

Abstract
Bardet–Biedl syndrome is a rare autosomal recessive disorder, recently categorized as ciliopathy characterized by dysfunction of primary cilia which results in myriad manifestations in various organ systems. Though renal abnormalities can occur in this syndrome, renal failure is a rare presentation. The author reports a case of 18-year-old female who presented with polydactyly, obesity, retinitis pigmentosa, learning disability and renal failure.
Keywords: Bardet–Biedl syndrome, ciliopathy, end stage renal disease
INTRODUCTION
“Ciliopathies” are recently categorized group of genetic disorders that occur due to dysfunction of primary cilia and can affect multiple organ systems. Bardet–Biedl syndrome (BBS) has been recently included in this category. The predominant features of this syndrome are retinal dystrophy, obesity, polydactyly, learning disability, hypogonadism, and renal abnormalities. Though structural renal abnormalities are common in these patients, chronic kidney disease as a feature of this syndrome has been reported rarely.
CASE REPORT
An 18-year-old female was referred to our hospital for evaluation of swelling of legs of 2 months duration. There was history of bone pain, polydipsia, and polyuria for the past 3 years. She was born of non-consanguineous marriage. Patient had delayed developmental milestones. She began to walk and speak at 3 and 5 years of age respectively. Patient had difficulty in night vision since 8 years of age and difficulty in distant vision since 9 years of age. She dropped out from school due to poor scholastic performance. Patient had not attained menarche. Her both younger siblings were normal. The family history was unremarkable.
On examination, her height and weight were 136 cm (<10th centile) and 58 kg respectively with a body mass index of 31.4 kg/m2. She had pallor with bilateral pitting pedal edema. Physical examination was notable for the absence of secondary sexual characters in the form of absence of axillary, pubic hair, and poor breast bud development. She had post axial polydactyly in both her legs, central polydactyly in her right hand and clinodactyly of left little finger [Figures 1 and 2]. Genital examination was normal. Her visual acuity was decreased to counting fingers at 1 m in both the eyes. Ophthalmic examination revealed features of retinitis pigmentosa [Figure 3]. Her psychological evaluation showed an IQ of 70.
Figure 1.
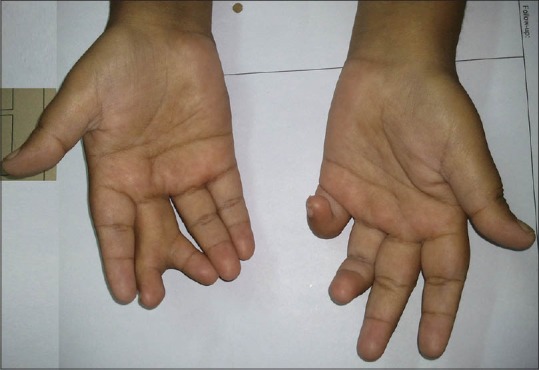
Picture showing central polydactyly in right hand and clinodctyly in left hand
Figure 2.
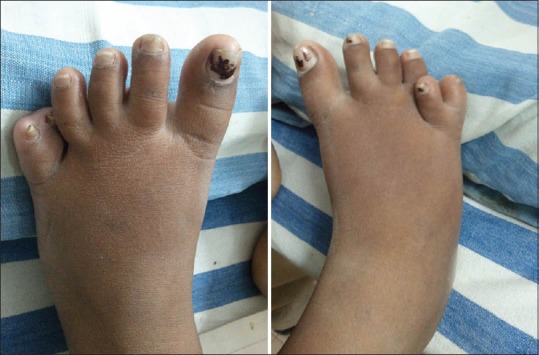
Picture showing post axial polydactyly in both feet
Figure 3.
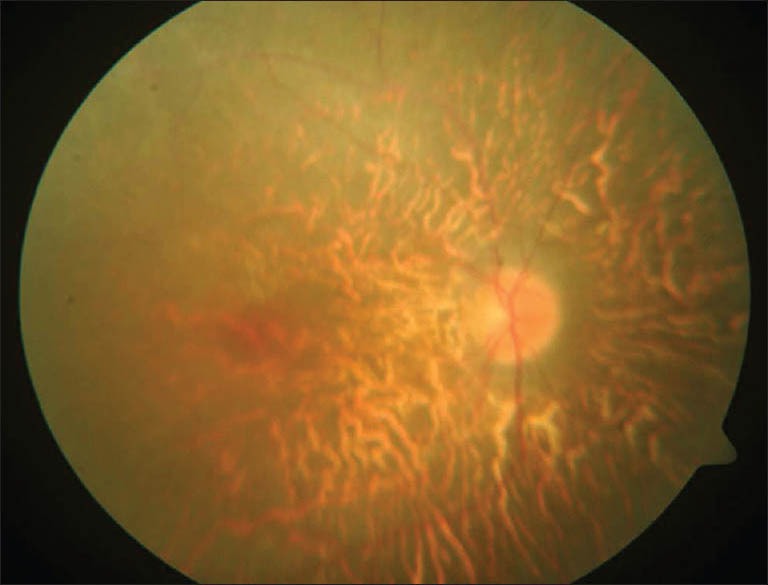
Fundus picture showing retinitis pigmentosa
Investigations revealed hemoglobin - 7.6 gm/dl, urea - 64 mg/dl, creatinine - 10.6 mg/dl, calcium - 7.0 mg/dl, inorganic phosphorus - 5.6 mg/dl, and serum albumin - 3.7 gm/l. Urine analysis revealed trace proteinuria with no active sediments. She had features of secondary hyperparathyroidism with serum alkaline phosphatase of 1157 U/l and intact PTH of 418.3 pg/ml. Her fasting and post prandial blood glucose were 57 and 87 mg/dl respectively. Ultrasound examination showed normal sized kidneys with increased echogenicity. Renal biopsy revealed interstitial fibrosis and tubular atrophy in 75% of the area examined suggestive of chronic interstitial nephritis [Figure 4].
Figure 4.
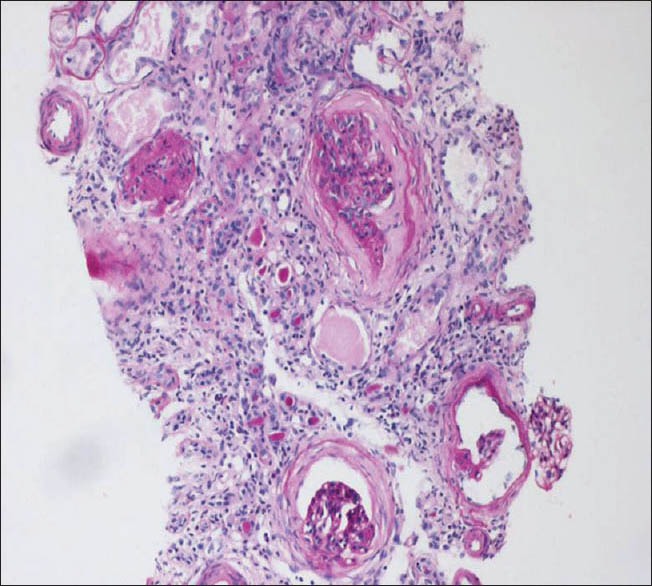
Kidney biopsy (PAS stain) showing features suggestive of chronic interstitial nephritis
The combination of pigmentary retinopathy, polydactyly, mild mental retardation, obesity, and renal failure fits well with the diagnosis of BBS. Patient was initiated on hemodialysis. At present, she is on maintenance hemodialysis in a peripheral dialysis center.
DISCUSSION
Georges Bardet and Arthur Biedl first described the syndrome in 1920. The primary features of this syndrome are early-onset retinal dystrophy, obesity, limb defects, mental retardation, hypogonadism, and renal abnormalities. It occurs throughout the world with prevalence rates of 1:140000–1:160000.[1]
Rod-cone dystrophy is the most common (93%) feature.[2] The onset is often earlier and progression more rapid than in isolated typical retinitis pigmentosa. Truncal obesity is reported to occur in 72% of cases.[2] It usually starts in childhood and severity increases with age.
Postaxial polydactyly is the most common limb defect, though brachydactyly, partial syndactyly, fifth finger clinodactyly, and a prominent gap between the first and second toes were also noted.[2] Mental retardation occurs with varying frequency and severity. Hypogonadism is more common in males while genital abnormalities are more common in females.[2]
The renal abnormalities as a part of this syndrome have been recognized only recently. In a seminal study by Beales et al.,[2] 26 (46%) out of 57 patients had renal structural abnormalities which included renal parenchymal cysts, communicating calyceal cysts, calyceal clubbing and blunting, fetal lobulation and scarring, dysplastic kidneys, unilateral agenesis, vesicoureteric reflux, bladder obstruction, horseshoe kidney, and ectopic kidney. Though only 5% had chronic renal failure, renal failure is the most common cause of mortality in these patients.[3] All the three modalities of renal replacement therapy namely hemodialysis, peritoneal dialysis, and transplantation can be optimally used in these individuals.[4]
The other secondary features are hearing loss, speech disturbances, pigmented naevi, hypertension, diabetes mellitus, congenital heart disease, cardiomyopathy, hepatic fibrosis, nephrogenic diabetes insipidus, hypothyroidism, Hirschsprung's disease and abnormal dentition.[2]
Bardet–Biedl syndrome is genetically heterogenous. Mutations in 16 genes (BBS1 to BBS12, MKS1, NPHP6/CEP290, SDCCAG8, and SEPT7) have been identified to cause the BBS phenotype.[5] BBS has been recently included in the broad category of “ciliopathies”, a class of genetic diseases that occur due to primary ciliary dysfunction. The primary cilium projects from the cell surface of most mammalian cells. Increasing evidence suggests that primary cilia are key coordinators of signaling pathways during development and in tissue homeostasis.[6]
CONCLUSION
To the best of our knowledge, only 13 cases of BBS have been reported from India. Out of the 13 reported cases, only two patients had end stage renal disease (ESRD). Our patient is probably the third with BBS to present with ESRDse. To conclude, BBS is a rare cause of ESRD. Elucidation of the genetics and pathogenesis of this disease may yield novel therapeutic options in the future.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Klein D, Ammann F. The syndrome of Laurence-Moon-Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9:479–513. doi: 10.1016/0022-510x(69)90091-4. [DOI] [PubMed] [Google Scholar]
- 2.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J Med Genet. 1999;36:437–46. [PMC free article] [PubMed] [Google Scholar]
- 3.Rathi M, Ganguli A, Singh SK, Kohli HS, Gupta KL, Sakhuja V, et al. Bardet-Biedl syndrome with end-stage kidney disease: A case report and review of literature. Indian J Nephrol. 2007;17:10–3. [Google Scholar]
- 4.Nordén G, Friman S, Frisenette-Fich C, Persson H, Karlberg I. Renal transplantation in the Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. Nephrol Dial Transplant. 1991;6:982–3. doi: 10.1093/ndt/6.12.982. [DOI] [PubMed] [Google Scholar]
- 5.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–43. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123:499–503. doi: 10.1242/jcs.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]