

Abstract
The success of tyrosine kinase inhibitors in treating chronic myeloid leukemia highlights the potential of targeting oncogenic kinases with small molecules. By using drug activity profiles and individual patient genotypes, one can guide personalized therapy selection for patients with resistance.
Introduction
Small molecules that inhibit oncogenic signaling pathways are redefining cancer therapy. Potential therapeutic targets have been identified in all physiological processes, reflecting the diversity of mechanisms that promote malignant transformation. In particular, tyrosine and serine/threonine kinases have attracted much attention, which is not surprising given their fundamental role in regulating eukaryotic cellular signaling [1]. Activating mutations in tyrosine and serine/threonine kinases have been identified in many types of cancer and associated with the malignant phenotype, providing a strong therapeutic rationale for the development of small molecule inhibitors that block their activity [2]. The biggest clinical successes to date are the BCR-ABL1 tyrosine kinase inhibitor (TKI) imatinib and its successor compounds, dasatinib, nilotinib, bosutinib and ponatinib (Figure 1). These drugs have transformed chronic-phase chronic myeloid leukemia (CML-CP) from a lethal cancer into a chronic disorder that is compatible with a largely normal span and quality of life.
Figure 1.
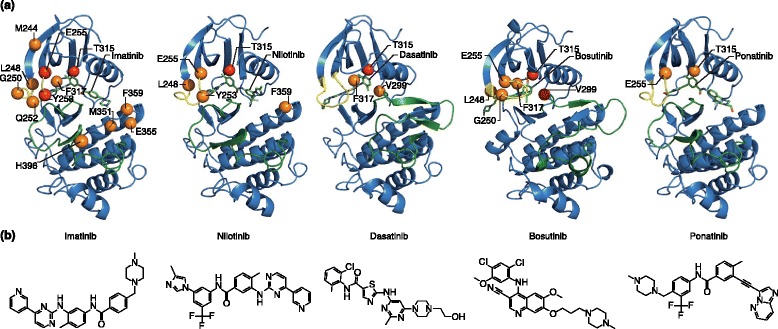
Tyrosine kinase inhibitors (TKIs) approved for the treatment of chronic myeloid leukemia. (a) The crystal structure of the ABL1 kinase domain is shown in complex with the indicated TKI. Highlighted residues indicate mutations that confer resistance to the indicated TKI in vitro. Orange (moderate) and red (severe) spheres indicate the level of TKI resistance. (b) The chemical structures of the TKIs. Adapted with permission from O’Hare et al. [3].
Chronic myeloid leukemia (CML) is caused by the chimeric tyrosine kinase BCR-ABL1, which results from the t(9;22)(q34;q11) chromosomal translocation and is visible cytogenetically as the Philadelphia chromosome [3]. Resistance to imatinib is frequently caused by mutations in the tyrosine kinase domain of BCR-ABL1, and because the approved TKIs differ in their activity against specific mutants, the clinical selection of TKIs can be driven by BCR-ABL1 genotype, providing a prime example of personalized therapy in oncology.
Here, we discuss TKI therapy for CML to illustrate the challenges of molecularly targeted cancer therapy, focusing on therapy individualization, the role of clonal evolution and complexity in therapy response and resistance, and how the lessons learned from CML may be applied to TKI therapy in other types of cancer.
Development of BCR-ABL1 TKIs for CML
Most patients are diagnosed in CML-CP, during which the myeloid cell compartment is expanded but cellular differentiation is maintained [4]. Without effective therapy, CML-CP inexorably progresses to blast phase CML (CML-BP), a disease that resembles an acute leukemia, with complete block of terminal differentiation and a poor prognosis. Murine models indicate that BCR-ABL1 is required and sufficient to induce CML-CP, whereas diverse additional mutations have been implicated in progression to CML-BP (Table 1) [3,5–16].
Table 1.
Mutations associated with CML-BP
| Mutation | Percentage prevalence | Reference |
|---|---|---|
| Double Ph chromosome | 38% | [6] |
| Isochromosome 17q | 30% (myeloid) | [7] |
| Trisomy 8 | 53% (myeloid) | [7] |
| Trisomy 19 | 23% (myleoid) | [7] |
| p53 mutations | 20-30% (myeloid) | [8] |
| p16 mutations | 50% (lymphoid) | [9] |
| NUP98-HOXA9 translocations | NR | [10] |
| AML-EVI1 translocations | NR | [11] |
| GATA-2 mutations | 18% (lymphoid) | [12] |
| RUNX1 mutations | 38% (myeloid) | [13] |
| CDKN2A/B mutations | 50% (lymphoid) | [14] |
| IKZF1 mutations | 55% (lymphoid) | [14] |
| ASXL1 mutations | 20.5% (myeloid) | [16] |
| TET2 mutations | 7.7% (myeloid) | [16] |
| WT1 mutations | 15.4% (myeloid) | [16] |
| NRAS/KRAS mutations | 5.1/ 5.1% (myeloid) | [16] |
Ph, Philadelphia; NUP98, nucleoporin 98 kDa; HOXA9, homeobox A9; AML, acute myeloid leukemia; EVI1, ecotropic viral integration site 1; GATA-2, GATA binding protein 2; RUNX1, runt-related transcription factor 1; CDKN2A/B, cyclin-dependent kinase inhibitor 2A/B; IKZF1, IKAROS family zinc finger 1; ASXL1, additional sex combs like transcription regulator 1; TET2, tet methylcytosine dioxygenase 2; WT1, wilms tumor 1; NRAS, neuroblastoma RAS viral oncogene homolog; KRAS, Kirsten rat sarcoma viral oncogene homolog; NR, not reported.
Clinical trials with the first BCR-ABL1 inhibitor, imatinib, were initiated in 1998. The striking activity of imatinib led to rapid regulatory approval for the treatment of patients with CML for whom interferon-α therapy had failed (in 2001), and subsequently to approval for the treatment of newly diagnosed patients (in 2003). Patients with CML-CP who begin treatment with imatinib at diagnosis have an 8-year overall survival of approximately 85%, with an acceptable quality of life [17,18]. Nevertheless, imatinib has limitations: imatinib treatment fails for some 25 to 30% of CML-CP patients because of primary or acquired resistance, and for additional patients due to intolerance [19].
To overcome resistance to imatinib, three second-generation inhibitors have been developed (Figure 1). Dasatinib, nilotinib and bosutinib provide durable salvage therapy for about half of the patients for whom imatinib fails in CML-CP, but not for those with progression to CML-BP [20,21]. Subsequent studies that compared dasatinib or nilotinib with imatinib in frontline CML-CP revealed more profound responses and reduced rates of transformation to CML-BP for the second-generation TKIs, but have yet to show differences in overall survival [22,23]. The most recent addition to the CML armamentarium is the third-generation TKI ponatinib [24]. This drug is highly active, even in patients with resistance to multiple TKIs. However, as for all other BCR-ABL1 inhibitors, although responses are durable in CML-CP, they are only transient in CML-BP [25]. In 2014, most patients diagnosed with CML-CP can expect to achieve durable responses to TKIs, and their long-term prognosis is good. A minority of patients, however, do not respond effectively to multiple TKIs or progress to CML-BP. Thus, although TKIs have improved the survival and quality of life for many CML patients, a better understanding of TKI resistance and the mechanisms leading to blastic transformation will be crucial for improving outcomes.
Resistance to TKIs
TKI resistance in CML involves two fundamentally different mechanisms. First, BCR-ABL1 kinase-dependent resistance is driven by reactivation of BCR-ABL1 kinase activity. This typically occurs as the result of missense mutations in the kinase domain that impair drug binding through steric hindrance or conformational changes, or through BCR-ABL1 genomic amplification [26]. Other mechanisms include impaired drug influx or increased drug efflux. For example, OCT-1, a cation transporter, has been implicated in transmembrane transport of imatinib, and reduced activity or expression of this protein is associated with drug resistance [27,28]. Conversely, high expression of MDR1 is associated with nilotinib resistance [29].
Second, BCR-ABL1 kinase-independent resistance is thought to occur when alternative signaling pathways are activated that maintain cell proliferation and viability despite continued suppression of BCR-ABL1 kinase activity (Figure 2) [3]. Evidence suggests that both extrinsic and intrinsic mechanisms are involved in BCR-ABL1 kinase-independent resistance and may activate the same downstream signaling molecules. Multiple extrinsic and intrinsic signals and pathways have been implicated, including JAK/STAT [30–32], phosphatidyl inositol 3′ kinase (PI3K) [33], Wnt/β-catenin [34–36], SHP-1 [37], SRC family kinases such as Lyn [38], and polymorphisms of the pro-apoptosis protein BIM [39]. The mechanistic heterogeneity of BCR-ABL1 kinase-independent TKI resistance poses a diagnostic and therapeutic challenge. Hence, individualized TKI therapy as it exists today centers on BCR-ABL1 kinase domain mutations, and BCR-ABL1 kinase-dependent resistance will be the focus of this review.
Figure 2.
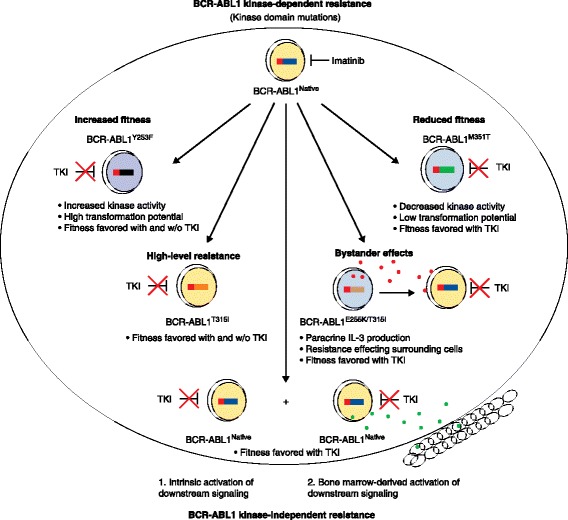
Multiple mechanisms of tyrosine kinase inhibitor (TKI) resistance in chronic myeloid leukemia. The schematic portrays multiple mechanisms of TKI resistance, including BCR-ABL1 kinase-dependent mechanisms (top) and BCR-ABL1 kinase-independent mechanisms (bottom). Certain tyrosine kinase mutations impart increased or decreased fitness on the BCR-ABL1 kinase. Other mutations such as T315I impart high-level resistance to first- and second-generation TKIs. Cells that carry resistance mutations may impart resistance on neighboring bystander cells by secretion of paracrine factors (such as the cytokine IL-3), so that even cells with native BCR-ABL1 become TKI resistant. Last, CML cells may acquire resistance through intrinsic activation of alternative signaling pathways or through interaction with the bone marrow microenvironment. Red and green dots denote paracrine factors produced by leukemic cells or the bone marrow microenvironment.
BCR-ABL1 kinase domain mutations
Differential TKI activity against BCR-ABL1 mutants
More than 50 different BCR-ABL1 mutations have been identified in patients with clinically manifest resistance to imatinib, but a much smaller set of mutations accounts for most acquired resistance [3]. Solving the structure of the ABL1 kinase domain crystallized with imatinib was critical to understanding mutation-based TKI resistance [40]. Unexpectedly, imatinib was found to bind an inactive conformation of ABL1, with the activation loop in a closed conformation and extensive downward displacement of the ATP-binding loop. Multiple residues are engaged by imatinib through hydrogen bonds or hydrophobic interactions, providing ample opportunity for point mutations to impair drug binding. In contrast to imatinib, which is vulnerable to a large number of different mutations, the spectrum of resistance mutations is much more limited for the second-generation TKIs, dasatinib, nilotinib and bosutinib [3]. For dasatinib, a type I inhibitor, resistance is reduced by binding to the active ABL1 conformation [41], which places less stringent requirements on inhibitor binding and hence is less liable to mutational escape. Although the conformation of ABL1 that is bound by nilotinib resembles that of the ABL1-imatinib complex, a much-improved topographic fit provides additional free energy, thereby moving many BCR-ABL1 mutants into the range of achievable nilotinib plasma concentrations [42]. Interestingly, bosutinib binds both the active and inactive conformations of ABL1 kinase [43].
Despite many improvements, all second-generation TKIs share a common vulnerability with imatinib, namely the T315I mutation of the ‘gatekeeper’ residue in ABL1 [3]. Substitution of threonine 315 with isoleucine prevents the formation of a key hydrogen bond (or van der Waals interaction in the case of bosutinib) between the kinase and the TKI drug, resulting in high-level resistance to multiple TKIs (Figure 2). Additionally, access to a hydrophobic pocket that is engaged by all first- and second-generation TKIs is blocked by this substitution. Ponatinib, the only third-generation TKI approved to date, is a type II inhibitor that was designed to avoid T315 by inclusion of a rigid triple carbon bond (Figure 1) [24]. Higher concentrations of ponatinib are required for inhibition of certain BCR-ABL1 mutants (for example, E255V), but these are still within the range of plasma concentrations achievable in patients, and clinical responses have been observed in patients who harbor these genotypes [24]. In vitro assays based on culturing cells that express randomly mutagenized BCR-ABL1 in the presence of TKIs are remarkably accurate in predicting clinically relevant BCR-ABL1 resistance mutations and contact points between TKIs and the kinase domains. Mutagenesis is achieved either by initial expression of a BCR-ABL1 plasmid in a mutagenic bacterial strain or by exposing the BCR-ABL1-expressing cells to N-nitroso-N-methylurea (ENU). Despite the fact that in vivo activity is dependent on multiple additional factors, including bioavailability, achievable plasma concentrations, transmembrane transport and protein binding, the in vitro drug sensitivity of cell lines (typically the pro-B cell line BaF/3, engineered to express BCR-ABL1 mutants in comparison to the native BCR-ABL1 kinase) is generally correlated with clinical activity (Figure 3). This allows rational TKI selection on the basis of the patient’s BCR-ABL1 genotype, and provides an example of how molecular knowledge can aid the personalization of cancer therapy.
Figure 3.
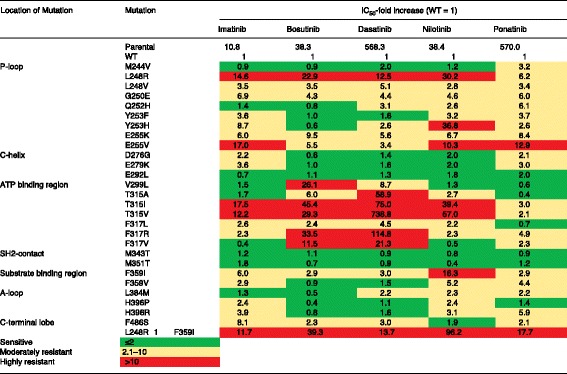
Activities of imatinib, bosutinib, dasatinib, nilotinib, and ponatinib against mutated forms of BCR-ABL1. Half maximal inhibitory concentration (IC50) values for cell proliferation of the indicated TKIs are shown against BCR-ABL1 single mutants. The color gradient demonstrates the IC50 sensitivity for each TKI relative to its activity against cells expressing native BCR-ABL1. Note that clinical activity is also dependent on additional factors, such as the drug concentrations achieved in the plasma of patients. Adapted with permission from Redaelli et al. [57].
Low-level BCR-ABL1 mutations
It seems logical that it would be beneficial to detect resistance mutations as early as possible, as appropriate changes can then be made to treatment strategies at an early stage to halt the expansion of a resistant clone. Given the low sensitivity of Sanger sequencing (approximately 20%), considerable effort has been dedicated to designing more sensitive assays that use a range of different technologies, including denaturing high-performance liquid chromatography (HPLC), allele-specific PCR, allele-specific ligation PCR, MassArray (Sequenom, San Diego, CA, USA) and most recently next-generation sequencing (NGS) [44–49]. These studies generally suggest that resistance mutations that are detected at low levels are predictive of less profound responses and subsequent relapse. Nevertheless, mutations detected at very low levels by allele-specific or ligation PCR were not predictive of subsequent TKI resistance [44,49]. Some of these low-level signals may be false-positive results, but an alternative explanation is that they might originate from cells that do not have full leukemogenic potential. Thus, expansion of a mutant clone to a biologically significant level may be required to validate its leukemogenic fitness. For instance, kinase domain mutations may be acquired by transiently expanding short-term leukemic stem cells (LSCs) that are unable to sustain leukemic hematopoiesis. If markers become available to select BCR-ABL1-positive LSCs, single-cell sequencing may supply critical information in this area in the future. On the other hand, low-level BCR-ABL1 kinase-domain mutations may be a marker of genetic instability, and thus the presence of multiple low-level mutations may predict a poor response to second generation TKIs [46]. Given these uncertainties, the clinical utility of high-sensitivity mutation screening is currently unclear and more prospective studies will be needed to clarify the value of this technique.
BCR-ABL1 kinase domain mutations and clonal fitness
Several common resistance mutations localize to critical structural elements of the BCR-ABL1 enzyme, such as the ATP-binding and activation loops, and have been shown to alter the catalytic activity of the kinase. For example, certain ATP-binding loop mutations such as Y253F can increase intrinsic kinase activity to levels above that of the native kinase [50,51]. Other mutations, such as M351T, reduce intrinsic kinase activity. Results for the T315I mutation are inconsistent, probably reflecting differences in the techniques used to purify the proteins that have been subjected to enzyme-kinetic assays [50,51]. The competitiveness of cells that express BCR-ABL1 kinase mutants in a given TKI environment will reflect a balance between the gains afforded by TKI resistance with changes in kinase catalytic activity (Figure 2). Reduced kinase activity may be a critical factor that limits the acquisition of additional mutations and requires further investigation.
Compound mutations
In Sanger sequencing traces, the presence of compound mutations (that is, two or more mutations in the same BCR-ABL1 molecule) is inferred if the percentages of mutant alleles combined, based on their peak height relative to that of the native sequence, exceed 100%. If the combined mutant alleles are less than 100%, Sanger sequencing cannot distinguish between compound mutations and polyclonal mutations (that is, multiple BCR-ABL1 mutant clones). A widely used method to ascertain that two mutations localize to the same BCR-ABL1 allele is shotgun cloning of BCR-ABL1 PCR products followed by sequencing of individual colonies; however, long-range NGS may provide a less tedious approach in the future [47].
Colony sequencing has been used to demonstrate linear clonal evolution in several patients who developed multidrug-resistant compound mutant clones [52]. Interestingly, the likelihood that an additional mutation is silent rather than missense increases with the total number of mutations in the BCR-ABL1 molecule (Figure 4). This suggests that the fitness of the BCR-ABL1 kinase must ultimately be compromised by the acquisition of successive missense mutations, leading to evolutionary dead ends. From a therapeutic standpoint, this is good news as it suggests that mutational escape of the primary target kinase is not unlimited. As the impact on kinase fitness of two mutations in the same BCR-ABL1 allele is unpredictable, experimental validation is required [53].
Figure 4.
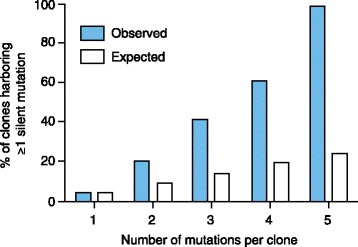
Silent mutations increase with the total number of mutations per cell clone. The graph represents the total number of silent mutations per clone (x-axis) and the percentage of clones with at least one silent mutation (blue bars). White bars represent the expected percentage of mutations. Adapted with permission from Khorashad et al. [52].
Compound mutations containing a T315I component confer high-level resistance to all approved TKIs, posing a considerable clinical challenge [54]. Fortunately, it seems that most compound mutations identified in patients are composed from a core set of single resistance mutations, suggesting that the number of catalytically viable combinations is limited [55]. The hope is that structural commonalities exist between subsets of possible mutations, which will allow the generation of TKIs that effectively target multiple compound mutants.
Bystander effects of BCR-ABL1 kinase mutant clones
Some patients develop clinical resistance, although only a minority of the BCR-ABL1 amplicons found in such patients are kinase domain mutants. Two explanations for this come to mind. First, multiple resistant clones may co-exist, some with kinase domain mutations and some with BCR-ABL1 kinase-independent TKI resistance. Second, kinase domain mutant subclones may generate paracrine factors such as IL-3 that promote the survival of bystander cells. Evidence for the latter has been found in vitro for clones carrying the E255K/T315I compound mutation [56]. If confirmed in vivo, this could add another level of complexity, as resistant clones could enhance the fitness of sensitive clones by altering their microenvironment (Figure 2).
Individualizing TKI therapy for CML
CML is one of few cancers with a close correlation between morphology and the causal genetic abnormality, which greatly facilitates the accrual of fairly homogenous patient populations for clinical studies. As imatinib, nilotinib and dasatinib are all approved for patients with newly diagnosed CML-CP, drug selection for initial therapy depends on disease risk and co-morbidities. Many attempts have been made to develop molecular prognostic markers, but the risk stratification of CML-CP patients is still largely based on clinical scoring systems such as the Sokal score, which is based on age, platelet count, spleen size and peripheral blood blast count [4]. Patients who have intermediate or high Sokal risk scores stand to benefit from second-generation TKIs, in terms of progression-free survival, whereas patients with low risk scores have excellent outcomes with all three TKIs [22]. Patients presenting with CML-BC should be treated with a second-generation TKI, typically combined with chemotherapy. Certain co-morbidities are absolute or relative contraindications for certain TKIs. For example, a prolonged heart-rate corrected QT (QTc) interval is a contraindication for the use of nilotinib, and a history of pleural effusions is a contraindication for the use of dasatinib [22].
Upon disease progression, BCR-ABL1 genotyping is crucial for selection of the optimal TKI as salvage therapy. Recommendations are based on activity comparisons in vitro, typically half maximal (IC50) or 90% of maximal (IC90) inhibitory concentration values determined in BaF/3 cells expressing BCR-ABL1 mutants. Most commonly, TKI activity against a mutant is semi-quantifiable in relation to the native kinase, which permits a relative ranking of TKI activities despite different dose ranges (Figure 3) [57]. Although these assays ignore important in vivo factors, such as protein binding, they are indeed clinically useful. For example, V299L predicts poor response to dasatinib, E255K/V poor response to nilotinib, and T315I failure with imatinib and all second-generation TKIs, making T315I-mutant CML a prime indication for selection of ponatinib [3]. It is worth noting, however, that the correlations are tight only toward the negative side (that is, prediction of resistance). By contrast, a substantial proportion of patients with ‘sensitive mutants’ fail to respond to the respective TKI, indicating that resistance is multifactorial and presumably involves BCR-ABL1 kinase-independent mechanisms that are not measured by the currently available diagnostic assays. Ex vivo screening of leukemia cells using short hairpin RNAs (shRNAs) that silence kinase sequences or kinase inhibitor library panels may uncover novel therapeutic targets [58,59]. Ironically, ponatinib as a ‘pan BCR-ABL1 inhibitor’ with activity against all single mutants, including T315I, appeared to avoid the complexity of selecting the appropriate TKI for salvage; but the drug’s recently reported cardiovascular toxicity now mandates a thorough balancing of its excellent activity against the risk of potentially serious adverse events [60].
Translating the CML paradigm to other malignancies
Kinase-targeted therapies have been approved for a range of malignancies, but few have shown activity that is comparable to that achieved in CML (Table 2). The most convincing results were seen in relatively indolent conditions, such as chronic lymphocytic leukemia (CLL) [61], hypereosinophilic syndrome [62], and myeloproliferative neoplasms with rearrangements of the platelet-derived growth factor receptors (PDGFRs) [63]. Hairy cell leukemia, which is almost universally positive for the V600E mutation in BRAF, may become another example, as profound responses have been reported even in chemotherapy-refractory cases [64,65]. Clinically, these conditions resemble CML in their chronic course and in the trend to progress to a more advanced stage. Biologically, the key similarity may be that constitutive activation of the target kinase is an early event in disease evolution, and is both necessary and sufficient for disease induction. Interestingly, point mutations in the target kinase BTK have been identified in CLL patients for whom ibrutinib has failed [66], and FIP1L1-PDGFα mutations in patients with hypereosinophilic syndrome for whom imatinib has failed [67]. Point mutations in FLT3 have also been reported in acute myeloid leukemia (AML) patients harboring FLT3 internal tandem duplications who relapsed after a transient response to quizartinib, a potent FLT3 inhibitor, suggesting that at least some AML patients may acquire these mutations early during disease evolution [68].
Table 2.
Approved indications for kinase-targeted therapies
| Disease | Kinase target | Approved inhibitors |
|---|---|---|
| Chronic myeloid leukemia (CML) | BCR-ABL1 | Imatinib, dasatinib, nilotinib, bosutinib, ponatinib |
| Ph acute lymphocytic leukemia (ALL) | BCR-ABL1 | Imatinib, dasatinib, nilotinib, bosutinib, ponatinib |
| Mastocytosis | KIT | Imatinib |
| Hypereosinophilic syndrome (HES) | FIP1L1-PDGFRα | Imatinib |
| Chronic eosinophilic leukemia (CEL) | FIP1L1-PDGFRα | Imatinib |
| PDGFRβ | Imatinib | |
| Gastrointestinal stromal tumors (GIST) | KIT; PDGFRα | Imatinib |
| Melanoma | BRAF | Vemurafenib |
| Non-small cell lung cancer (NSCLC) | EGFR1 | Gefinitinib, erlotinib |
| ALK | Crizotinib, ceritinib | |
| Chronic lymphocytic leukemia (CLL) | BTK | Ibrutinib |
| Mantle cell lymphoma | BTK | Ibrutinib |
BCR, breakpoint cluster region; ABL1, Abelson murine leukemia viral oncogene homolog 1; KIT, c-kit proto-oncogene; FIP1L1, FIP1-like 1; PDGFRa, platelet-derived growth factor receptor alpha; PDGFRb, platelet-derived growth factor receptor beta; BRAF, B-Raf proto-oncogene; EGFR1, epidermal growth factor receptor 1; ALK, anaplastic lymphoma kinase; BTK, Bruton’s tyrosine kinase.
At the opposite end of the spectrum of kinase-targeted therapy in hematologic malignancies is myelofibrosis. Activation of JAK/STAT signaling is universal in this disease as a result of mutations in JAK2 [69], calreticulin [70,71], or MPL [72], and JAK2 inhibitors improve clinical symptoms and possibly survival. Nevertheless, these drugs have to date failed to induce profound responses that include reduction of the malignant clone or disease burden [73,74]. Several explanations may account for the relatively disappointing results, including the genetic complexity of myelofibrosis, suppression of residual normal hematopoiesis as a result of JAK2 inhibition and the relatively low potency of available JAK2 inhibitors [75,76].
The situation is similar in solid tumors. While imatinib is active in metastatic gastrointestinal stromal tumors (GISTs), which are characterized by mutations in KIT or PDGFRα, complete responses are rare and resistance typically develops after 1 to 2 years [77]. Most melanomas with BRAF mutations are responsive to RAF inhibitors, such as vemurafenib, but complete responses are uncommon and remissions are typically transient [78]. Similarly, non-small cell lung cancers (NSCLCs) with EGFR1 mutations respond to gefitinib or erlotinib [79], and those with ALK mutations respond to crizotinib or ceritinib [80,81], but most responses are incomplete and not sustained [82]. A plethora of mechanisms have been implicated in the kinase inhibitor resistance of solid tumors. Although point mutations in the target kinase do occur (for example, in KIT and PDGFRα in GISTs [77], or EGRF and ALK in NSCLCs [83,84]), they are generally less common than alternative pathway activation, and selection of rational salvage therapies poses a greater challenge.
Why some malignancies are much more likely than others to acquire resistance by reactivation of the target kinase is unknown, but the reason is likely to be multifactorial. For example, in the case of BRAF, the specific mechanism of kinase activation promotes resistance through heterodimer formation and subsequent RAS activation [85]. Another factor may be the complexity of the signaling network operated by the activated kinase. In the case of BCR-ABL1 in CML, it may be challenging for the leukemia cells to adequately replace a large multi-domain protein with alternative signaling pathways, driving resistance toward BCR-ABL1 mutational escape [3]. In other cancers, such as AML, the presence of multiple fully oncogenic but genetically diverse clones may lead to resistance through clonal selection on therapy; alternatively, a high level of genetic instability may promote linear clonal evolution toward a drug-resistant phenotype [86]. In the future, detailed knowledge of the likely escape mechanisms for a given therapy may impact drug selection and the sequencing of active targeted agents.
Conclusions and future directions
Current therapy of CML involves five approved TKIs that are used according to risk, disease stage, co-morbidities and BCR-ABL1 genotype, reflecting the high level of personalization that has already been achieved in this disease. Kinase domain mutants, with their differential sensitivity to TKIs, were key drivers for this development. Clonal fitness in a given TKI environment and the intrinsic transforming capacity of the BCR-ABL1 genotype are important determinants of drug response and resistance, providing insights into the complex interplay between drugs, malignant cells and the host that ultimately determines clinical outcomes. Current approaches to identifying resistance mechanisms to targeted cancer therapy are focused on tests that are simple to standardize for routine diagnostics, such as testing for kinase domain mutations in BCR-ABL1. Nevertheless, the detection and interpretation of low-level mutations, particularly low-level compound mutations, may be limited by the recent discovery of artifacts produced by PCR-mediated recombination events [87], a challenge that has yet to be overcome.
Characterizing drug resistance that is driven by mechanisms outside of the primary drug target is much more difficult and will predictably require genome-wide scanning technologies, such as whole-genome sequencing, or function-first assays, such as inhibitor library screens or those involving shRNAs or short interfering RNAs (siRNAs) [58]. Perhaps the greatest challenge is determining clonal complexity at diagnosis as much as at emergence of resistance. Ultra-deep sequencing and sophisticated mathematical modeling allow for reconstruction of the clonal architecture, but the resolution of this approach is ultimately limited by the error rate of the sequencing technology [88]. Overcoming this limitation will require single-cell analysis on a large scale, which is currently prohibitively expensive. In solid tumors, this is further complicated by topographic heterogeneity, which implies that multiple samples are needed to generate a representative genetic picture. Isolation and analysis of tumor cells from the blood might solve this problem in the future. Once these roadblocks have been cleared, targeted therapy will predictably move to the next level, bringing another round of fundamental change to the practice of oncology.
Abbreviations
- AML
Acute myeloid leukemia
- CLL
Chronic lymphocytic leukemia
- CML
Chronic myeloid leukemia
- CML-BP
Blast phase CML
- CML-CP
Chronic-phase CML
- ENU
N-nitroso-N-methylurea
- GIST
Gastrointestinal stromal tumor
- HPLC
High-performance liquid chromatography
- IC50
Half maximal inhibitory concentration
- IC90
90% of maximal inhibitor concentration
- IL-3
Interleukin-3
- LSC
Leukemic stem cells
- NGS
Next-generation sequencing
- NSCLC
Non-small cell lung cancer
- PDGFR
Platelet-derived growth factor receptor
- PI3K
Phosphatidyl inositol 3′ kinase
- shRNA
Short hairpin RNA
- siRNA
Short interfering RNA
- TKI
Tyrosine kinase inhibitor
Footnotes
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Anna M Eiring, Email: anna.eiring@hci.utah.edu.
Michael W Deininger, Email: michael.deininger@hci.utah.edu.
References
- 1.Lopez-Otin C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer. 2010;10:278–292. doi: 10.1038/nrc2823. [DOI] [PubMed] [Google Scholar]
- 2.Brognard J, Hunter T. Protein kinase signaling networks in cancer. Curr Opin Genet Dev. 2011;21:4–11. doi: 10.1016/j.gde.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12:513–526. doi: 10.1038/nrc3317. [DOI] [PubMed] [Google Scholar]
- 4.Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes JE, Guilhot F, Hjorth-Hansen H, Hughes TP, Kantarjian HM, Kim DW, Larson RA, Lipton JH, Mahon FX, Martinelli G, Mayer J, Muller MC, Niederwieser D, Pane F, Radich JP, Rousselot P, Saglio G, Saussele S, Schiffer C, Silver R, Simonsson B, Steegmann JL, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872–884. doi: 10.1182/blood-2013-05-501569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120:2254–2264. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stich W, Back F, Dormer P, Tsirimbas A. Double Philadelphia chromosome and isochromosome 17 in the terminal phase of chronic myeloid leukemia. Klin Wochenschr. 1966;44:334–337. doi: 10.1007/BF01752282. [DOI] [PubMed] [Google Scholar]
- 7.Parreira L, Kearney L, Rassool F, Babapulle VB, Matutes E, Parreira A, Tavares de Castro J, Goldman JM, Catovsky D. Correlation between chromosomal abnormalities and blast phenotype in the blast crisis of Ph-positive CGL. Cancer Genet Cytogenet. 1986;22:29–34. doi: 10.1016/0165-4608(86)90134-2. [DOI] [PubMed] [Google Scholar]
- 8.Feinstein E, Cimino G, Gale RP, Alimena G, Berthier R, Kishi K, Goldman J, Zaccaria A, Berrebi A, Canaani E. p53 in chronic myelogenous leukemia in acute phase. Proc Natl Acad Sci U S A. 1991;88:6293–6297. doi: 10.1073/pnas.88.14.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sill H, Goldman JM, Cross NC. Homozygous deletions of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood. 1995;85:2013–2016. [PubMed] [Google Scholar]
- 10.Nakamura T, Largaespada DA, Lee MP, Johnson LA, Ohyashiki K, Toyama K, Chen SJ, Willman CL, Chen IM, Feinberg AP, Jenkins NA, Copeland NG, Shaughnessy JD., Jr Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet. 1996;12:154–158. doi: 10.1038/ng0296-154. [DOI] [PubMed] [Google Scholar]
- 11.Cuenco GM, Ren R. Cooperation of BCR-ABL and AML1/MDS1/EVI1 in blocking myeloid differentiation and rapid induction of an acute myelogenous leukemia. Oncogene. 2001;20:8236–8248. doi: 10.1038/sj.onc.1205095. [DOI] [PubMed] [Google Scholar]
- 12.Zhang SJ, Ma LY, Huang QH, Li G, Gu BW, Gao XD, Shi JY, Wang YY, Gao L, Cai X, Ren RB, Zhu J, Chen Z, Chen SJ. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2008;105:2076–2081. doi: 10.1073/pnas.0711824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deluche L, Joha S, Corm S, Daudignon A, Geffroy S, Quief S, Villenet C, Kerckaert JP, Lai JL, Preudhomme C, Roche-Lestienne C. Cryptic and partial deletions of PRDM16 and RUNX1 without t(1;21)(p36;q22) and/or RUNX1-PRDM16 fusion in a case of progressive chronic myeloid leukemia: a complex chromosomal rearrangement of underestimated frequency in disease progression? Genes Chromosomes Cancer. 2008;47:1110–1117. doi: 10.1002/gcc.20611. [DOI] [PubMed] [Google Scholar]
- 14.Mullighan CG, Williams RT, Downing JR, Sherr CJ. Failure of CDKN2A/B (INK4A/B-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev. 2008;22:1411–1415. doi: 10.1101/gad.1673908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, Relling MV, Shurtleff SA, Downing JR. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 16.Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, Schnittger S, Kern W, Muller MC, Hochhaus A, Haferlach T, Haferlach C. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25:557–560. doi: 10.1038/leu.2010.298. [DOI] [PubMed] [Google Scholar]
- 17.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 18.Deininger M, O’Brien SG, Guilhot F, Goldman JM, Hochhaus A, Hughes TP, Radich JP, Hatfield AK, Mone M, Filian J, Reynolds J, Gathmann I, Larson RA, Druker BJ. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009;114:426. [Google Scholar]
- 19.Hehlmann R, Lauseker M, Jung-Munkwitz S, Leitner A, Muller MC, Pletsch N, Proetel U, Haferlach C, Schlegelberger B, Balleisen L, Hanel M, Pfirrmann M, Krause SW, Nerl C, Pralle H, Gratwohl A, Hossfeld DK, Hasford J, Hochhaus A, Saussele S. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-α in newly diagnosed chronic myeloid leukemia. J Clin Oncol. 2011;29:1634–1642. doi: 10.1200/JCO.2010.32.0598. [DOI] [PubMed] [Google Scholar]
- 20.Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, Cortes J, Alland L, Ottmann OG. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 21.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, Blackwood-Chirchir MA, Iyer V, Chen TT, Huang F, Decillis AP, Sawyers CL. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 22.Kantarjian HM, Shah NP, Cortes JE, Baccarani M, Agarwal MB, Undurraga MS, Wang J, Ipina JJ, Kim DW, Ogura M, Pavlovsky C, Junghanss C, Milone JH, Nicolini FE, Robak T, Van Droogenbroeck J, Vellenga E, Bradley-Garelik MB, Zhu C, Hochhaus A. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION) Blood. 2012;119:1123–1129. doi: 10.1182/blood-2011-08-376087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cortes JE, Kim DW, Kantarjian HM, Brummendorf TH, Dyagil I, Griskevicius L, Malhotra H, Powell C, Gogat K, Countouriotis AM, Gambacorti-Passerini C. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012;30:3486–3492. doi: 10.1200/JCO.2011.38.7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA, III, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ, Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, DiPersio J, DeAngelo DJ, Abruzzese E, Rea D, Baccarani M, Muller MC, Gambacorti-Passerini C, Wong S, Lustgarten S, Rivera VM, Clackson T, Turner CD, Haluska FG, Guilhot F, Deininger MW, Hochhaus A, Hughes T, Goldman JM, Shah NP, Kantarjian H, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783–1796. doi: 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 27.White DL, Saunders VA, Dang P, Engler J, Zannettino AC, Cambareri AC, Quinn SR, Manley PW, Hughes TP. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108:697–704. doi: 10.1182/blood-2005-11-4687. [DOI] [PubMed] [Google Scholar]
- 28.White DL, Dang P, Engler J, Frede A, Zrim S, Osborn M, Saunders VA, Manley PW, Hughes TP. Functional activity of the OCT-1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J Clin Oncol. 2010;28:2761–2767. doi: 10.1200/JCO.2009.26.5819. [DOI] [PubMed] [Google Scholar]
- 29.Agrawal M, Hanfstein B, Erben P, Wolf D, Ernst T, Fabarius A, Saussele S, Purkayastha D, Woodman RC, Hofmann WK, Hehlmann R, Hochhaus A, Muller MC. MDR1 expression predicts outcome of Ph + chronic phase CML patients on second-line nilotinib therapy after imatinib failure. Leukemia. 2014;28:1478–1485. doi: 10.1038/leu.2014.6. [DOI] [PubMed] [Google Scholar]
- 30.Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Holbl A, Gleixner KV, Dworzak M, Mayerhofer M, Hoermann G, Herrmann H, Sillaber C, Egger G, Valent P, Moriggl R, Sexl V. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117:3409–3420. doi: 10.1182/blood-2009-10-248211. [DOI] [PubMed] [Google Scholar]
- 31.Traer E, MacKenzie R, Snead J, Agarwal A, Eiring AM, O’Hare T, Druker BJ, Deininger MW. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26:1140–1143. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eiring AM, Page BD, Kraft IL, Mason CC, Vellore NA, Resetca D, Zabriskie MS, Zhang TY, Khorashad JS, Engar AJ, Reynolds KR, Anderson DJ, Senina A, Pomicter AD, Arpin CC, Ahmad S, Heaton WL, Tantravahi SK, Todic A, Moriggl R, Wilson DJ, Baron R, O’Hare T, Gunning PT, Deininger MW: Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia.Leukemia 2014, doi:10.1038/leu.2014.245. [DOI] [PubMed]
- 33.Quentmeier H, Eberth S, Romani J, Zaborski M, Drexler HG. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011;4:6. doi: 10.1186/1756-8722-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, Shultz L, Bhatia R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood. 2013;121:1824–1838. doi: 10.1182/blood-2012-02-412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, Ferenchak G, Dorrance AM, Paisie CA, Eiring AM, Ma Y, Mao HC, Zhang B, Wunderlich M, May PC, Sun C, Saddoughi SA, Bielawski J, Blum W, Klisovic RB, Solt JA, Byrd JC, Volinia S, Cortes J, Huettner CS, Koschmieder S, Holyoake TL, Devine S, Caligiuri MA, Croce CM, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123:4144–4157. doi: 10.1172/JCI68951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eiring AM, Anderson DJ, Zhang ZT, Kraft IL, Reynolds KR, Pomicter AD, Mason CC, O’Hare T, Deininger MW. Direct contact with bone marrow stromal cells protects CML progenitors from imatinib through cytoplasmic stabilization of beta-catenin. Blood. 2013;122:3982. doi: 10.1182/blood-2013-02-483255. [DOI] [PubMed] [Google Scholar]
- 37.Esposito N, Colavita I, Quintarelli C, Sica AR, Peluso AL, Luciano L, Picardi M, Del Vecchio L, Buonomo T, Hughes TP, White D, Radich JP, Russo D, Branford S, Saglio G, Melo JV, Martinelli R, Ruoppolo M, Kalebic T, Martinelli G, Pane F. SHP-1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome-positive cells derived from patients with chronic myeloid leukemia. Blood. 2011;118:3634–3644. doi: 10.1182/blood-2011-03-341073. [DOI] [PubMed] [Google Scholar]
- 38.Gioia R, Leroy C, Drullion C, Lagarde V, Etienne G, Dulucq S, Lippert E, Roche S, Mahon FX, Pasquet JM. Quantitative phosphoproteomics revealed interplay between Syk and Lyn in the resistance to nilotinib in chronic myeloid leukemia cells. Blood. 2011;118:2211–2221. doi: 10.1182/blood-2010-10-313692. [DOI] [PubMed] [Google Scholar]
- 39.Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, Soh S, Lee WH, Huang JW, Allen JC, Jr, Woo XY, Nagarajan N, Kumar V, Thalamuthu A, Poh WT, Ang AL, Mya HT, How GF, Yang LY, Koh LP, Chowbay B, Chang CT, Nadarajan VS, Chng WJ, Than H, Lim LC, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18:521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 40.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 41.Vajpai N, Strauss A, Fendrich G, Cowan-Jacob SW, Manley PW, Grzesiek S, Jahnke W. Solution conformations and dynamics of ABL kinase-inhibitor complexes determined by NMR substantiate the different binding modes of imatinib/nilotinib and dasatinib. J Biol Chem. 2008;283:18292–18302. doi: 10.1074/jbc.M801337200. [DOI] [PubMed] [Google Scholar]
- 42.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Levinson NM, Boxer SG. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS One. 2012;7:e29828. doi: 10.1371/journal.pone.0029828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lange T, Ernst T, Gruber FX, Maier J, Cross M, Muller MC, Niederwieser D, Hochhaus A, Pfirrmann M. The quantitative level of T315I mutated BCR-ABL predicts for major molecular response to second-line nilotinib or dasatinib treatment in patients with chronic myeloid leukemia. Haematologica. 2013;98:714–717. doi: 10.3324/haematol.2012.068890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker WT, Lawrence RM, Ho M, Irwin DL, Scott HS, Hughes TP, Branford S. Sensitive detection of BCR-ABL1 mutations in patients with chronic myeloid leukemia after imatinib resistance is predictive of outcome during subsequent therapy. J Clin Oncol. 2011;29:4250–4259. doi: 10.1200/JCO.2011.35.0934. [DOI] [PubMed] [Google Scholar]
- 46.Parker WT, Ho M, Scott HS, Hughes TP, Branford S. Poor response to second-line kinase inhibitors in chronic myeloid leukemia patients with multiple low-level mutations, irrespective of their resistance profile. Blood. 2012;119:2234–2238. doi: 10.1182/blood-2011-08-375535. [DOI] [PubMed] [Google Scholar]
- 47.Soverini S, De Benedittis C, Machova Polakova K, Brouckova A, Horner D, Iacono M, Castagnetti F, Gugliotta G, Palandri F, Papayannidis C, Iacobucci I, Venturi C, Bochicchio MT, Klamova H, Cattina F, Russo D, Bresciani P, Binotto G, Giannini B, Kohlmann A, Haferlach T, Roller A, Rosti G, Cavo M, Baccarani M, Martinelli G. Unraveling the complexity of tyrosine kinase inhibitor-resistant populations by ultra-deep sequencing of the BCR-ABL kinase domain. Blood. 2013;122:1634–1648. doi: 10.1182/blood-2013-03-487728. [DOI] [PubMed] [Google Scholar]
- 48.Ernst T, Erben P, Muller MC, Paschka P, Schenk T, Hoffmann J, Kreil S, La RP, Hehlmann R, Hochhaus A. Dynamics of BCR-ABL mutated clones prior to hematologic or cytogenetic resistance to imatinib. Haematologica. 2008;93:186–192. doi: 10.3324/haematol.11993. [DOI] [PubMed] [Google Scholar]
- 49.Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, Stoffregen EP, McWeeney S, Kovacs I, Park B, Druker BJ, Deininger MW. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106:2128–2137. doi: 10.1182/blood-2005-03-1036. [DOI] [PubMed] [Google Scholar]
- 50.Griswold IJ, MacPartlin M, Bumm T, Goss VL, O’Hare T, Lee KA, Corbin AS, Stoffregen EP, Smith C, Johnson K, Moseson EM, Wood LJ, Polakiewicz RD, Druker BJ, Deininger MW. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol. 2006;26:6082–6093. doi: 10.1128/MCB.02202-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, Komisopoulou E, Brown LM, Loo JA, Landaw EM, Sawyers CL, Graeber TG. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc Natl Acad Sci U S A. 2006;103:19466–19471. doi: 10.1073/pnas.0609239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khorashad JS, Kelley TW, Szankasi P, Mason CC, Soverini S, Adrian LT, Eide CA, Zabriskie MS, Lange T, Estrada JC, Pomicter AD, Eiring AM, Kraft IL, Anderson DJ, Gu Z, Alikian M, Reid AG, Foroni L, Marin D, Druker BJ, O’Hare T, Deininger MW. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood. 2013;121:489–498. doi: 10.1182/blood-2012-05-431379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–2569. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gibbons DL, Pricl S, Posocco P, Laurini E, Fermeglia M, Sun H, Talpaz M, Donato N, Quintas-Cardama A. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc Natl Acad Sci U S A. 2014;111:3550–3555. doi: 10.1073/pnas.1321173111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, Khoury HJ, Larson RA, Konopleva M, Cortes JE, Kantarjian H, Jobbour EJ, Kornblau SM, Lipton JH, Rea D, Stenke L, Barbany G, Lange T, Hernandez-Boluda J, Ossenkoppele GJ, Press RD, Chuah C, Soverini S, Rosti G, Rousselot P, Friedman R, Deininger M, Reynolds KR, Heaton WL, Eiring AM, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;ᅟ:ᅟ. doi: 10.1016/j.ccr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, Joha S, Idziorek T, Corm S, Hetuin D, Philippe N, Preudhomme C, Quesnel B. BCR-ABL mutants spread resistance to non-mutated cells through a paracrine mechanism. Leukemia. 2008;22:791–799. doi: 10.1038/leu.2008.3. [DOI] [PubMed] [Google Scholar]
- 57.Redaelli S, Mologni L, Rostagno R, Piazza R, Magistroni V, Ceccon M, Viltadi M, Flynn D, Gambacorti-Passerini C. Three novel patient-derived BCR/ABL mutants show different sensitivity to second and third generation tyrosine kinase inhibitors. Am J Hematol. 2012;87:E125–E128. doi: 10.1002/ajh.23338. [DOI] [PubMed] [Google Scholar]
- 58.Tyner JW, Yang WF, Bankhead A, 3rd, Fan G, Fletcher LB, Bryant J, Glover JM, Chang BH, Spurgeon SE, Fleming WH, Kovacsovics T, Gotlib JR, Oh ST, Deininger MW, Zwaan CM, Den Boer ML, van den Heuvel-Eibrink MM, O’Hare T, Druker BJ, Loriaux MM. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res. 2013;73:285–296. doi: 10.1158/0008-5472.CAN-12-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khorashad JS, Mason CC, Kraft IL, Reynolds KR, Pomicter AD, Eiring AM, Zabriskie MS, Iovino AJ, Heaton W, Tantravahi SK, Kauffman M, Schacham S, Chenchik A, Bonneau K, O’Hare T, Deininger MW. An unbiased shRNA library screen identifies nucleocytoplasmic transport as a potential target for treatment of chronic myeloid leukemia. Blood. 2013;122:2707. [Google Scholar]
- 60.Groarke JD, Cheng S, Moslehi J. Cancer-drug discovery and cardiovascular surveillance. N Engl J Med. 2013;369:1779–1781. doi: 10.1056/NEJMp1313140. [DOI] [PubMed] [Google Scholar]
- 61.Byrd JC, O’Brien S, James DF. Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:1278–1279. doi: 10.1056/NEJMoa1215637. [DOI] [PubMed] [Google Scholar]
- 62.Metzgeroth G, Schwaab J, Gosenca D, Fabarius A, Haferlach C, Hochhaus A, Cross NC, Hofmann WK, Reiter A. Long-term follow-up of treatment with imatinib in eosinophilia-associated myeloid/lymphoid neoplasms with PDGFR rearrangements in blast phase. Leukemia. 2013;27:2254–2256. doi: 10.1038/leu.2013.129. [DOI] [PubMed] [Google Scholar]
- 63.Cheah CY, Burbury K, Apperley JF, Huguet F, Pitini V, Gardembas M, Ross DM, Forrest D, Genet P, Rousselot P, Patton N, Smith G, Dunbar CE, Ito S, Aguiar RC, Odenike O, Gimelfarb A, Cross NC, Seymour JF. Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood. 2014;123:3574–3577. doi: 10.1182/blood-2014-02-555607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci R, Elliott O, Liso A, Ambrosetti A, Pulsoni A, Forconi F, Trentin L, Semenzato G, Inghirami G, Capponi M, Di RF, Patti C, Arcaini L, Musto P, Pileri S, Haferlach C, Schnittger S, Pizzolo G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dietrich S, Glimm H, Andrulis M, von Kalle C, Ho AD, Zenz T. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366:2038–2040. doi: 10.1056/NEJMc1202124. [DOI] [PubMed] [Google Scholar]
- 66.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, Dave SS, Zhang J, Yilmaz AS, Jaglowski SM, Blum KA, Lozanski A, Lozanski G, James DF, Barrientos JC, Lichter P, Stilgenbauer S, Buggy JJ, Chang BY, Johnson AJ, Byrd JC. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, Tefferi A, Malone J, Alam R, Schrier SL, Schmid J, Rose M, Vandenberghe P, Verhoef G, Boogaerts M, Wlodarska I, Kantarjian H, Marynen P, Coutre SE, Stone R, Gilliland DG. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 68.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 70.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, Martincorena I, Van Loo P, Jones AV, Guglielmelli P, Tarpey P, Harding HP, Fitzpatrick JD, Goudie CT, Ortmann CA, Loughran SJ, Raine K, Jones DR, Butler AP, Teague JW, O’Meara S, McLaren S, Bianchi M, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant’Antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schonegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 72.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, DeAngelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL. MPLW515L is anovel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:1140–1151. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH, Jr, Arcasoy MO, Hexner E, Lyons RM, Paquette R, Raza A, Vaddi K, Erickson-Viitanen S, Koumenis IL, Sun W, Sandor V, Kantarjian HM. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger MW, Miller CB, Silver RT, Talpaz M, Winton EF, Harvey JH, Jr, Arcasoy MO, Hexner EO, Lyons RM, Paquette R, Raza A, Vaddi K, Erickson-Viitanen S, Sun W, Sandor V, Kantarjian HM. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-I. Haematologica. 2013;98:1865–1871. doi: 10.3324/haematol.2013.092155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Milosevic JD, Kralovics R. Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol. 2013;97:183–197. doi: 10.1007/s12185-012-1235-2. [DOI] [PubMed] [Google Scholar]
- 76.Tam CS, Verstovsek S. Investigational Janus kinase inhibitors. Expert Opin Investig Drugs. 2013;22:687–699. doi: 10.1517/13543784.2013.774373. [DOI] [PubMed] [Google Scholar]
- 77.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–878. doi: 10.1038/nrc3143. [DOI] [PubMed] [Google Scholar]
- 78.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, Group B-S. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gazdar AF. Personalized medicine and inhibition of EGFR signaling in lung cancer. N Engl J Med. 2009;361:1018–1020. doi: 10.1056/NEJMe0905763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, Michellys PY, Awad MM, Yanagitani N, Kim S, Pferdekamper AC, Li J, Kasibhatla S, Sun F, Sun X, Hua S, McNamara P, Mahmood S, Lockerman EL, Fujita N, Nishio M, Harris JL, Shaw AT, Engelman JA. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, Riely GJ, Solomon BJ, Wolf J, Thomas M, Schuler M, Liu G, Santoro A, Lau YY, Goldwasser M, Boral AL, Engelman JA. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 83.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, Sequist LV, Iafrate AJ, Engelman JA. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 86.Klco JM, Spencer DH, Miller CA, Griffith M, Lamprecht TL, O’Laughlin M, Fronick C, Magrini V, Demeter RT, Fulton RS, Eades WC, Link DC, Graubert TA, Walter MJ, Mardis ER, Dipersio JF, Wilson RK, Ley TJ. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25:379–392. doi: 10.1016/j.ccr.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parker WT, Phillis SR, Yeung DT, Hughes TP, Scott HS, Branford S. Many BCR-ABL1 compound mutations reported in chronic myeloid leukemia patients may actually be artifacts due to PCR-mediated recombination. Blood. 2014;124:153–155. doi: 10.1182/blood-2014-05-573485. [DOI] [PubMed] [Google Scholar]
- 88.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O’Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]