

Abstract
Phosphodiesterase 4B (PDE4B) is a member of the phosphodiesterase family of proteins that plays a critical role in regulating intracellular levels of cyclic adenosine monophosphate (cAMP) by controlling its rate of degradation. It has been demonstrated that this isoform is involved in the orchestra of events which includes inflammation, schizophrenia, cancers, chronic obstructive pulmonary disease, contractility of the myocardium, and psoriatic arthritis. Phosphodiesterase 4B has constituted an interesting target for drug development. In recent years, a number of PDE4B inhibitors have been developed for their use as therapeutic agents. In this review, an up-to-date status of the inhibitors investigated for the inhibition of PDE4B has been given so that this rich source of structural information of presently known PDE4B inhibitors could be helpful in generating a selective and potent inhibitor of PDE4B.
Keywords: Phosphodiesterases (PDE) enzymes, Cyclic adenosine monophosphate, Selective PDE inhibitors, Chronic obstructive pulmonary disease, Antiproliferative activity, PDE4B
Introduction
Phosphodiesterases (PDEs) are a diverse family of enzymes involved in phosphoric diester hydrolytic cleavage. The term, however, is usually applied to phosphodiesterases that cleave cyclic nucleotides that are important for transmitting signals within the cell. These enzymes are known as cyclic nucleotide phosphodiesterases (PDEs). There are other families of phosphodiesterases, including phospholipases C and D, autotaxin, sphingomyelin phosphodiesterase, DNases, RNases, and restriction endonucleases. Cyclic nucleotide PDEs constitute a superfamily of enzymes which catalyze the hydrolysis of 3',5'-cyclic adenosine monophosphate (cAMP) and 3',5'-cyclic guanosine monophosphate (cGMP) to their inactive 5′-AMP and 5’-GMP forms, respectively. In mammals, PDE enzymes are classified into 11 families, namely PDE1-PDE11. PDE4 is differentiated from other PDEs by its high sensitivity towards the inhibitors, and because of its potential use in the treatment of depression and schizophrenia [1, 2].
PDE4 is the major cAMP-metabolizing enzyme found in inflammatory and immune cells [3, 4]. The PDE4 family encompasses four genes coding the subtypes PDE4A, PDE4B, PDE4C, and PDE4D [5], but the PDE4B subtype is believed to play a central role in inflammation [6], being the predominant subtype in monocytes and neutrophils. All four PDE4 subtypes comprise a related structural organization with a highly conserved catalytic domain in the C-terminal region and upstream-conserved regions in the N-terminal portion of the protein [5]. The high-resolution structures of PDE4B complexes are presented and provide an insight into understanding the substrate and inhibitor binding as well as the critical function of metal ions [7, 8]. PDE4B comprises three domains: an N-terminal regulatory domain, a catalytic domain of about 300 amino acids, and a C-terminal domain. The catalytic domain is the most conserved domain among the PDE families and consists of 17 α-helices. Several reviews on PDE4 have appeared in articles [9–16] covering the role of phosphodiesterase-4 inhibitors in the treatment of asthma, chronic obstructive pulmonary, psoriasis, psoriatic arthritis, chronic inflammatory, autoimmune, and inflammatory bowel diseases. In this review, we present the most significant examples of PDE4B inhibitors that exhibit various biological activities reported in literature.
Selective PDE4B Inhibitors
The design of selective PDE inhibitors started in 1958 when Sutherland and Rall [17] identified the enzymatic activity of phosphodiesterases and its biochemical importance. These cyclic nucleotide levels play an important role in many mammalian physiologies ranging from immune and inflammatory responses [18], regulation of the contractility of the myocardium and smooth muscles [19], to depression and cognition [20]. These centric roles of cAMP initiated research efforts in developing selective inhibitors of PDEs such as vinpocetin for PDE1, inamrinone, milrinone, enaximone for PDE2, rolipram, cilomilast, roflumilast for PDE4, and sildenafil for PDE5 [21]. A pharmacological study in mice proved that PDE4B mediates antipsychotic effects [22, 23] by its role in dopamine-associated and stress-related processes. PDE4B is the only subtype of PDE4 expressed in the locus coeruleus, a region in the brain that is rich in nonadrenergic neurons that mediates some anti-depressant effects [24] and it is the only isoform of PDE4 expressed in the white matter of the brain. PDE4B is also involved in schizophrenia [25] and anxiety [26]. In contrast to PDE4B, PDE4D is expressed in the area postrema and nucleus of the solitary tract [27, 28], which are responsible for emesis.
The first generation the PDE4 inhibitor rolipram was withdrawn for its potent action on the PDE4D isoform, which is likely to be implicated in emesis [30]. PDE4B is abundant in inflammatory, immune, and airway smooth muscle cells [31] and thoroughly investigated for treating inflammatory pulmonary disorders [11]. Further, gene knockout studies proved that PDE4B could suppress TNF-α production [32] and has been investigated as an attractive and excellent therapeutic target due to its sensitivity to selective inhibitors [33–35]. In the year 2004, a high throughput screening resulted in the identification of the lead compound 1a (Figure 1), which exhibited significant PDE4B inhibition (pIC506.8) [36]. A high temperature reaction between N-1-ethyl-5-aminopyrazole and diethyl ethoxymethylenemalonate resulted in the formation of the intermediate 4-hydroxypyrazolopyridine, which upon treatment with phosphoryl chloride gave the 4-chloro-derivative. Treatment of the 4-chloro-derivative with a diverse range of amines yielded 2a–g (Figure 1) via thermal displacement [37]. Structure-activity data showed a significant increase in the potency with a branched or cycloalkyl amino group at fourth position of the pyridine nucleus, with 6-membered saturated rings being more potent than 5-membered rings. Tertiary amines such as pyrrolidine in 1c and the N-methylated analogue such as 1e were less tolerated and suggested the importance of the NH group. These observations were further supported by crystal structures of the pyrazolopyridine analogue 3a (Figure 1) bound to PDE4B 152-503 (PDB ID: 3D3P), which showed the tetrahydropyran moiety towards the metal ions. An intramolecular hydrogen bond between the 4-amino NH group and the carbonyl of the amide was observed, which maintained the co-planarity with the template. Unbranched alkyl amines gave poor selectivity in comparison to the branched analogues, which routinely gave >100-fold selectivity.
Fig. 1.
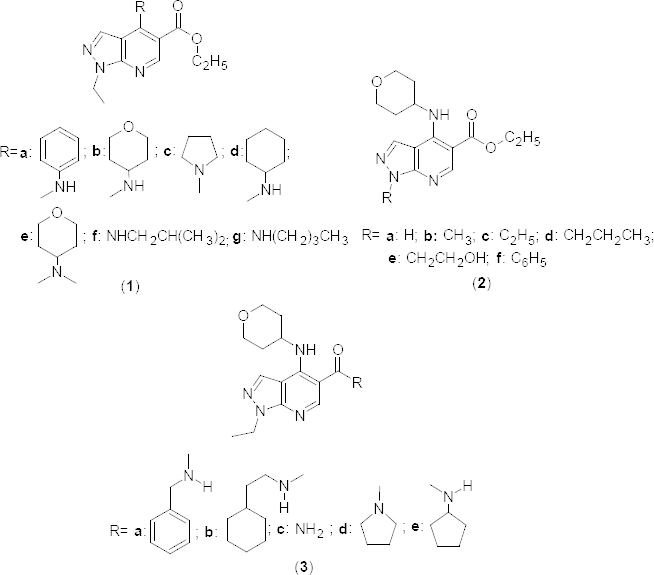
Structure of various congeners of pyrazolopyridine with PDE4B inhibitory activity
The structure-activity relationship is further exemplified by varying the substituent at first position of the pyrazolopyridine ring 2. However, the simple alkyl and aryl substituent on first position did not yield selective PDE4B inhibitors, except for the hydroxyethyl substituent in the pyrazolopyridine derivative 2e, which exhibited a similar pIC50 of 6.8 as that of the lead molecule. Introduction of the amide function at fifth position retained PDE4B selectivity and secondary amides were well-tolerated because of their lipophilic nature, while benzyl amide derivative 3a and saturated analogues such as 3b showed enhanced inhibitory activity on PDE4B with pIC50 values of 8.5 and 8.1, respectively. Primary amide 3c showed significant inhibition (pIC50 6.2), but tertiary amide 3d and cyclic amide 3e exhibited low inhibitory potential towards PDE4B with pIC50 values of 7.3 and 6.8, respectively.
In the year 2009, 2-arylpyrimidine derivative 4a (Figure 2) was identified as a selective and potent PDE4B inhibitor with an IC50 of sub-micromolar range (0.19 µM) and a potential 10-fold selectivity over PDE4D [38]. Authors further investigated the structure-activity relationship to optimize this lead compound which afforded a series of potent PDE4B inhibitors with >100-fold selectivity over the PDE4D isozyme. With a good pharmacokinetic profile, the selected compound exhibited potent anti-inflammatory properties. The compounds bearing allyl (4a), ethyl (4b), cyano (4c), or formyl (4d) groups at fifth position of the pyrimidine ring were found to be equipotent for PDE4B inhibition (pIC50190, 140, 120, and 300 nM, respectively) and 4B/4D selectivity. Bulkier groups at this position reduced the potency in derivatives 4f (PDE4B pIC50 1300 nM), 4g, 4h, and 4j (PDE4B pIC50>1000 nM in all three compounds). Derivative 4k, having an ethyl group at sixth position of the pyrimidine ring, showed enhanced potency (PDE4B pIC50 34 nM), but less selectivity compared with the methyl analog 4a. Authors also explored the SAR at second position of the pyrimidine ring. Potency and selectivity towards PDE4B was retained when the phenyl ring at second position of the pyrimidine ring was substituted with 2-thienyl 5a (Figure 2) (PDE4B pIC50 120 nM) or 3-thienyl 4b (PDE4B pIC50 68 nM) groups. However, substitution of thiazol-4-yl 5c (PDE4B pIC50 2800 nM), 2-pyridyl 5d (PDE4B pIC50 3700 nM), or pyradinyl 5e (PDE4B pIC50 860 nM) groups at the same position markedly reduced the potency. Similar results were observed when the phenyl ring was substituted at second position of the pyrimidine ring and a carboxymethyl group at fourth position of the aminophenyl moiety present at fourth position of the pyrimidine ring (6a) (Figure 2). However, compounds 6c and 6d, possessing a substituted thienyl group at R1 and a carboxymethyl group at second position of pyrimidine ring, showed more than a 25-fold selectivity towards PDE4B. Further modification of the above compounds resulted in compounds 7a–d (Figure 2), which showed potent inhibitory activity on PDE4B (pIC50 15 nM, 6.8 nM, and 15 nM, respectively) and >100-fold selectivity over PDE4D (PDE4B pIC50 1700 nM, 2900 nM, 3100 nM, respectively).
Fig. 2.

Analogues of 2-arylpyrimidines with PDE4B inhibitory activity
In another communication, the discovery of 3-aminocarboxy-4-anilino quinoline 8 (Figure 3) as a potent and selective PDE4 inhibitor (PDE4B pIC50 8.4) is described [39]. Further structural modification of compound 8 has been carried out and the structure-activity relationship of a novel series of quinoline-3-carboxamides 9a–d (Figure 3) at fourth, sixth, and 8-methyl compounds is described by the authors. A further compound, 9d (GSK256066), was identified as a highly potent PDE4 inhibitor suitable for inhaled administration, which also inhibited the LPS-induced production of TNF-α from isolated human peripheral blood mononuclear cells with a pIC50 of 11.1, and negligible oral bioavailability in rats. The crystal structure of GSK256066 bound to PDE4B was also obtained to 1.75 Å resolution (PDB ID: 3GWT).
Fig. 3.
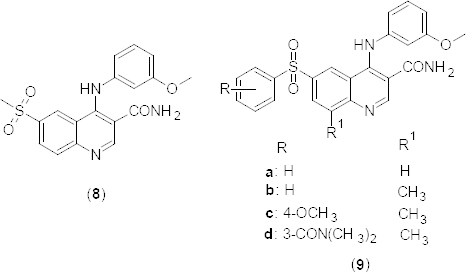
Analogues of 3-aminocarboxy-4-anilinoquinoline with PDE4B inhibitory activity
By high throughput screening, tetrahydrobenzothiophene (THBT) bisamide (10) (Figure 4) was discovered to be a potent and modest PDE4B over the 4D-selective inhibitor (PDE4B/4D pIC50 6.7/6.5) [40]. Authors investigated the HBT binding mode with the ligand/PDE4B catalytic domain of the available co-crystal structure (PBD ID: 3HMV) [41] to design the analogue having PDE4B selectivity over PDE4D. Furthermore, based on the binding mode at the active site of PDE4B, disubstituted-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamides 11 (Figure 4) were synthesized and evaluated for their binding affinity to the PDE4B isoenzyme. Authors disclosed the structure-activity relationship at 3-carboxy and around the tetrahydrobenzo groups. Potencies significantly reduced when the primary amide was methylated and indicated a clear requirement for the primary carboxamide at third position. Structural modification around the tetrahydrobenzo group confirmed the 4B-subtype selectivity and indicated a preference for small C6 substituents. Moreover, the maximum potency and 10-fold selectivity was observed in the compound where R2 = 6-ethyl.
Fig. 4.
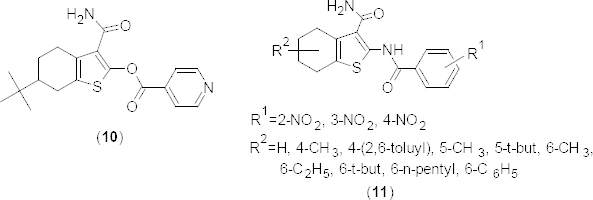
Structures of various congeners of 1-benzothiophene-3-carboxamides with PDE4B inhibitory activity
In search of new compounds to treat inflammation, the pyridazino[4,5-b]indolizine analogue 12a (Figure 5) was found to possess a 23-fold selectivity for PDE4B (Ki 2.60 lM) over PDE4D [42]. In order to improve potency and selectivity, the substituted phenyl analogues 12b–g were prepared and comparable results were obtained from a cell-based adenylyl cyclase functional assay (PDE4B, IC50 13.2 µM; PDE4D, IC50 >100 µM). Interestingly, the fluorine-substituted analogue 12e was found to have high selectivity towards PDE4B compared to the derivative 12a. In N-acetyl piperazine analogue 13 (Figure 6), the electron-withdrawing substituent on the 3-position of benzene significantly increased the PDE4B inhibitory potency (PDE4B Ki 1.90 µM). 1,2-Dimethyl-1H-pyrrolo[2,3-d]pyridazine analogues 14a–d (Figure 6) were synthesized and evaluated for the selective affinity for PDE4B compared to PDE4D. Results revealed that -Cl and -Br substituents, respectively, at second and fourth position of the phenyl ring reduced the PDE4B affinity.
Fig. 5.
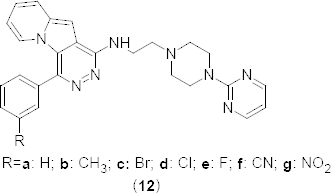
Analogues of pyridazino[4,5-b]indolizine with PDE4B inhibitory activity
Fig. 6.
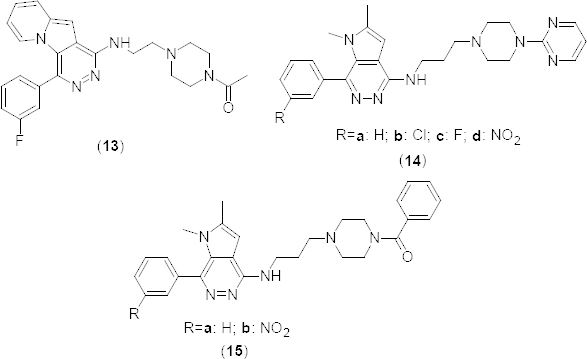
Analogues of 1,2-dimethyl-1H-pyrrolo[2,3-d]pyridazine with PDE4B inhibitory activity
In continuation of the above work [42], two 1,2-dimethyl-1H-pyrrolo[2,3-d]pyridazine analogues, 15a and 15b (Figure 6) have been reported to possess greater affinity as well as selectivity for PDE4B. However, the analogue 15b having the 3-NO2-C6H5 substituent on seventh position of the pyrrolo[2,3-d]pyridazine ring showed poor oral bioavailability in the pharmacokinetic study. On the other hand, unsubstituted analogue 15a exhibited an impressive 205-fold selectivity for PDE4B when compared to PDE4D.
In relation to the development of selective PDE4B inhibitors, a series of novel 1-(arylmethylidyne)-1,2,3,4-tetrahydro-1λ5-quinolines 16a–f (Figure 7), structurally related, respectively, to nimesulide and their 2-oxo analogues and 17a–f (Figure 7), have been designed and synthesized using Sonogashira coupling as a key step [43]. Synthesized compounds were evaluated in vitro for their PDE4B inhibitory activity at 30 µM. Compounds 16c and 17d, having 3-nitrophenyl and 2-chlorophenyl substituents, respectively, on the alkynyl chain attached to the nitrogen of the tetrahydroquinoline ring showed significant PDE4B inhibition (41.64 and 54.37%, respectively). Authors also presented in silico docking study results and interestingly, derivative 17c showed both electrostatic and hydrophobic interactions with the PDE4B enzyme.
Fig. 7.
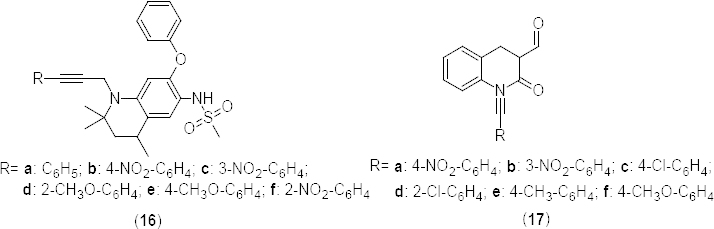
Analogues of 1-(arylmethylidyne)-1,2,3,4-tetrahydro-1λ5-quinoline with PDE4B inhibitory activity
Further investigation established a basis for the development of potent PDE4-selective and dual PDE3/4-selective inhibitors [44]. In a separate communication [45] authors disclosed (−)-6-[7-methoxy-2-(trifluoromethyl)pyrazolo[1,5-a]pyridin-4-yl]-5-methyl-4,5-dihydro-3(2H)-pyridazinone (18) (Figure 8) as a dual PDE3/4 inhibitor (PDE4B IC50 0.47) with potent bronchodilatory and anti-inflammatory activities and an improved therapeutic window over roflumilast in a number of in vitro and in vivo models used for pharmacological profiling. Based on the structure of compound 18 and in continuation of the above work, authors [46] disclosed a novel series of 4,4-dimethylpyrazolones 19a-h (Figure 8) as dual PDE3/4 inhibitors. Potential bicyclic heteroaromatic replacement subunits for the pyrazolo[1,5-a]pyridine core of this series has also been undertaken and the activity of the resulting compounds is described. Synthesized compounds were evaluated to optimize the effect of substituents on seventh position of the pyrazolo[1,5-a]-pyridine nucleus. Compounds 19a-h showed promising in vitro PDE4B inhibition (IC50 0.16, 0.017 µM, 0.41 µM, 0.079 µM, 0.015 µM, 0.035 µM, 0.0034 µM, 0.0075 µM, respectively). Interestingly, the 7-ethyl derivative (19b) was found to be 25-fold more effective as a PDE4 inhibitor than the 7-methoxy analog (19c), revealing that strict catechol ether is not required and indeed potentially detrimental to PDE4-inhibitory activity.
Fig. 8.
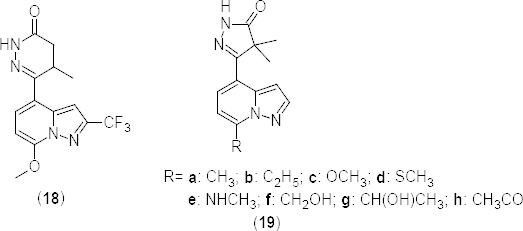
Analogues of pyrazolo[1,5-a]pyridine with PDE4B inhibitory activity
A novel series of 3,5-disubstituted-1,2,4-oxadiazoles 20a-g (Figure 9) has been prepared and evaluated for PDE4B2 inhibitory activity [47]. Among the synthesized compounds, 20a exhibited maximum PDE4B2 inhibition (IC50 5.28 lM). A structure-activity relationship study revealed that the substituents 3-cyclopentyloxy-4-methoxyphenyl group at third position and the cyclic ring bearing heteroatoms at fifth position of the 1,2,4-oxadiazole ring are important for activity. The molecular modeling study showed similar interactions of the 3-cyclopentyloxy-4-methoxyphenyl group compared to the piclamilast. However, the heteroatom ring was found to be slightly deviated compared to piclamilast. Compound 20a exhibited significant analgesic and anti-inflammatory activities in the formalin-induced pain in mice and carrageenan-induced paw edema model in rats.
Fig. 9.
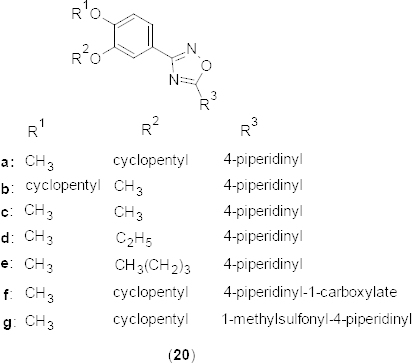
Analogues of 3,5-disubstituted-1,2,4-oxadiazole with promising PDE4B2 inhibitory activity
Inspired by the development of multifunctional drugs, a new series of phthalazine analogues 21 and 22a–d (Figure 10) is described [48] for the treatment of both asthma and COPD, which could target the β2-adrenoceptor and PDE4B2. Synthesized compounds 21 and 22a–d exhibited PDE4B2 IC50 values of 0.520, 0.280, 0.278, 0.257, and 0.251 µM, respectively. Authors disclosed that the hexyl chain linking the hexahydrophthalazinone and N-[5-(2-aminoethyl)-2-hydroxyphenyl]formamide moieties (22d) is optimum for PDE4B2 inhibition. The synthesized compounds were also tested for guinea pig tracheal chain relaxation. Derivative (R)-(22b) showed significant relaxant effects on histamine-induced guinea pig tracheal chain contractions (pEC50 9.3) compared to the reference drug isoprenaline (pEC50 7.5).
Fig. 10.
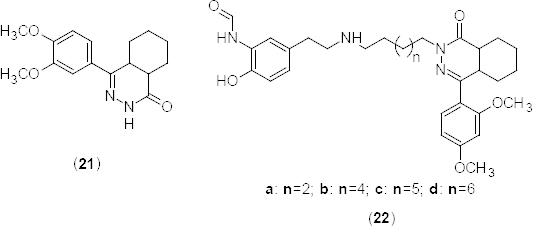
Analogues of hexahydrophthalazinone that have shown promising PDE4B2 inhibitory activity
In a similar work, PDE4B inhibitors bearing formoterol-phthalazinone hybrids were synthesized and evaluated as potent PDE4B2 inhibitors with β-agonist activity [49]. Three derivatives 23a-c (Figure 11) exhibited potent PDE4B2 inhibition (IC50 0.117 µM, 0.118 µM, and 0.104 µM, respectively). These compounds also displayed moderate–to-high β2-adrenoceptor agonist potency on isolated guinea pig tracheal rings pre-contracted with histamine. Compound (R,R)-23c showed the most potent agonist activity with a pEC50 value of approximately 9.0. Moreover, the same compound displayed strong PDE4B2 inhibitory activity with an IC50 of 0.092 µM.
Fig. 11.
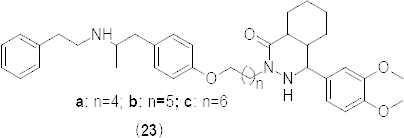
Formoterol–phthalazinone hybrids that have shown promising PDE4B2 inhibitory activity
Ethyl 4,5-disubstituted-4,5-dihydropyrazole-3-carboxylates 24–27 (Figure 12) were synthesized through Pd/C–Cu catalysis as a new class of PDE4B inhibitors [50]. Synthesized compounds were evaluated against the PDE4B enzyme isolated from Sf9 cells and results were compared with the reference compound rolipram, a well-known inhibitor of PDE4. All compounds showed significant inhibition (>70%) of PDE4B when tested at 30 µM. A dose-response study was also carried out using compound 27 which showed dose-dependent inhibition of PDE4B. It is well-known that inhibition of the PDE4D subtype is linked to the emetic response [51]; hence, compounds 24-27 were tested for their PDE4B selectivity over PDE4D. Tested compounds showed 20–30% inhibition at 30 µM indicating their approximately two-fold selectivity towards PDE4B. The in silico docking study result of compound 25 showed the interaction of the hydroxyl function of Tyr233 with pyrazole nitrogen, the π–π stacking interaction between the pyrazole ring and His234, coordinate interaction between the Mg+2 ion and the ester carbonyl function, and coordinate interaction between the Zn+2 ion and pyrazole nitrogen. On the other hand, the π–π stacking interaction between pyrazole and His234 and the coordinate interaction between Mg++ and the ester carbonyl group was observed in compound 26.
Fig. 12.
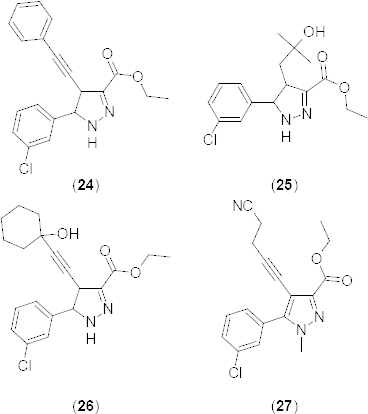
Analogues of ethyl 4,5-disubstituted-4,5-dihydropyrazole-3-carboxylate with PDE4B inhibitory activity
Green synthesis of functionalized cyano pyridines 28a–e (Figure 13) as novel PDE4B inhibitors has been described via the montmorillonite K-10-mediated multi-component reaction in a chemo- and regioselective manner [52]. The synthesized compounds were evaluated for their PDE4B inhibitory potency at 30 µM using rolipram as the standard. The in vitro results revealed that a strong electron donating group such as OCH3 at the para position of the benzene ring in 28a (percentage inhibition 64.10) and 28b (percentage inhibition 65.0) was optimum for the activity compared to other milder electron donating groups such as F or Br (28c–e) (percentage inhibition <45). The di- or tri- substitution on the C-4 benzene ring resulted in the loss of PDE4B inhibitory activity. In silico docking studies showed the binding affinities of compounds 28a and 28c with a docking score of −25.47 and −17.8 kcal/mol, respectively. The cyano group of compound 28c formed H-bonding with HIS234 of the PDE4B isoenzyme. However, it was the OCH3 group in 28a that formed H-bonding with the Glu443 residue of PDE4B. In addition, it also showed an arene-arene interaction with the Phe446 residue.
Fig. 13.
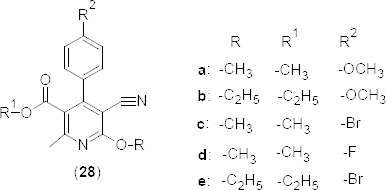
Cyano pyridine derivatives with PDE4B inhibitory activity
1,3,4-oxadiazole and 1,2,4-oxadiazole containing 1-ethyl-1H-pyrazolo[3,4-b]pyridines 29a–d and 30a–d (Figure 14) have been explored as potent PDE4B inhibitors [39] (pIC508.0–8.7 and 7.8–9.4, respectively) taking into account the discovery of 4-(substituted amino)-1-alkyl-pyrazolo[3,4-b]pyridine-5-carboxamides as potent and selective PDE4B inhibitors [37]. The structure-activity relationship of fifth 5-position was studied [53] further. Preliminary structure–activity studies were conducted against isolated PDE4B, the predominant PDE4 subtype in inflammatory cells of interest. Results indicated that both oxadiazole isomers were tolerated at fifth position. Different oxadiazole substituents showed significant potency in each series, but unfortunately no structure-activity relationship was established. Further di-substituted and mono-substituted oxazoles 31a and 31b (Figure 14) were also synthesized. The di-substituted oxazole 31a demonstrated sub-nM activity in the isolated enzyme and isolated human peripheral blood mononuclear cells (PBMC) assay and gave at least a 10-fold increase in potency over 1,3,4-oxadiazole 13c and 1,2,4-oxadiazole 29b. Moreover, in compound 31a, the introduction of a second substituent showed an increase in potency in comparison to the mono-substituted oxazole 31b.
Fig. 14.
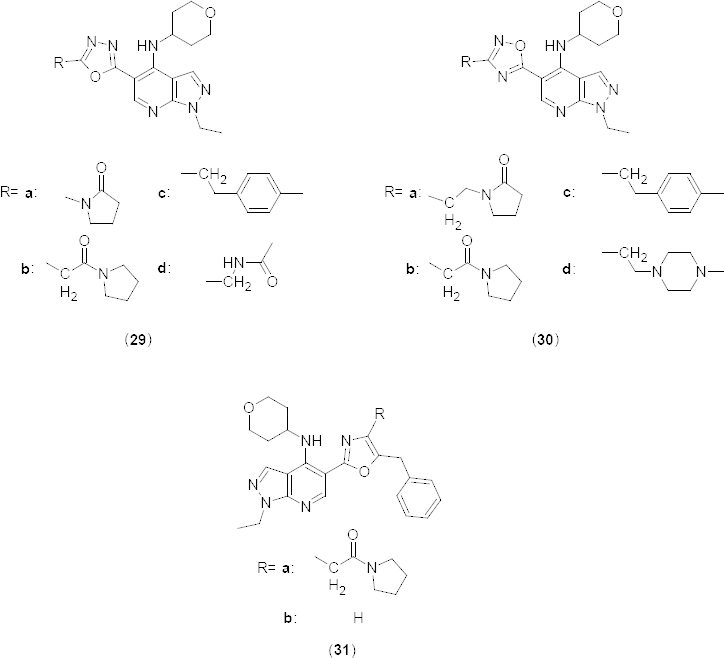
1,3,4-oxadiazole and 1,2,4-oxadiazole containing 1-ethyl-1H-pyrazolo[3,4-b]-pyridines with PDE4B inhibitory activity
In the year 2013, compound 32 (Figure 15) was identified as an orally active PDE4B-selective inhibitor over PDE4D both in humans (80-fold selective) and mice (29-fold selective) [54]. The therapeutic effect of compound 32 was evaluated on lipopolysaccaride (LPS) injection–induced plasma TNF-α elevation and on LPS inhalation-induced pulmonary neutrophilia in mice. The therapeutic index for TNF-α production (TITNF = ID50 in gastric emptying/ID50 in LPS injection-induced plasma TNF-α elevation) of compound 32 was larger than roflumilast (9.0 and 0.2, respectively), whereas the therapeutic index for pulmonary neutrophilia of compound 32 was comparable to roflumilast (1.0 and 0.5, respectively). Authors disclosed that the TITNF of compound 32 was not superior compared to that of roflumilast in spite of its high selectivity for PDE4B over PDE4D in mice.
Fig. 15.
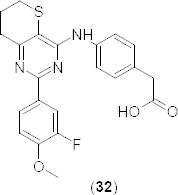
Structure of 7,8-dihydro-6H-thiopyrano[3,2-d]pyrimidine analogue, an orally active PDE4B-selective inhibitor
In a separate communication, 2-phenylpyrimidine 33 and 5-carbamoyl-2-phenylpyrimidine 34 derivatives (Figure 16) were reported as potent PDE4B (IC50 25 nM and 200 nM, respectively) and mTNF-α (IC50 390 and 690 nM, respectively) inhibitors [55]. Compounds were also evaluated in vivo against LPS-induced pulmonary neutrophilia in mice. In compound 34, substitution of methyl (35a) (PDE4B IC50 4000 nM) or methylthio (35b) (PDE4B IC50 2000 nM) groups (Figure 16) on sixth position of the pyrimidine ring markedly reduced the PDE4B inhibitory activity. Moreover, substitution of N-methylacetamide (36a) (PDE4B IC50 8200 nM), N,N-dimethylacetamide (36b) (PDE4B IC50 8% at 10 µM), and N-hydroxyacetamide (36c) (PDE4B IC50 42% at 3 µM) groups (Figure 16) on fifth position of the pyrimidine ring resulted in a marked decrease in potency, whereas derivative 37 (Figure 16) with the N-(2,2-dimethylpropyl)acetamide substituent on fourth position of the phenyl ring exhibited maximum in vitro inhibitory activities against PDE4B (IC50 8.3 nM) and TNF-α (IC50 3.0 nM). The 5-carbamoyl moiety of compound 37 exhibited a water-bridged hydrogen bonding network with Asn395 and Gln443 residues in an X-ray crystallography study. In derivatives 36a–c, substituents hindered these interactions and this may be the reason for the lowered potency. The higher potency of 37 is further supported by the lengthy neopentyl substituent which fits into the lipophilic pocket.
Fig. 16.
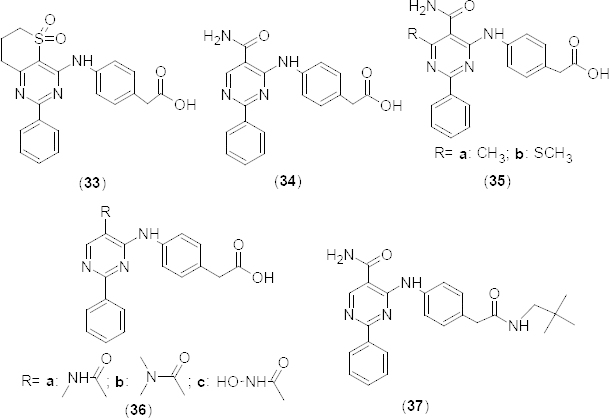
Structure of 2-phenylpyrimidine analogues showing selective PDE4B inhibitory activity
A novel series of spirooxindoles have been synthesized in the one-pot efficient methodology using Cu-mediated 1,3-dipolar cycloaddition of azomethineylides with dipolarophiles and evaluated for in vitro inhibition of PDE4B [56] using rolipram as a reference compound. The structure-activity relationship study revealed compound 38 (Figure 17) as a potent PDE4B inhibitor with >40% inhibition at 30 µM. In the in silico docking result, compound 38 C=O function showed two hydrogens bonding with His278 and Met347 residues of the active site. Additionally, hydrophobic interactions with the hydrophobic clamp residue of the Q-pocket were also observed.
Fig. 17.

Structure of spirooxindole that showed promising PDE4B inhibitory activity
One-pot synthesis of benzofuran fused N-heterocycles 39 and 40a–g (Figure 18) has been achieved by AlCl3-mediated C–C followed by C–O bond formation between 2,3-dichloropyrazine or its derivatives and phenols. Synthesized compounds were tested in vitro for their PDE4B inhibitory activity [57]. Compound 39 showed significant PDE4B inhibition at 30 µM in comparison to the reference compound rolipram. Compounds possessing a substituent on the benzofuran moiety showed moderate activities and the presence of a smaller substituent was well-tolerated compared to a larger or bulky group in terms of PDE4 inhibition.
Fig. 18.
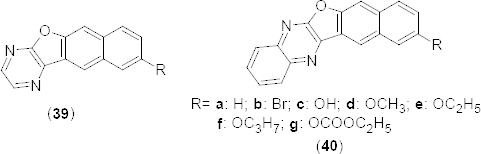
Structure of benzofuran analogues that showed promising PDE4B inhibitory activity
A number of novel imidazophenoxazine-4-sulfonamides 41a-c (Figure 19) have been designed as potential inhibitors of PDE4. Designed compounds were synthesized in a multi-step reaction process involving the construction of a 1-nitro-10H-phenoxazine ring and then fusion with an imidazole ring as the key steps [58]. Some of these compounds showed promising in vitro PDE4B and D inhibition when tested. Compounds bearing 2-hydroxyphenyl (41a), 2-bromophenyl (41b), and 5-bromo-2-fluorophenyl (41c) exhibited dose-dependent inhibition of PDE4B with IC50 values of 3.31 ± 0.62, 1.23 ± 0.18, and 0.53 ± 0.18 µM, respectively. The interaction of compound 41a with the PDE4B was mainly contributed by H-bonding between the amino group of 41a with His-278,H-bonding between the hydroxyl group of 41a and Asp392, and two π–π stacking interactions between the benzimidazole moiety and His234. Similarly, the interaction of compound 41b with the PDE4B was contributed by H-bonding between the NH2-group of 41b and Thr345, as well as Asp392, π–π stacking interactions between the central 1,4-oxazine ring and Tyr233, and π–π stacking interactions between the phenyl group of 41b and Phe446. In compound 41c, H-bonding between the amino group and Asp275 and π–π stacking interactions between the aromatic ring system and Tyr233, His234, and Phe446 residues of PDE4B was observed.
Fig. 19.
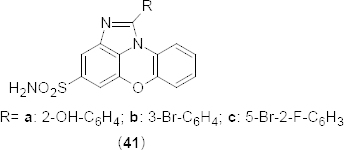
Novel series of imidazophenoxazine-4-sulfonamides that have shown promising PDE4B inhibitory activity
Yet in another publication [59], authors explored the effect of N-alkylation of the pyridazinone ring in compound 18 (Figure 8) on PDE4B inhibition and selectivity. N-alkylation of the pyridazinone ring resulted in a marked enhancement of potency against PDE4, but suppressed PDE3 inhibition. Addition of a 6-aryl-4,5-dihydropyridazin-3(2H)-one extension to the N-alkyl group in derivatives 42a–f (Figure 20) facilitated both the enhancement of PDE4-inhibitory activity and the restoration of potent PDE3 inhibition. These modifications of structural changes afforded potent dual PDE3/4 inhibitors and most of the synthesized compound suppressed histamine-induced bronchoconstriction in vivo and exhibited promising anti-inflammatory activity via intratracheal administration.
Fig. 20.
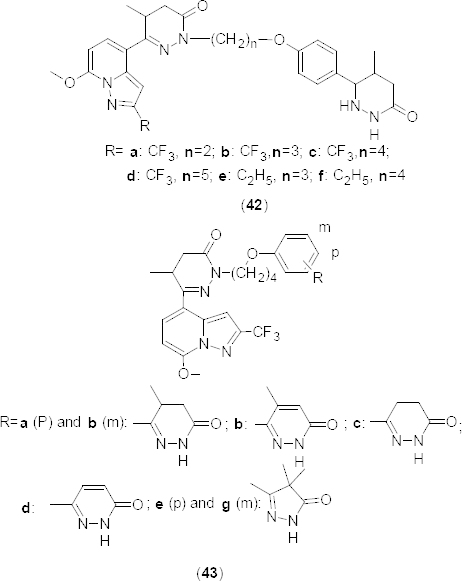
Novel series of N-alkylated pyridazinones that have shown selective PDE4B inhibitory activity
The selectivity towards PDE4B increased by 23- to 350-fold in the trifluoromethyl substituted series 42a–d (IC50 0.66, 3, 0.3, 0.2 nM, respectively) compared to the lead compound 18 (PDE4B IC50 70 nM), whereas the ethyl substituted derivatives 42e and 42f exhibited a lower PDE4B selectivity than its congeners (IC50 5.9 and 1.7 nM, respectively). Considering the variations in selectivity of compounds possessing tetramethylene (42c) and pentamethylene (42d), linkers were further explored for the structure-activity relationship with 5-methyl-3,4-dihydropyridazinone and 4,4-dimethylpyrazolone subunits (43a–g) (Figure 20). In vitro screening results revealed a reduced PDE3A selectivity with moderate inhibition of PDE4B in compounds 43b and 43d (PDE4B IC50 6 nM and 20 nM, respectively) having a fully unsaturated pyridazanone nucleus [59]. Varying the 5-methyl-3,4-dihydropyridazinone ring from fourth position in 43a (PDE4B IC50 0.3 nM) to third position in 43b (PDE4B IC50 0.5 nM) retained the selectivity for PDE4B at the cost of PDE43A selectivity. Based upon the in vitro results, compounds having 4,5-dihydropyridazin-3(2H)-one (43c, PDE4B IC50 0.1 nM) and 4,4-dimethyl-1H-pyrazol-5(4H)-one (43f, PDE4B IC50 0.5 nM) rings were found to be promising in PDE4B inhibition.
Compound 44 (Figure 21) was discovered as a virtual hit in a docking study where the benzoxazinone moiety carbonyl function formed a H-bond with His234 of the metal binding pocket and the N-methanesulfonyl oxygen formed a H-bond with Gln443 of the Q pocket in the active site. These interactions were similar to that of the known inhibitor rolipram [60]. 2H-1,3-benzoxazin-4(3H)-one derivatives 45a-d and 46 (Figure 21) containing respectively 1-(methylsulfonyl)-1H-indole or benzofuran moieties were synthesized by Pd/C–Cu-mediated coupling-cyclization. The o-iodoanilides or o-iodophenol were coupled with 3-{2-(prop-2-ynyloxy)ethyl}-2H-benzo[e][1,3]oxazin-4(3H)-one using 10% Pd/C–CuI–PPh3 as the catalyst and triethylamine as the base. Synthesized compounds were tested in vitro for their PDE4B inhibitory potential using a cell-based cAMP reporter assay. The tested compounds exhibited potent inhibition of PDE4B at 30 µM. Compound 44 showed a fivefold increase over the control, whereas rolipram showed a nine-fold increase. In the dose-response curve, compound 44 also showed a dose-dependent increase in cAMP levels.
Fig. 21.
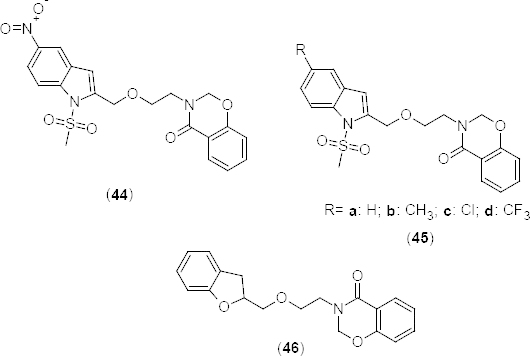
Structure of benzoxazinone derivatives that have shown potent PDE4B inhibitory activity
In search of highly selective PDE4B inhibitors, a novel series of 5,5-dioxo-7,8-dihydro-6H-thiopyrano[3,2-d]pyrimidines were synthesized and evaluated for their PDE4B subtype selectivity using human PDE4B2 and PDE4D2 full-length enzymes [61]. The molecules were optimized by varying the substituents on the pyrimidine ring and the phenyl ring present over the side chain. The in vitro screening of the synthesized compounds resulted in the identification of 2-(3-chloro-4-methoxyphenyl)-5,5-dioxo-7,8-dihydro-6H-thiopyrano[3,2-d]pyrimidine (47) (Figure 22) as a highly selective PDE4B inhibitor, which showed PDE4B inhibitory activity with an IC50 value of 3.0 nM and 433-fold PDE4B selectivity over PDE4D.
Fig. 22.
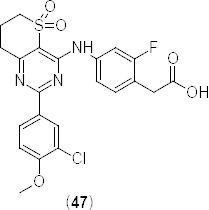
Structure of the 5,5-dioxo-7,8-dihydro-6H-thiopyrano[3,2-d]pyrimidine analogue that has shown highly selective PDE4B inhibition
Selective PDE4 inhibitors exhibit antiproliferative activity against T-cells and B-cells and this finding helped researchers to investigate the selective PDE4 inhibitors as novel anticancer agents. This was further supported by the antiproliferative activity of PDE4 inhibitors against murine carcinoma cells [62]. PDE4 inhibitor rolipram induced cell cycle arrest and apoptosis [63], and overexpression of PDE4B resulted in diffuse large cell lymphoma (DLBCL); a fatal malignancy indicating that PDE4B inhibitors could be useful anticancer agents [64]. Moreover, PDE4B selective inhibitors were reported for selective apoptosis in malignant cells without affecting the normal cells. Based on these facts, novel PDE4B inhibitors were developed and screened for anticancer activity [65].
In the year 2013, novel N-[(1-substituted-1H-1,2,3-triazol-4-yl)methyl]-N-2,2,4-trimethyl-7-phenoxy-1,2-dihydroquinolinemethanesulfonamides 48a–e (Figure 23) were synthesized as selective PDE4B inhibitors. The reaction was carried out in aqueous DMF via a multi-step sequence consisting of copper-catalyzed azide-alkyne cycloaddition (CuAAC) as the key step [66]. The compounds were evaluated in vitro for their cytotoxic activity using the lung adenocarcinoma epithelial cell line (A549), prostate cancer cell line (DU145), cervical cancer cell line (HeLa), and hepatocellular liver carcinoma cell line (HepG2) in an MTT assay. Among the synthesized inhibitors, 48a and 48b showed promising activity against the human lung cancer cell line (A549) with an IC50 of 8–9 µM. Although compounds 48b, 48d, and 48e were identified as potent PDE4B inhibitors, they showed no cytotoxic activity towards the tested cancer cell lines. The docking results of the synthesized compounds revealed a high binding affinity of compound 48d with 1XMY (PBD ID). Hydrogen bonding with HIE-234 and π-π stacking interactions with PHE446 residues were also observed.
Fig. 23.
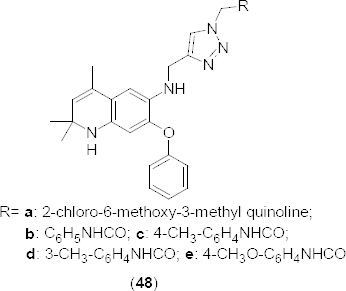
Structure of 1,2-dihydroquinolinemethanesulfonamide analogues that have shown potent PDE4B inhibition and anticancer activity
The novel nimesulide-based new class of triazole derivatives 49a–d (Figure 24) were synthesized as PDE4B inhibitors [67]. The synthesized compounds were tested against cancer cells, keeping in view the reported anticancer activity of nimesulide [68] and the anti-inflammatory activity of 1,2,4-triazoles [69]. Some of the synthesized compounds exhibited significant in vitro PDE4B inhibitory properties with >50% inhibition at 30 µM. Moreover, results were also supported by the docking studies. The in vitro anticancer activity was evaluated using the HCT-15 human colon cancer cell lines with doxorubicin [IC50 50 µg/mL, (0.09 µM)], an anthracyclin antibiotic as the reference compound. Two of the synthesized compounds 49c and 49d exhibited IC50 values in the range 21-22 µg/mL. Docking results of synthesized compounds revealed similar interactions with Gln443 as that of rolipram. Apart from Gln443, compound 49a also exhibited interactions with His234 and His278, whereas compound 49b and 49c showed binding affinity for Asn283.
Fig. 24.
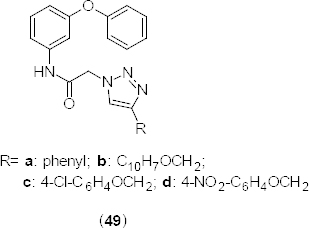
Structure of 1,2,4-triazole analogues that have PDE4B inhibition and anticancer activity
During cardiac action potential, Ca2+ influx through the sarcolemma L-type Ca2+ channels (LTCC) triggers Ca2+ release from ryanodine receptor 2 located in the sarcoplasmic reticulum; this results in elevated Ca2+ throughout the cell leading to contraction of the myocardium. PDE4 is one of the main PDEs expressed in the heart. PDE4 becomes predominantly active upon β-adrenergic receptor stimulation and regulates LTCC and cAMP levels in cardiomyocytes [70–75]. In PDE4B-deficient mice, increased expression of the β-adrenoreceptor along with LTCC was observed leading to elevated intracellular Ca2+ and cell contraction [76].
In another study, a novel series of 1,4-dihydropyridine-based PDE4B inhibitors were synthesized [77] and evaluated for their in vitro Ca2+ channel blocking and PDE4B inhibitory activities. Compounds 50a-g (Figure 25) exhibited >80% inhibition of PDE4B. Among the synthesized inhibitors, 50d, having an indaloyl moiety, exhibited significant selectivity towards PDE4B with a docking score of −8.1 kcal/mol. The in silico docking result of compound 50d supported the reason for the selectivity, where the indole moiety exists in two different orientations in PDE4D, and in both cases, it makes a stronger hydrogen bond with Glu396 or Glu505 residues of PDE4D, while its orientation in PDE4B is completely different than that in PDE4D. The dimethoxy group of 50d interacted with the metal atoms in PDE4B, while the indole moiety was shown to be H-bonded with the Ser442 backbone.
Fig. 25.
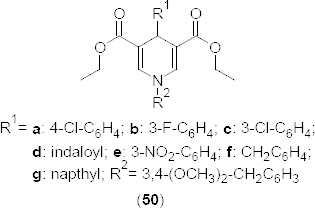
Structure of 1,4-dihydropyridine analogues that have Ca2+ channel blocking and
In recent research [78], authors disclosed that PDE4B selectivity can be achieved by the capture of a C-terminal regulatory helix, known as CR3 (control region 3), across the active site in a conformation that closes access by cAMP. PDE4B selectivity is driven by a single amino acid polymorphism in CR3 (Leu674 in PDE4B1 versus Gln594 in PDE4D). A new co-crystal structure with a bound ligand was developed which provided a guide map for the design of PDE4B-selective anti-inflammatory drugs. The comparative binding modes of the 2-arylpyrimidine derivative with PDE4D (PDB: 3G58), NVW-PDE4B (PDB: 3W5E), and OCP-PDE4B (PDB: 3KKT) structures showed that small molecules can interact with different residues of the CR3 helix resulting in multiple closed conformations. The study highlighted the significance of (PDE4B Leu674/PDE4DGln594 and PDE4B Met675/PDE4D Thr595) that is essential to engage the ligand and/or catalytic domain.
Conclusion
Advancements in PDE4B selective-targeted therapies have shown promise in recent years for the treatment of inflammation, chronic obstructive pulmonary disorders, cancers, and myocardium contractility disorder. After the development of the prototypic PDE4 inhibitor rolipram, more selective inhibitors targeting the PDE4B isozyme have been widely investigated. However, the biological activity of this class of compounds deserves further investigation. This is evident when inflammatory diseases are considered. Although the research on this subject is incipient, the number of reports disclosing the effects of PDE4B inhibitors on TNF-α production and Ca2+ channel blockage has recently been increasing. PDE4B selective inhibitors have been shown to be promising, which calls for the design of more efficient anti-inflammatory, anticancer, and cardio-protective agents. Although some patterns appear relating the structure and the pharmacological field, this also should be considered when designing new selective PDE4B inhibitors.
Acknowledgement
The authors are grateful to the Science and Engineering Research Board (SERB), Government of India for the financial support (No. SR/S0/HS-0264/2012).
Glossary
Abbreviations
- PDE
phosphodiesterases;
- cAMP
cyclic adenosine monophosphate;
- cGMP
cyclic guanosine monophosphate;
- pIC50
negative logarithm of the half maximal inhibitory concentration;
- COPD
chronic obstructive pulmonary disease;
- TNF
tumor necrosis factor;
- nM
nanomol;
- PDB
protein data bank;
- IC50
half maximal inhibitory concentration;
- pEC50
negative logarithm of half maximal effective concentration;
- µM
micromol;
- LPS
lipopolysaccharide;
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;
- LTCC
sarcolemma L-type Ca2+ channels;o
- SAR
structure-activity relationship;
- CR3
control region 3;
- NVW
N-tert-butyl-2-{4-[(5,5-dioxido-2-phenyl-7,8-dihydro-6H-thiopyrano[3,2-d]-pyrimidin-4-yl)amino]phenyl}acetamide;
- OCP
5-{3-[(1S,2S,4R)-bicyclo[2.2.1]hept-2-yloxy]-4-methoxyphenyl}-tetrahydropyrimidin-2(1H)-one;
Authors’ Statement
Competing Interests
The authors declare no conflict of interest.
References
- 1.O’Donnell JM, Zhang HT. Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4). Trends Pharmacol Sci. 2004;25:158–163. http://dx.doi.org/10.1016/j.tips.2004.01.003 [DOI] [PubMed] [Google Scholar]
- 2.Kanes SJ, Tokarczyk J, Siegel SJ, Bilker W, Abel T, Kelly MP. Rolipram: a specific phosphodiesterase 4 inhibitor with potential antipsychotic activity. Neuroscience. 2007;144:239–246. http://dx.doi.org/10.1016/j.neuroscience.2006.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumer W, Hoppmann J, Rundfeldt C, Kietzmann M. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets. 2007;6:17-26. http://dx.doi.org/10.2174/187152807780077318 [DOI] [PubMed] [Google Scholar]
- 4.Souness JE, Aldous D, Sargent C. Immunosuppressive and anti-inflammatory effects of cyclic AMP phosphodiesterase (PDE) type 4 inhibitors. Immunopharmacology. 2000;47:127–162. http://dx.doi.org/10.1016/S0162-3109(00)00185-5 [DOI] [PubMed] [Google Scholar]
- 5.Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278:5493–5496. http://dx.doi.org/10.1074/jbc.R200029200 [DOI] [PubMed] [Google Scholar]
- 6.Wang P, Ohleth KM, Wu P, Billah MM, Egan RW. Phosphodiesterase 4B2 is the predominant phosphodiesterase species and undergoes differential regulation of gene expression in human monocytes and neutrophils. Mol Pharmacol. 1999;56:170–174. http://www.ncbi.nlm.nih.gov/pubmed/10385698 [DOI] [PubMed] [Google Scholar]
- 7.Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Nolte RT. Crystal Structures of the Catalytic Domain of Phosphodiesterase 4B Complexed with AMP, 8-Br-AMP, and Rolipram. J Mol Biol. 2004;337:355–365. http://dx.doi.org/10.1016/j.jmb.2004.01.040 [DOI] [PubMed] [Google Scholar]
- 8.Xu RX, Hassell AM, Vanderwall D, Lambert MH, Holmes WD, Luther MA, Rocque WJ, Milburn MV, Zhao Y, Ke H, Nolte RT. Atomic structure of PDE4: insights into phosphodiesterase mechanism and specificity. Science. 2000;288:1822–1825. http://dx.doi.org/10.1126/science.288.5472.1822 [DOI] [PubMed] [Google Scholar]
- 9.Sanz MJ, Cortijo J, Morcillo EJ. PDE4 inhibitors as new anti-inflammatory drugs: effects on cell trafficking and cell adhesion molecules expression. Pharmacol Ther. 2005;106:269–297. http://dx.doi.org/10.1016/j.pharmthera.2004.12.001 [DOI] [PubMed] [Google Scholar]
- 10.Lipworth BJ. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet. 2005;8–14:365:167–175. http://dx.doi.org/10.1016/S0140-6736(05)17708-3 [DOI] [PubMed] [Google Scholar]
- 11.Fan CK. Phosphodiesterase inhibitors in airways disease. Eur J Pharmacol. 2006;533:110–117. http://dx.doi.org/10.1016/j.ejphar.2005.12.059 [DOI] [PubMed] [Google Scholar]
- 12.Keshavarzian A, Mutlu E, Guzman JP, Forsyth C, Banan A. Phosphodiesterase 4 inhibitors and inflammatory bowel disease: emerging therapies in inflammatory bowel disease. Expert Opin Investig Drugs. 2007;16:1489–1506. http://dx.doi.org/10.1517/13543784.16.9.1489 [DOI] [PubMed] [Google Scholar]
- 13.Page CP, Spina D. Selective PDE inhibitors as novel treatments for respiratory diseases. Curr Opin Pharmacol. 2012;12:275–286. http://dx.doi.org/10.1016/j.coph.2012.02.016 [DOI] [PubMed] [Google Scholar]
- 14.Jin SL, Ding SL, Lin SC. Phosphodiesterase 4 and Its Inhibitors in Inflammatory Diseases. Chang Gung Med J. 2012;35:197–210. http://www.ncbi.nlm.nih.gov/pubmed/22735051 [DOI] [PubMed] [Google Scholar]
- 15.Wittmann M, Helliwell PS. Phosphodiesterase 4 Inhibition in the Treatment of Psoriasis, Psoriatic Arthritis and Other Chronic Inflammatory Diseases. Dermatol Therap. 2013;3:1–15. http://dx.doi.org/10.1007/s13555-013-0023-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar N, Goldminz AM, Kim N, Gottlieb AB. Phosphodiesterase 4-targeted treatments for autoimmune diseases. BMC Medicine. 2013;11:96 http://dx.doi.org/10.1186/1741-7015-11-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem. 1958;232:1077–1091. http://www.ncbi.nlm.nih.gov/pubmed/13549488 [PubMed] [Google Scholar]
- 18.Schett G, Sloan VS, Stevens RM, Schafer P. Apremilast: A Novel PDE4 inhibitor in the treatment of autoimmune and inflammatory diseases. Ther Adv Musculoskelet Dis. 2010;2:271–278. http://dx.doi.org/10.1177/1759720X10381432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Page CP, Spina D. Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handbook Exp Pharmacol. 2011;204:391–414. http://dx.doi.org/10.1007/978-3-642-17969-3_17 [DOI] [PubMed] [Google Scholar]
- 20.Uretsky BF, Generalovich T, Reddy PS, Spangenberg RB, Follansbee WP. The acute hemodynamic effects of a new agent, MDL 17,043, in the treatment of congestive heart failure. Circulation. 1983;67:823–828. http://www.ncbi.nlm.nih.gov/pubmed/6218939 [DOI] [PubMed] [Google Scholar]
- 21.Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am J Respir Crit Care Med. 1998;157:351–370. http://dx.doi.org/10.1164/ajrccm.157.2.9708012 [DOI] [PubMed] [Google Scholar]
- 22.Siuciak JA, Chapin DS, McCarthy SA, Martin AN. Antipsychotic profile of rolipram: efficacy in rats and reduced sensitivity in mice deficient in the phosphodiesterase-4B (PDE4B) enzyme. Psychopharmacology (Berl). 2007;192:415–424. http://dx.doi.org/10.1007/s00213-007-0727-x [DOI] [PubMed] [Google Scholar]
- 23.Siuciak JA, McCarthy SA, Chapin DS, Martin AN. Behavioural and neurochemical characterization of mice deficient in the phosphodiesterase-4B (PDE4B) enzyme. Psychopharmacology (Berl). 2008;197:115–126. http://dx.doi.org/10.1007/s00213-007-1014-6 [DOI] [PubMed] [Google Scholar]
- 24.Cherry JA, Davis RL. Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J Comp Neurol. 1999;407:287–301. http://dx.doi.org/10.1002/(SICI)1096-9861(19990503)407:2%3C287::AID-CNE9%3E3.0.CO;2-R [PubMed] [Google Scholar]
- 25.Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ, Porteous DJ. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signalling. Science. 2005;310:1187–1191. http://dx.doi.org/10.1126/science.1112915 [DOI] [PubMed] [Google Scholar]
- 26.Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, Dlaboga D, Jin SL, Conti M, O'Donnell JM. Anxiogenic like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology. 2008;33:1611–1623. http://dx.doi.org/10.1038/sj.npp.1301537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres PS, Miro X, Palacios JM, Cortes R, Puigdomenech P, Mengod G. Phosphodiesterase type 4 isozymes expression in human brain examined by in-Situ hybridization histochemistry and[3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J Chem Neuroanat. 2000;20:349–374. http://dx.doi.org/10.1016/S0891-0618(00)00097-1 [DOI] [PubMed] [Google Scholar]
- 28.Lamontagne S, Meadows E, Luk P, Normandin D, Muise E, Bou-let L, Pon DJ, Robichaud A, Robertson GS, Metters KM, Nantel F. Localization of phosphodiesterase-4 isoforms in the medulla and nodose ganglion of the squirrel monkey. Brain Res. 2001;920:84–96. http://dx.doi.org/10.1016/S0006-8993(01)03023-2 [DOI] [PubMed] [Google Scholar]
- 29.Ariumi H, Saito R, Nago S, Hyakusoku M, Takano Y, Kamiya H. The role of tachykinin NK-1 receptors in the area postrema of ferrets in emesis. Neurosci Lett. 2000;286:123–126. http://dx.doi.org/10.1016/S0304-3940(00)01113-7 [DOI] [PubMed] [Google Scholar]
- 30.Heaslip RJ, Evans DY. Emetic, central nervous system, and pulmonary activities of rolipram in the dog. Eur J Pharmacol. 1995;286:281–290. http://dx.doi.org/10.1016/0014-2999(95)00457-2 [DOI] [PubMed] [Google Scholar]
- 31.Yuasa K, Kanoh Y, Okumura K, Omori K. Genomic organization of the human phosphodiesterase PDE11A gene. Evolutionary relatedness with other PDEs containing GAF domains. Eur J Biochem. 2001;268:168–178. http://dx.doi.org/10.1046/j.1432-1327.2001.01866.x [DOI] [PubMed] [Google Scholar]
- 32.Jin SL, Conti M. Induction of the cyclic nucleotide Phosphodiesterase PDE4B is essential for LPS-activated TNF-α response. Proc Natl Acad Sci U S A. 2002;99:7628–7633. http://dx.doi.org/10.1073/pnas.122041599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prabha NI, Bremer R, Gillette S, Chao H, Nepsi M, Mamo S, Zhang C, Artis DR, Lee B, Zuckerman R. PDE4B inhibitors. US patent 7605168B2, Oct., 2009. Application No. 11/219635, Sept. 2005.
- 34.Prabha NI, Cho, Hanna C, Bruce E, Sam G, Dean RA, Chao RZ. PDE4B inhibitors and uses therefor. US Patent 7585859B2, Sept. 2009. Application No. 11/123893, May, 2005.
- 35.Prabha NI, England B, Gillette S, Artis DR, Zuckerman R, Zhang C, Chao RZ. PDE4B inhibitors and uses therefor. US Patent 8470821, June 2013. Application No. 12/506222, July, 2009.
- 36.Allen DG, Coe DM, Cook CM, Dowle MD, David Edlin CD, Hamblin JN, Johnson MR, Jones PS, Knowles RG, Lindvall MK, Mitchell CJ, Redgrave AJ, Trivedi N, Ward P. Pyrazolo[3,4-b]pyridine compounds, and their use as phosphodiesterase inhibitors. WO2004024728 A3, October 21, 2004;Application No. PCT/EP2003/011814, Sep. 12, 2003.
- 38.Hamblin JN, Angell TDR, Ballantine SP, Cook CM, Cooper AWJ, Dawson J, Delves CJ, Jhones PS, Lindvall M, Lucas FS, Mitchell CJ, Neu MY, Ranshaw LE, Solanke YE, Somers DO, Wiseman JO. Pyrazolopyridines as a novel structural class of potent and selective PDE4 inhibitors. Bioorg Med Chem Lett. 2008;18:4237–4241. http://dx.doi.org/10.1016/j.bmcl.2008.05.052 [DOI] [PubMed] [Google Scholar]
- 39.Naganuma K, Omura A, Maekawara N, Saitoh M, Ohkawa N, Kubota T, Nagumo H, Kodama T, Takemura M, Ohtsuka Y, Nakamura J, Tsujita R, Kawasaki K, Yokoi H, Kawanishi M. Discovery of selective PDE4B inhibitors. Bioorg Med Chem Lett. 2009;19:3174–3176. http://dx.doi.org/10.1016/j.bmcl.2009.04.121 [DOI] [PubMed] [Google Scholar]
- 40.Woodrow MD, Ballantine SP, Barker MD, Clarke BJ, Dawson J, Dean TW, Delves CJ, Evans B, Gough SL, Guntrip SB, Holman S, Holmes DS, Kranz M, Lindvaal MK, Lucas FS, Neu M, Ranshaw LE, Solanke YE, Somers DO, Ward P, Wiseman JO. Quinolines as a novel structural class of potent and selective PDE4 inhibitors. Optimisation for inhaled administration. Bioorg Med Chem Lett. 2009;19:5261–5265. http://dx.doi.org/10.1016/j.bmcl.2009.04.012 [DOI] [PubMed] [Google Scholar]
- 41.Kranz M, Wall M, Evans B, Miah A, Ballantine S, Delves C, Dombroski B, Gross J, Schneck J, Villa JP, Neu M, Somers DO. Identification of PDE4B Over 4D subtype-selective inhibitors revealing an unprecedented binding mode. Bioorg Med Chem. 2009;17:5336–5341. http://dx.doi.org/10.1016/j.bmc.2009.03.061 [DOI] [PubMed] [Google Scholar]
- 42.Zhang KYJ, Card GL, Suzuki Y, Artis DR, Fong D, Gillette S, Hsieh D, Neiman J, West BL, Zhang C, Milburn MV, Kim SH, Schlessinger J, Bollag G. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol Cell. 2004;15:279–286. http://dx.doi.org/10.1016/j.molcel.2004.07.005 [DOI] [PubMed] [Google Scholar]
- 43.Donnell AF, Dollings PJ, Butera JA, Dietrich AJ, Lipinski KK, Ghavami A, Hirst WD. Identification of pyridazino[4,5-b]indolizines as selective PDE4B inhibitors. Bioorg Med Chem Lett. 2010;20:2163–2167. http://dx.doi.org/10.1016/j.bmcl.2010.02.044 [DOI] [PubMed] [Google Scholar]
- 44.Pal S, Durgadas S, Nallapati SB, Mukkanti K, Kapavarapu R, Meda CL, Parsa KVL, Pal M. Novel 1-alkynyl substituted 1,2-dihydroquinoline derivatives from nimesulide(and their 2-oxo analogues): A new strategy to identify inhibitors of PDE4B. Bioorg Med Chem Lett. 2011;21:6573–6576. http://dx.doi.org/10.1016/j.bmcl.2011.08.033 [DOI] [PubMed] [Google Scholar]
- 45.Allcock RW, Blakli H, Jiang Z, Johnston KA, Morgan KM, Rosair GM, Iwase K, Kohno Y, Adams DR. Phosphodiesterase inhibitors. Part 1: Synthesis and structure-activity relationships of pyrazolopyridine-pyridazinone PDE inhibitors developed from ibudilast. Bioorg Med Chem Lett. 2011;21:3307–3312. http://dx.doi.org/10.1016/j.bmcl.2011.04.021 [DOI] [PubMed] [Google Scholar]
- 46.Ochiai K, Ando N, Iwase K, Kishi T, Fukuchi K, Ohinata A, Zushi H, Yasue T, Adams DR, Kohno Y. Phosphodiesterase inhibitors. Part 2: Design, synthesis, and structure-activity relationships of dual PDE3/4-inhibitory pyrazolo[1,5-a]pyridines with anti-inflammatory and bronchodilatory activity. Bioorg Med Chem Lett. 2011;21:5451–5456. http://dx.doi.org/10.1016/j.bmcl.2011.06.118 [DOI] [PubMed] [Google Scholar]
- 47.Ochiai K, Takita S, Eiraku T, Kojima A, Iwase K, Kishi T, Fukuchi K, Yasue T, Adams DR, Allcock RW, Jiang Z, Kohno Y. Phosphodiesterase inhibitors. Part 3: Design, synthesis and structure-activity relationships of dual PDE3/4-inhibitory fused bicyclic heteroaromatic-dihydropyridazinones with anti-inflammatory and bronchodilatory activity. Bioorg Med Chem. 2012;20:1644–1658. http://dx.doi.org/10.1016/j.bmc.2012.01.033 [DOI] [PubMed] [Google Scholar]
- 48.Kumar D, Patel G, Vijayakrishnan L, Dastidar SG, Ray A. Design and Synthesis of 3,5-Disubstituted-1,2,4-Oxadiazoles as Potent Inhibitors of Phosphodiesterase 4B2. Chem Biol Drug Des. 2012;79:810–818. http://dx.doi.org/10.1111/j.1747-0285.2011.01304 [DOI] [PubMed] [Google Scholar]
- 49.Shan WJ, Huang L, Zhou Q, Jiang HL, Luo ZH, Lai KF, Li XS. Dual β2-adrenoceptor agonists-PDE4 inhibitors for the treatment of asthma and COPD. Bioorg Med Chem Lett. 2012;22:1523–1526. http://dx.doi.org/10.1016/j.bmcl.2012.01.013 [DOI] [PubMed] [Google Scholar]
- 50.Huang L, Shan W, Zhou Q, Xie J, Lai K, Li X. Design, synthesis and evaluation of dual pharmacology β2-adrenoceptor agonists and PDE4 inhibitors. Bioorg Med ChemLett. 2014;24:249–253. http://dx.doi.org/10.1016/j.bmcl.2013.11.028 [DOI] [PubMed] [Google Scholar]
- 51.Gorja DR, Kumar SK, Kandale A, Meda CL, Parsa KV, Mukkanti K, Pal M. Design and synthesis of 4-alkynyl pyrazoles as inhibitors of PDE4: A practical access via Pd/C-Cu catalysis. Bioorg Med Chem Lett. 2012;22:2480–2487. http://dx.doi.org/10.1016/j.bmcl.2012.02.008 [DOI] [PubMed] [Google Scholar]
- 52.Robichaud A, Stamatiou PB, Catherine Jin SL, Lachance N, Mac Donald D, Laliberte F, Liu S, Huang Z, Conti M, Chan CC. Deletion of Phosphodiesterase4D in mice shortens α2-adrenoreceptor mediated anesthesia, a behavioural correlate to emesis. J Clin Invest. 2002;110:1045–1052. http://dx.doi.org/10.1172/JCI15506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reddy TR, Reddy GR, Reddy LS, Jamula S, Lingappa Y, Kapavarapu R, Meda CLT, Parsa KVL, Pal M. Montmorillonite K-10 mediated green synthesis of cyano pyridines: Their evaluation as potential inhibitors of PDE4. Eur J Med Chem. 2012;48:265–274. http://dx.doi.org/10.1016/j.ejmech.2011.12.024 [DOI] [PubMed] [Google Scholar]
- 54.Mitchell CJ, Ballantine SP, Coe DM, Cook CM, Delves CJ, Dowle MD, Edlin CD, Hamblin JN, Holman S, Johnson MR, Jones PS, Keeling SE, Kranz M, Lindvall M, Lucas FS, Neu M, Solanke YE, Somers DO, Trivedi NA, Wiseman JO. Pyrazolopyridines as potent PDE4B inhibitors: 5-Heterocycle SAR. Bioorg Med Chem Lett. 2010;20:5803–5806. http://dx.doi.org/10.1016/j.bmcl.2010.07.136 [DOI] [PubMed] [Google Scholar]
- 55.Suzuki O, Mizukami K, Etori M, Sogawa Y, Takagi N, Tsuchida H, Morimoto K, Goto T, Yoshino T, Mikkaichi T, Hirahara K, Nakamura S, Maeda H. Evaluation of the Therapeutic Index of a Novel Phosphodiesterase 4B-Selective Inhibitor Over Phosphodiesterase 4D in Mice. J Pharmacol Sci. 2013;123:219–226. http://dx.doi.org/10.1254/jphs.13103FP [DOI] [PubMed] [Google Scholar]
- 56.Goto T, Yoshino T, Mizukami K, Hirahara K, Suzuki O, Sogawa Y, Takahashi T, Mikkaichi T, Nakao N, Takahashi M, Hasegawa M, Sasaki S. Synthesis and biological evaluation of 5-carbamoyl-2-phenylpyrimidine derivatives as novel and potent PDE4 inhibitors. Bioorg Med Chem. 2013;21:7025–7037. http://dx.doi.org/10.1016/j.bmc.2013.09.013 [DOI] [PubMed] [Google Scholar]
- 57.Singh SN, Regati S, Paul AK, Layek M, Jayaprakash S, Reddy VK, Deora GS, Mukherjee S, Pal M. Cu-mediated 1,3-dipolar cycloaddition of azomethineylides with dipolarophiles: a faster access to spirooxindoles of potential Pharmacological interest. Tetrahedron Lett. 2013;54:5448–5452. http://dx.doi.org/10.1016/j.tetlet.2013.07.126 [Google Scholar]
- 58.Kumar SK, Adepu R, Kapavarapu R, Rambabu D, Krishna RG, Reddy MC, Priya KK, Parsa KVL, Pal M. AlCl3 induced C-arylation/cyclization in a single pot: a new route to benzofuran fused N-heterocycles of pharmacological interest. Tetrahedron Lett. 2012;53:1134–1138. http://dx.doi.org/10.1016/j.tetlet.2011.12.096 [Google Scholar]
- 59.Reddy SV, Rao GM, Kumar BV, Meda CLT, Deora GS, Kumar KS, Parsa KVL, Pal M. Novel imidazophenoxazine-4-sulfonamides: Their synthesis and evaluation as potential inhibitors of PDE4. Bioorg Med Chem 2013;21:1952–1963. http://dx.doi.org/10.1016/j.bmc.2013.01.023 [DOI] [PubMed] [Google Scholar]
- 60.Ochiai K, Takita S, Kojima A, Eiraku T, Iwase K, Kishi T, Ohinata A, Yageta Y, Yasue T, Adams DR, Yasushi Kohno Y. Phosphodiesterase inhibitors. Part 5: Hybrid PDE3/4 inhibitors as dual bronchorelaxant/anti-inflammatory agents for inhaled administration. Bioorg Med Chem Lett. 2013;23:375–381. http://dx.doi.org/10.1016/j.bmcl.2012.08.121 [DOI] [PubMed] [Google Scholar]
- 61.Rao RM, Luther BJ, Rani CS, Suresh N, Kapavarapu R, Parsa KV, Rao MV, Pal M. Synthesis of 2H-1,3-benzoxazin-4(3H)-one derivatives containing indole moiety: Their in vitro evaluation against PDE4B. Bioorg Med Chem Lett. 2014;24:1166–1171. http://dx.doi.org/10.1016/j.bmcl.2013.12.117 [DOI] [PubMed] [Google Scholar]
- 62.Goto T, Shiina A, Murata T, Tomii M, Yamazaki T, Yoshida K, Yoshino T, Suzuki O, Sogawa Y, Mizukami K, Takagi N, Yoshitomi T, Etori M, Tsuchida H, Mikkaichi T, Nakao N, Takahashi M, Takahashi H, Sasaki S. Identification of the 5,5-dioxo-7,8-dihydro-6H-thiopyrano[3,2-d]pyrimidine derivatives as highly selective PDE4B inhibitors. Bioorg Med Chem Lett. 2014;24:893–899. http://dx.doi.org/10.1016/j.bmcl.2013.12.076 [DOI] [PubMed] [Google Scholar]
- 63.Marko D, Romanakis K, Zankl H, Fürstenberger G, Steinbauer B, Eisenbrand G. Induction of apoptosis by an inhibitor of cAMP-specific PDE in malignant murine carcinoma cells overexpressing PDE activity in comparison to their non-malignant counterparts. Cell Biochem Biophys. 1998;28:75–101. http://dx.doi.org/10.1007/BF02737806 [DOI] [PubMed] [Google Scholar]
- 64.Ogawa R, Streiff MB, Bugayenko A, Kato GJ. Inhibition of PDE4 phosphodiesterase activity induces growth suppression, apoptosis, glucocorticoid sensitivity, p53, and p21 (WAF1/CIP1) proteins in human acute lymphoblastic leukemia cells. Blood. 2002;99:3390–3397. http://www.ncbi.nlm.nih.gov/pubmed/11964308 [DOI] [PubMed] [Google Scholar]
- 65.Smith PG, Wang F, Wilkinson KN, Savage KJ, Klein U, Neuberg DS, Bollag G, Shipp MA, Aguiar RC. The phosphodiesterase PDE4B limits cAMP-associated PI3K/AKT dependent apoptosis in diffuse large B-cell lymphoma. Blood. 2005;105:308–316. http://www.ncbi.nlm.nih.gov/pubmed/15331441 [DOI] [PubMed] [Google Scholar]
- 66.Mentz F, Mossalayi MD, Ouaaz F, Baudet S, Issaly F, Ktorza S, Semichon M, Binet JL, Merle-Beral H. Theophylline synergizes with chlorambucil in inducing apoptosis of chronic B-lymphocytic leukemia cells. Blood. 1996;88:2172–2182. http://www.ncbi.nlm.nih.gov/pubmed/8822937 [PubMed] [Google Scholar]
- 67.Praveena KSS, Durgadas S, Suresh Babu N, Akkenapally S, Ganesh Kumar C, Deora GS, Murthy NY, Mukkanti K, Pal S. Synthesis of 2,2,4-trimethyl-1,2-dihydroquinolinyl substituted 1,2,3-triazole derivatives: Their evaluation as potential PDE 4B inhibitors possessing cytotoxic properties against cancer cells. Bioorg Chem. 2014;53:8–14. http://dx.doi.org/10.1016/j.bioorg.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 68.Mareddy J, Nallapati SB, Anireddy J, Devi YP, Mangamoori LN, Kapavarapu R, Pal S. Synthesis and biological evaluation of nimesulide based new class of triazole derivatives as potential PDE4B inhibitors against cancer cells. Bioorg Med Chem Lett. 2013;23:6721–6727. http://dx.doi.org/10.1016/j.bmcl.2013.10.035 [DOI] [PubMed] [Google Scholar]
- 69.Chen B, Su B, Chen S. A COX-2 inhibitor nimesulideanalog selectively induces apoptosis in Her2 overexpressing breast cancer cells via cytochrome c dependent mechanisms. Biochem Pharmacol. 2009;77:1787–1794. http://dx.doi.org/10.1016/j.bcp.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shafi S, Alam MM, Mulakayala N, Mulakayala C, Vanaja G, Kalle AM, Pallu R, Alam MS. Synthesis of novel 2-mercaptobenzothiazole and 1,2,3-triazole based bis-heterocycles: Their anti-inflammatory and anti-nociceptive activities. Eur J Med Chem. 2012;49:324–333. http://dx.doi.org/10.1016/j.ejmech.2012.01.032 [DOI] [PubMed] [Google Scholar]
- 71.Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different G-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006;98:1081–1088. http://dx.doi.org/10.1161/01.RES.0000218493.09370.8e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet JL, Cooper DM, Fischmeister R. Negative feedback exerted by PKA and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes. An in vivo study using adenovirus-mediated expression of CNG channels. J Biol Chem. 2004;279:52095–52105. http://dx.doi.org/10.1074/jbc.M405697200 [DOI] [PubMed] [Google Scholar]
- 73.Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. http://www.ncbi.nlm.nih.gov/pubmed/15178638 [DOI] [PubMed] [Google Scholar]
- 74.Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4D is required for ß adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci U S A. 2005;102:909–914. http://dx.doi.org/10.1073/pnas.0405263102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of ß-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: Role of phosphodiesterases. Circ Res. 2008;102:1091–1100. http://dx.doi.org/10.1161/CIRCRESAHA.107.167817 [DOI] [PubMed] [Google Scholar]
- 76.Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ, Engelhardt S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching ß-adrenergic but locally confined ß-2 adrenergic receptor-mediated signaling. Circ Res. 2006;99:1084–1091. http://dx.doi.org/10.1161/01.RES.0000250046.69918.d5 [DOI] [PubMed] [Google Scholar]
- 77.Leroy J, Richter W, Mika D, Castro LRV, Abi-Gerges A, Xie M, Scheitrum C, Lefebvre F, Schittl J, Mateo P, Westenbroek R, Catterall WA, Charpentier F, Conti M, Fischmeister R, Vandecasteele G. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+current and protects against ventricular arrhythmias in mice. J Clin Invest. 2011;121:2651–2661. http://dx.doi.org/10.1172/JCI44747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poondra RR, Nallamelli RV, Meda CL, Srinivas BN, Grover A, Muttabathula J, Voleti SR, Sridhar B, Pal M, Parsa KV. Discovery of novel 1,4-dihydropyridine-based PDE4 inhibitors. MedChem Lett. 2013;23:1104–1109. http://dx.doi.org/10.1016/j.bmcl.2012.11.121 [DOI] [PubMed] [Google Scholar]
- 79.Fox D 3rd, Burgin AB, Gurney ME. Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell Signal. 2014;26:657–663. http://dx.doi.org/10.1016/j.cellsig.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]