.Olsen JV, Vermeulen M, Santamaria A, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 47.Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299:1228–31. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 48.Matsuzaki K, Shinohara A, Shinohara M. Forkhead-associated domain of yeast Xrs2, a homolog of human Nbs1, promotes nonhomologous end joining through interaction with a ligase IV partner protein, Lif1. Genetics. 2008;179:213–25. doi: 10.1534/genetics.107.079236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuzaki K, Terasawa M, Iwasaki D, Higashide M, Shinohara M. Cyclin-dependent kinase-dependent phosphorylation of Lif1 and Sae2 controls imprecise nonhomologous end joining accompanied by double-strand break resection. Genes Cells. 2012;17:473–93. doi: 10.1111/j.1365-2443.2012.01602.x. [DOI] [PubMed] [Google Scholar]
- 50.Herrmann G, Lindahl T, Schar P. Saccharomyces cerevisiae LIF1: a function involved in DNA double-strand break repair related to mammalian XRCC4. EMBO J. 1998;17:4188–98. doi: 10.1093/emboj/17.14.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–4. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–3. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peterson SE, Li Y, Chait BT, Gottesman ME, Baer R, Gautier J. Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J Cell Biol. 2011;194:705–20. doi: 10.1083/jcb.201103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Przewloka MR, Pardington PE, Yannone SM, Chen DJ, Cary RB. In vitro and in vivo interactions of DNA ligase IV with a subunit of the condensin complex. Mol Biol Cell. 2003;14:685–97. doi: 10.1091/mbc.E01-11-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Douglas P, Ye R, Trinkle-Mulcahy L, et al. Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci Rep. 2014;34:e00113. doi: 10.1042/BSR20140051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lees-Miller SP. DNA double strand break repair in mitosis is suppressed by phosphorylation of XRCC4. PLoS Genet. 2014;10:e1004598. doi: 10.1371/journal.pgen.1004598. [DOI] [PMC free article] [PubMed] [Google Scholar]