

Abstract
Phakomatosis pigmentovascularis (PPV) is a rare cutaneous disorder characterized by combination of capillary malformation and other pigmented naevi. Four types and two subtypes have been described where subtype ‘a’ present only with cutaneous form and subtype ‘b’ also with systemic association like in Sturge-Weber syndrome or Klippel-Trenaunay syndrome. Hereby, we report a case where our patient presented with port-wine stain, Nevus of Ota, Sturge-Weber syndrome, and Klippel-Trenaunay syndrome; which has made it a rare combination.
Keywords: Klippel-Trenaunay syndrome, phakomatosis, Sturge-Weber syndrome
What was known?
Phakomatosis pigmentovascularis along with systemic involvement like Sturge-Weber syndrome has been reported previously in literature.
Introduction
Phakomatosis pigmentovascularis (PPV) is a rare condition first described by Ota et al., in 1947.[1] It is defined by simultaneous presentation of capillary malformation and pigmented anomaly. There is no sex predilection, but the Japanese are found to be affected more.[2] It arises sporadically. Though four main types have been described, a fifth type with cutis marmorata and aberrant Mongolian blue spot have also been added to the list.
Case Report
A 13-year-old female presented with port-wine stain of left side of the body including face along the distribution of 5th cranial nerve, upper limb, trunk, and lower limb [Figure 1]. Additionally, this lesion was present on right side of the back as well. There was associated hypertrophy of the left lower limb with a visible vessel along the lateral aspect of the leg [Figure 2]. The left sclera showed blue discoloration [Figure 3]. There was no history of seizures, but she had subnormal intelligence. Ophthalmological examination showed raised intraocular tension in left eye but the right eye was normal. X-ray of upper and lower limb showed no bony hypertrophy. The color Doppler study of left lower limb showed absence of great and short saphenous vein. An abnormal dilated vein on lateral aspect of leg and thigh was present which was seen to drain into superficial circumflex iliac vein [Figure 4]. Superficial femoral vein was hypoplastic and popliteal vein was absent. Non-contrast computed tomography (CT) scan of brain showed focal parenchymal atrophy in the left parietal lobe region with foci of gyral calcification. Calvarial thickening predominantly in frontal and occipital region was seen. Frontal and sphenoid sinuses were found to be dilated.
Figure 1.
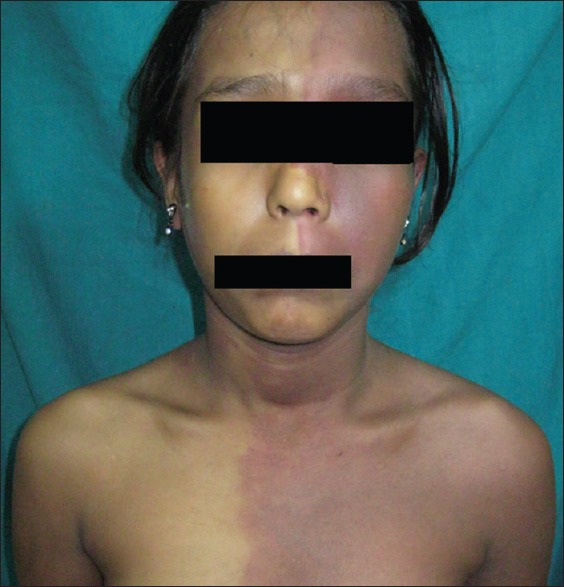
Widespread port-wine stain involving left side of face and chest
Figure 2.
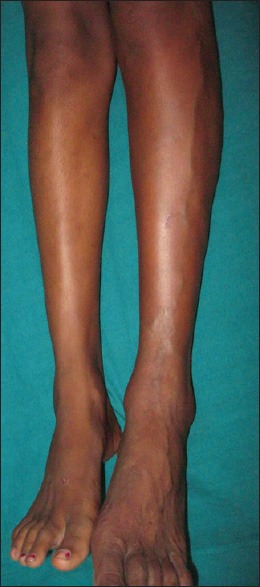
Engorged vein over lateral aspect of left leg along with hypertrophy of lower limb
Figure 3.
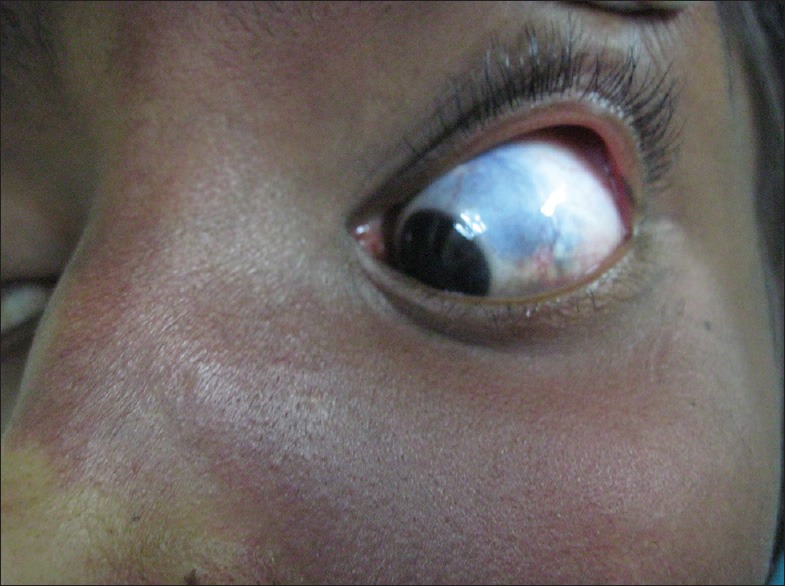
Bluish discoloration of sclera of left eye
Figure 4.
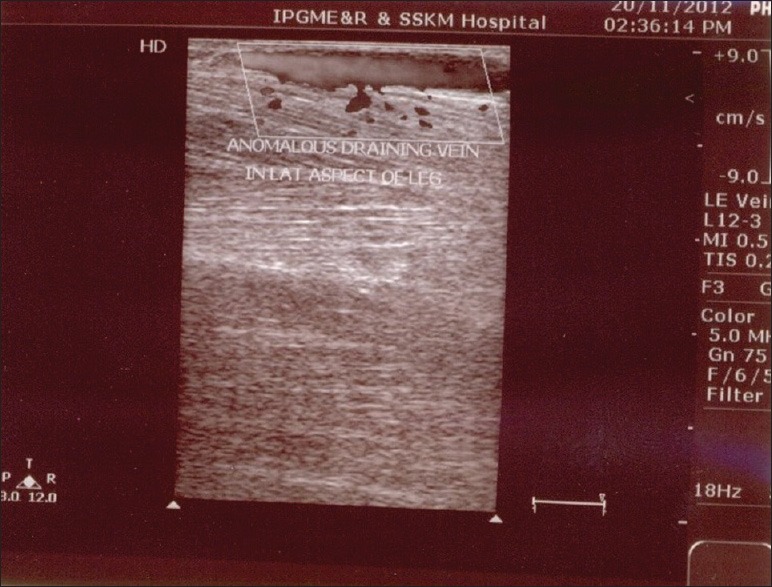
Color Doppler picture showing anomalous draining vein on lateral aspect of left leg
Discussion
Phakomatosis is a developmental abnormality simultaneously involving eye, central nervous system, and skin. Both, males and females are affected. Happle and Steijen proposed the genetic concept of twin spotting phenomenon to explain its etiology.[3] PPV were classified into four types namely; type I: Nevus flammeus and epidermal nevus, type II: Nevus flammeus, Mongolian spots, ± nevus anemicus, type III: Nevus flammeus, nevus spilus, ± nevus anemicus, type IV: Nevus flammeus, Mongolian spots, nevus spilus, ± nevus anemicus. Each type is subdivided into two subtypes; subtype ‘a’, cutaneous involvement only; and subtype ‘b’, both cutaneous and systemic involvement. PPV type I, III, and IV have very rarely been reported in literature.[4] Systemic syndromes which may be associated with it are Sturge-Weber syndrome, Nevus of Ota, and Klippel-Trenaunay syndrome. Our patient had port-wine stain and Nevus of Ota and also had systemic involvement which tilted the diagnosis in favor of PPV subtype ‘b’ variety.
Sturge-Weber syndrome is defined as a facial port-wine stain with associated ipsilateral vascular malformation of leptomeninges and eye. Eye involvement is not mandatory for meeting the diagnosis. Cutaneous changes most commonly involve the distribution of trigeminal nerve as in our patient. Brain changes show increased vascularity of leptomeninges which is usually unilateral and long-term pathological changes show calcification and atrophy.[5] Here our patient's CT scan changes showed long-term changes like atrophy and calcification of parietal lobe of brain. Eye changes in the form of glaucoma and buphthalmos may occur.
Though definition of Klippel-Trenaunay syndrome include cutaneous capillary malformation of a limb and soft tissue swelling with or without bone hypertrophy, original description indicate that varicosities of vein on affected limb is the usual finding and lateral venous anomaly is the most common abnormality.[6] Clinical picture showing widespread port-wine stain and hypertrophy of lower limb along with abnormal color Doppler finding, suggests the diagnosis of Klippel-Trenaunay syndrome.
Thus, our patient fulfilled all the criteria for the diagnosis of PPV along with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Few cases of PPV along with Sturge-Weber syndrome have been reported in literature, but PPV associated with both Sturge-Weber syndrome and Klippel-Trenaunay syndrome has been reported very rarely.[7,8]
What is new?
In our case report we are presenting phakomatosis pigmentovascularis with Sturge-Weber syndrome and Klippel-Trenaunay syndrome which is so far very rare.
Footnotes
Source of support: Nil
Conflict of Interest: Nil.
References
- 1.Ota M, Kawamura T, Ito N. Phacomatosis pigmentovascularis. Dermatol Surg. 1947;52:1–3. [Google Scholar]
- 2.Vidaurri-de la Cruz H, Tamayo-Sánchez L, Durán McKinster C, Orozco-Covarrubias Mde L, Ruiz-Maldonado R. Phakomatosis pigmentovascularis II A and II B: Clinical findings in 24 patients. J Dermatol. 2003;30:381–8. doi: 10.1111/j.1346-8138.2003.tb00403.x. [DOI] [PubMed] [Google Scholar]
- 3.Happle R. Stejlen PM: Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spots. Hautarzt. 1989;40:721–24. [PubMed] [Google Scholar]
- 4.Du LC, Delaporte E, Catteau B, Destee A, Piette F. Phakomatosis pigmentovascularis type II. Eur J Dermatol. 1998;8:569–72. [PubMed] [Google Scholar]
- 5.Marti-Bonmati L, Menor F, Mulas F. The Sturge-Weber syndrome: Correlation between the clinical status and radiological CT and MRI findings. Childs Nerv Syst. 1993;9:107–9. doi: 10.1007/BF00305319. [DOI] [PubMed] [Google Scholar]
- 6.Young AE. Combined vascular malformations. In: Mulliken JB, Young AE, editors. Vascular Birthmarks: Haemangiomas and Malformations. Philadelphia: WB Saunders; 1988. pp. 246–74. [Google Scholar]
- 7.Gupta A, Dubey S, Agarwal M. A case of Sturge-Weber syndrome in association with phacomatosis pigmentovascularis and developmental glaucoma. J AAPOS. 2007;11:398–9. doi: 10.1016/j.jaapos.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Goyal T, Varshney A. Phacomatosis cesioflammea: First case report from India. Indian J Dermatol Venereol Leprol. 2010;76:307. doi: 10.4103/0378-6323.62973. [DOI] [PubMed] [Google Scholar]