

Abstract
Enhanced in vivo gene expression using non-viral vectors is a critical issue in gene therapy in general. Among the many potential utilities of non-viral vector-mediated gene delivery, its application in DNA-based vaccination is an attractive approach with several practical advantages over conventional vaccination. We have previously shown that the endosomolytic bacterial protein listeriolysin O (LLO) is capable of facilitating transfection in vitro using the LPDII (anionic liposome-polycation-DNA complexes) delivery system. In the present study we have extended and investigated the DNA delivery of LLO-containing LPDII to in vivo and evaluated its utility in DNA vaccination in mice. We further investigated the ability of this non-viral gene delivery system to elicit an immune response to a model antigen ovalbumin (OVA), particularly focusing on the OVA-specific CD8+ cytotoxic T lymphocyte (CTL) response, after delivery of a plasmid containing the OVA cDNA. A DNA prime and protein boost protocol was employed to generate cytotoxic T cell responses. Our results show that increased in vitro and in vivo transfection efficiencies were observed when LLO was incorporated into LPDII. This LLO-LPDII formulation produced an enhanced functional antigen-specific CD8+ T cell response in vivo compared to the heat-inactivated LLO-containing LPDII (HI-LLO-LPDII) formulation. Furthermore, a significantly higher CTL frequency was observed in the splenocytes isolated from the mice primed with LLO-LPDII by an enzyme-linked immunosorbent spot assay. Interferon-γ production upon specific stimulation by OVA-specific CD8+ peptide was also significantly stronger with the inclusion of LLO into LPDII. These findings suggest that the LLO-containing LPDII system possesses noteworthy potential as a candidate carrier for DNA vaccine delivery.
Keywords: listeriolysin O, LPDII, DNA delivery, vaccine, CTL response
1. Introduction
The field of gene therapy has progressed steadily yet is still in need of safer and more effective delivery systems. The utility of non-viral gene delivery vectors, despite their superior safety characteristics, has been limited primarily due to their lower transfection efficiency in vivo in comparison with their viral vector-mediated counterparts [1, 2]. Among the many potential therapeutic uses of non-viral vector-mediated gene delivery, DNA-based vaccination is an emerging application that has great potential for reducing infectious disease-induced morbidity and mortality worldwide. Since their inception, DNA-based vaccines have been tested and used to stimulate protective immunity against many infectious pathogens, malignancies, and autoimmune disorders in animal models [3]. DNA vaccines exhibit many advantages over traditional vaccines, including their safety, stability, cost-effectiveness of manufacturing, and flexibility in design via the incorporation of recombinant DNA technology [3–5]. However, while exogenous plasmid DNA (pDNA) can be used to elicit cellular immune responses against the DNA-encoded immunogens in small laboratory animals, they have proven less potent in human clinical trials [6]. Numerous efforts have been made to enhance the potency of DNA vaccines in animals and humans. Recently, DNA vaccines have made noteworthy progress in the area of animal health care products; there are currently at least four licensed DNA-based products for animals on the market, including one for porcine growth hormone releasing hormone supplementation and a vaccine against equine West Nile virus [7–9]. Of significant relevance, because of the pathology's similarities to the human counterpart, is one xenogeneic DNA-based vaccine employed with promising results in the treatment of canine melanoma [8, 10]. Currently, various approaches, such as combinations with adjuvant or cytokines and particulating techniques, have been applied to enhance immune responses to the encoded antigens [11]. The particulate delivery approach is promising because of enhanced pDNA stability and immunogenicity, and particularly auspicious when it is used in combination with molecular adjuvants.
LPDII (anionic liposome-polycation-DNA complexes) is a liposomal non-viral gene delivery system that was first developed by Lee and Huang et al. [12, 13] to overcome the disadvantages of cationic liposomal vectors such as cytotoxicity, lack of tissue specificity and non-specific binding to negatively charged extracellular matrix components, lipoproteins and cells in the biological environment [12]. LPDII contains an anionic lipid shell and a highly condensed core, in which DNA is compacted by a cationic polymer such as poly-lysine or protamine. Compared with traditional anionic and neutral liposomal vectors, DNA is highly condensed in LPDII and is quantitatively encapsulated without the requirement of using excess amounts of lipids [14]. However, the relatively low transfection efficiency of the anionic LPDII vectors calls for further modifications with other functional components, such as targeting moieties or endosomolytic agents.
Listeriolysin O (LLO) is a 58 kDa pore-forming protein and essential virulence factor of the intracellular pathogen Listeria monocytogenes (LM). After LM enters endocytic compartments, the secreted monomeric LLO binds to cholesterol-containing endosomal and lysosomal membranes, followed by LLO oligomerization and permeabilization of the membrane to promote the escape of LM into the cytosol [15]. LLO has been utilized in drug delivery systems and has demonstrated its powerful utility as an endosomolytic agent in delivering exogenous macromolecules into the cytosol [16–20]. Furthermore, LLO has optimal activity at the acidic pH of endosomes [21, 22], making it ideal for applications that require efficient release from endosomes. In our previous study, enhanced in vitro gene expression was observed when LLO was incorporated into the LPDII gene delivery system [23]. In the present study we have extended the in vitro transfection results of LLO-LPDII to an in vivo scenario using a reporter gene and a model antigen in a mouse model. In our effort to develop an improved non-viral gene delivery system incorporating the LLO mechanism that has higher gene expression in animals and particularly for DNA-based vaccine delivery systems, we selected the pDNA encoding ovalbumin (OVA) as a model antigen; OVA-encoding plasmid DNA was formulated into an LLO-containing LPDII delivery system and a DNA prime-protein boost vaccination protocol was employed to investigate the effectiveness of DNA vaccine-based prime in the LLO-containing LPDII system by monitoring the resulting antigen-specific cytotoxic T cell response. One of the promising vaccination strategies is the `prime-boost'; i.e., priming with DNA vaccine and boosting with protein subunit vaccine, peptides or live attenuated viruses [24]. A number of studies have demonstrated that immune responses using this strategy were more robust than with the DNA prime/ DNA boost protocol [25–27].
Our study design therefore focused on the mode of DNA prime, keeping all of the protein boosts constant (i.e., protein-LLO-liposome that has been demonstrated to be effective) and changed the parameters/delivery systems of DNA primes. This strategy is in keeping with the primary goal of this study; not necessarily to find the best vaccination protocol but more so to test the hypothesis that the LLO-containing liposome-based LPDII DNA delivery system works effectively in DNA delivery and serves as a significantly efficient DNA prime.
2. Materials and Methods
2.1. Mice, cell line and peptide
C57BL/6 mice (female) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were in accordance with and approved by the University of Michigan's Committee on the Use and Care of Animals (UCUCA). The murine macrophage cell line P388D1 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 100 U/ml penicillin, 100 U/ml streptomycin, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 1 mM sodium pyruvate (complete RPMI) at 37 °C in a 5% CO2 humidified atmosphere. OVA peptide SIINFEKL (amino acids 257–264) was synthesized (AnaSpec Corp., Fremont, CA, USA), dissolved in PBS and kept at −80 °C in aliquots. Enzymes used in constructing plasmids were purchased from New England Biolabs (Beverly, MA, USA). All chemicals and reagents were purchased from Thermo Scientific (Rockford, IL, USA) unless otherwise noted.
2.2. Construction and preparation of plasmid DNA
The plasmid pNGVL3 (~7 kb) encoding firefly luciferase under the control of the cytomegalovirus promoter was a gift from Dr. Gary Nabel (Vaccine Research Center, National Institutes of Health, MD, USA). pNGVL3-OVA was constructed by replacing the luciferase cDNA in pNGVL3 between the EcoRI site and XbaI sites with the ovalbumin cDNA fragment (kindly provided by Dr. Kenneth Rock at University of Massachusetts Medical School, MA, USA). Both plasmids were propagated in E.coli and purified using QIAGEN Giga EndoFree plasmid purification kits (Qiagen, Valencia, CA, USA). Concentrations of pDNA were determined spectrophotometrically using absorbance at 260 nm, with 260/280 ratios consistently being > 1.8.
2.3. Recombinant LLO purification
Recombinant LLO was produced in E. coli strain BL21 (DE3) transformed with the pET29b vector expressing LLO with a C-terminal six-histidine tag as previously described [18]. His-tagged LLO was affinity-purified using a Ni-NTA agarose column (Qiagen). Protein purity was analyzed by SDS-PAGE, followed by Sypro Red (Invitrogen) staining and visualization with a Typhoon 9200 fluorescence scanner (GE Healthcare, Piscataway, NJ, USA). Protein concentrations were determined by the bicinchoninic acid (BCA) assay (Thermo Scientific) using bovine serum albumin as a standard. The membrane pore-forming activity of LLO was monitored using an in vitro sheep red blood cell (RBC) hemolysis assay as previously described [18].
2.4. Preparation of anionic liposomes and LPDII
Liposomes were prepared using the lipid thin film hydration and freeze/thaw technique. Briefly, phosphatidylethanolamine (PE) (Avanti, Alabaster, AL, USA) and cholesteryl hemisuccinate (CHEMS) (Sigma-Aldrich, St. Louis, MO, USA) dissolved in chloroform or chloroform/methanol (1:1), respectively, were mixed at a 2:1 molar ratio and dried to a thin film in a Büchi Rotavapor R-200 rotary evaporator (Büchi Labortechnik AG, Flawil, Switzerland) under vacuum (consistently < 10 mm Hg) at 25 °C. The lipid film was hydrated using 0.5 ml of HEPES-buffered glucose (HBG; 10 mM HEPES, 280 mM glucose, pH 8.4) containing 100 μg LLO, and vortexed. The liposomes were subjected to five freeze/thaw cycles in an ethanol bath at −80 °C and sonicated 5 times 30 seconds in a bath-type sonicator (Laboratory Supplies Company, Hicksville, NY, USA). Liposomes were then passed through a 1 × 25 cm Sepharose CL-4B gel filtration column (GE Healthcare) equilibrated with HBG to remove unencapsulated LLO. Encapsulation efficiency of LLO was generally 15–25%. The concentration of total phospholipids was determined by measuring the concentration of phosphate using the method of Bartlett [28].
LPDII were prepared as described previously [23]. Briefly, DNA and protamine sulfate (grade III, Clupeine, Sigma-Aldrich) were both diluted from stocks with HBG and equal volumes of each were mixed to achieve a final DNA concentration of 150 μg/mL. After mixing, the solution was briefly vortexed and the resulting DNA-polycation complexes were incubated for 10 minutes at room temperature. Preformed LLO-containing anionic liposomes were subsequently added to the DNA/protamine polyplexes with mild vortexing to achieve the desired final component concentrations and ratios.
2.5. Preparation of OVA/LLO Liposomes
OVA/LLO liposomes were prepared as previously described [18]. Briefly, OVA (Sigma-Aldrich) and LLO were encapsulated inside PE/CHEMS liposomes at 20 mg/mL and 0.25 mg/mL, respectively. Unencapsulated protein was removed by purification using a 1 × 25 cm Sepharose CL-4B gel filtration column. The amount of encapsulated protein was determined by quantitative SDS-PAGE with Sypro Red stain and quantified using ImageQuant software (GE Healthcare) after acquisition of data on the Typhoon 9200.
2.6. In vitro transfection
P388D1 cells were seeded at a density of 1.5 × 105 cells per well in 24-well plates and cultured for 16–24 hr prior to transfection. Cells were typically ~70% confluent at the time of transfection. In all transfection assays, 200 μl of the sample containing 1 μg pDNA in serum-free or 10% serum-containing RPMI-1640 was added dropwise into each well. After 6 hr of incubation at 37 °C, the transfection complexes were replaced by fresh complete medium and cells were incubated for an additional 40 hr. Lipofectamine (Invitrogen) was used as a positive control in the in vitro transfection assay. The transgene expression of luciferase was analyzed by a luminometer equipped with an automated injector (VICTOR™ X, PerkinElmer, Waltham, MA, USA) using a luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer's protocol. The total protein content in each well was measured by the BCA assay. The transfection results were expressed as relative light units (RLU) per mg total cellular protein. All assays were performed in triplicate.
2.7. In vivo transfection efficiency
C57BL/6 mice (4–5 weeks old, female) in groups of four were i.v. injected via tail vein with 50 μg of pNGVL3 plasmid formulated in LLO-LPDII or heat-inactivated LLO-LPDII (HI-LLO-LPDII), and sacrificed 24 hr following injection. Heart, lung, spleen, liver and kidney were collected and washed twice with PBS at 4 °C, and the organs were homogenized with CCLR lysis buffer (Promega). The homogenates were centrifuged at 12,000 × g for 10 min at 4 °C and 20 μL of the supernatant was analyzed for luciferase activity as described above. The total protein concentrations in tissue lysates were determined using the BCA assay, and the transfection results were expressed as RLU per mg total tissue protein.
2.8. Immunization of mice
Groups of four female C57BL/6 mice (7–8 weeks old) were subcutaneously injected with PBS, HI-LLO-LPDII, LLO-LPDII or the protein formulation OVA/LLO liposomes on day 0. On day 12, all four groups of mice were boosted with the OVA protein formulation (OVA/LLO liposomes). The amount of pNGVL3-OVA DNA in the HI-LLO-LPDII or LLO-LPDII formulation was 50 or 75 μg over four independent sets of experiments, and was kept constant in each set. The dose of the OVA protein in OVA/LLO liposomes was 50 μg for each mouse. On day 24, mice were sacrificed for the in vivo cytotoxic T lymphocyte (CTL) assay and enzyme-linked immunosorbent spot (ELISPOT) assay.
2.9. In vivo CTL assay
The cytolytic activity of antigen-specific CD8+ T cells generated in the vaccinated mice was examined by a sensitive in vivo CTL killing assay, as previously described [29, 30]. Briefly, splenocytes from naive C57BL/6 mice were first depleted of RBCs using ACK buffer (Invitrogen) and then pulsed with 5 μM SIINFEKL peptide or culture medium at 37 °C for 1 hr. The two populations of splenocytes, peptide-pulsed (specific) and non-pulsed (control) target, were labeled at 37 °C for 8 min with 4 μM (high) and 0.4 μM (low) carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen), respectively. After labeling, excess CFSE was quenched by the addition of fetal bovine serum (FBS) to a final concentration of 20% (v/v). After washing the labeled cells 3 times, equal numbers of peptide-pulsed target cells (CFSEhigh) and non-pulsed cells (CFSElow) were mixed together, and a total of 107 labeled cells were intravenously administered into naive or vaccinated mice. At 16 hr after injection, splenocytes from the recipient mice were analyzed by flow cytometry in a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA) to determine the relative percentage of the injected labeled CFSEhigh and CFSElow cells. The two target populations, CFSEhigh and CFSElow cells, were distinguished based on the differences in their CFSE intensity. The percentage of specific lysis was calculated by the following formula: 100 × [1− (ratio of CFSEhigh /CFSElow cells recovered from naive mice/ ratio of CFSEhigh and CFSElow cells recovered from immunized mice)].
2.10. Interferon-γ (IFN-γ) ELISPOT assay
The frequency of antigen-specific IFN-γ-secreting cells was analyzed using the IFN-γ ELISPOT Mouse Set (BD Biosciences) following manufacturer's instructions. Briefly, a 96-well ELISPOT plate was coated with 5 μg/ml anti-mouse IFN-γ capture antibody overnight at 4 °C, then washed and blocked with complete culture medium for 2 hr at room temperature. Splenocytes from immunized mice were added to microwells along with antigen-specific peptide (SIINFEKL) and were incubated at 37 °C and 5% CO2 for 16 hr. Control cells were incubated either without peptide or with the nonspecific stimulator concanavalin A (Con A; Sigma-Aldrich) at a concentration of 1 μg/ml. The wells were extensively washed with PBS containing 0.05% Tween 20 and subsequently incubated with 2 μg/ml biotinylated anti-mouse IFN-γ detection antibody for 2 hr at room temperature. After washing, wells were incubated with 5 μg/ml streptavidin-horseradish peroxidase for 1 hr at room temperature, after which the wells were washed again and the final substrate was added. Color development was monitored, and stopped by washing with ddH2O. After drying overnight at room temperature, the number of spot-forming units (SFU) in each well was determined using a computerized ImmunoSpot Image Analyzer (Cellular Technology Ltd., Shaker Heights, OH, USA). Data are presented as mean SFU ± standard deviation (SD).
2.11. Antigen-Specific IFN-γ secretion from spleen cells
Antigen-specific cytokine response was determined by culturing the splenocytes (5 × 106 /ml) from the immunized mice in the presence or absence of the OVA peptide SIINFEKL (5 μM) in 96-well plates for 72 hr at 37 °C. IFN-γ concentration in the culture supernatant was measured by enzyme-linked immunosorbent assay (ELISA) performed in duplicate using the paired monoclonal antibodies (BD Biosciences). IFN-γ concentration was calculated based on recombinant mouse IFN-γ (BD Biosciences) standards and expressed as IFN-γ units/ml.
2.12. Statistical analysis
Any statistical differences were analyzed by paired two-tail Student's t-tests, and a p-value less than 0.05 was considered statistically significant.
3. Results
3.1. Transfection activity of LLO-LPDII in P388D1 cells
The transfection efficiency of the LLO-LPDII was first tested and confirmed in cell culture systems prior to performing animal studies. The macrophage-like cell line P388D1 was used to evaluate the in vitro transfection efficiency of the LLO-enhanced LPDII gene delivery system. To monitor the enhancement of delivery specifically mediated by LLO in the LPDII delivery system, a heat-inactivated LLO-LPDII (HI-LLO-LPDII) formulation was prepared as a negative control. In all experiments, half of the LLO-containing liposomes were heated at 75 °C for 10 min prior to complex formation; the LLO activity is abolished under these conditions as monitored by a hemolysis assay (data not shown). Typically the average diameter of LLO-LPDII determined by quasi-elastic light scattering using a Nicomp 380 ZLS instrument (Particle Sizing Systems, Santa Barbara, CA, USA) was ~179 ± 32 nm. Zeta potential measurements, also performed with a Nicomp 380 ZLS, showed that LLO-LPDII were negatively charged with a potential of −28.5 ± 2.8 mV.
Transfection using the anionic LPDII delivery platform was significantly enhanced by the incorporation of LLO as shown in Figure 1. Under serum-free conditions, the transfection efficiency of LLO-LPDII in P388D1 cells was approximately 120-fold higher than that of protamine-DNA complexes, and approximately 40-fold higher in comparison with that of HI-LLO-LPDII (Fig. 1). In order to investigate whether the LLO-LPDII delivery system is affected by the presence of serum, we also used the condition of 10% serum-containing media for comparison with the transfection in the absence of serum. As shown in Fig. 1, no significant difference was observed between the presence and absence of serum during the 6 hr incubation period using either the LLO-LPDII or HI-LLO-LPDII formulation. In comparison, the transfection efficiencies in serum-containing media were significantly decreased for both protamine-DNA complexes and Lipofectamine. In the presence of serum, transfection levels using the LLO-containing LPDII system were comparable to the commercially available cationic transfection reagent Lipofectamine, suggesting that the LLO-LPDII system is serum-compatible.
Fig. 1.
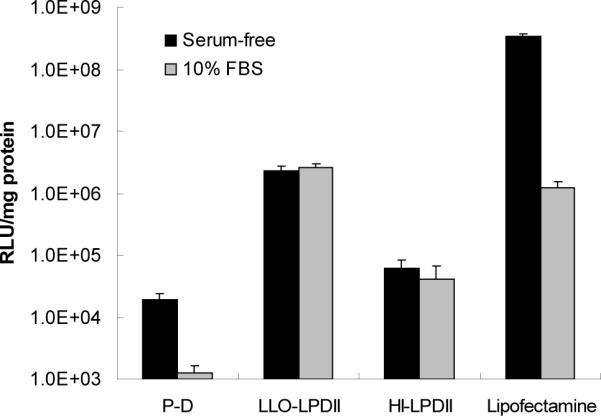
The presence of serum does not negatively impact LPDII-mediated transfection in a macrophage-like cell line. Plasmid DNA was condensed with protamine at a 1.2:1 weight ratio and incubated with P388D1 cells either alone (P-D), or after further complexation with either LLO-containing liposomes (LLO-LPDII) or Lipofectamine. As a negative control, heat-inactivated LPDII (HI-LPDII) were formulated by heating LLO-containing liposomes at 75 °C for 10 min. Plasmid DNA in various forms was incubated with cells at 1 μg per well under all conditions. The results are expressed as relative light units (RLU) of luciferase reporter gene expression per milligram of total cellular protein (n = 3).
3.2. Enhanced in vivo gene expression of LPDII by LLO incorporation
To further investigate the function of LLO in the LPDII gene delivery system in vivo, LLO-LPDII or heat-inactivated LLO-LPDII was administered to C57BL/6J mice. After intravenous injection, luciferase activity of the LLO-LPDII group was detectable in spleen, liver and lung, with the highest activity in spleen, followed by liver and then lung (Fig. 2). Consistent with the in vitro transfection results, the in vivo transfection efficiency of LLO-LPDII was much higher than that of heat-inactivated (HI) LLO-LPDII. The in vitro and in vivo transfection data suggest that LLO, as an endosomolytic agent, plays a very important role in the LLO-LPDII delivery system.
Fig. 2.
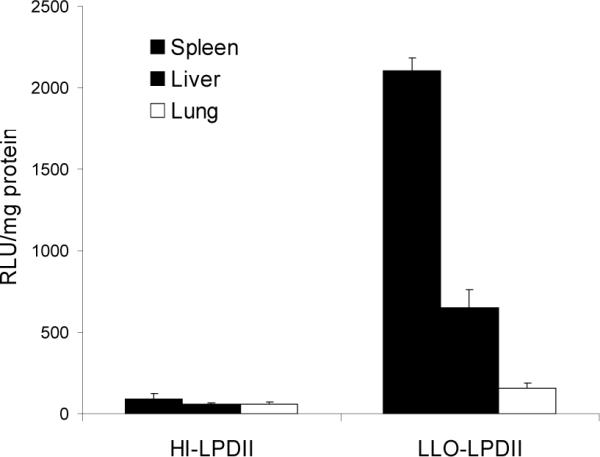
LLO facilitates the in vivo gene expression of LPDII-based plasmid DNA delivery. 50 μg of pNGVL3 formulated in LLO-LPDII or in heat-inactivated (HI) LLO-LPDII (HI-LPDII) was injected intravenously into each mouse. Twenty-four hours following injection, mice were sacrificed and major organs collected. Tissues were homogenized in lysis buffer and the supernatants were assayed for luciferase activity. The results are expressed as relative light units per milligram of total protein (n = 4).
3.3. Enhanced induction of OVA-specific CTL response by LLO-LPDII
Once enhanced gene expression was demonstrated in mice with the incorporation of LLO in LPDII, we further investigated its utility in DNA priming of vaccination by incorporating the pNGVL3-OVA plasmid DNA, which carries the ovalbumin cDNA under the control of the cytomegalovirus promoter, in the priming step. The expression of OVA protein from the constructed pNGVL3-OVA plasmid was first confirmed in HEK293 cells (ATCC) by Western blot analysis using an anti-OVA antibody (data not shown). Then, instead of the classical 51Cr release assay to detect CTL activity in vitro, we evaluated CTLs induced in vivo by using the intravital fluorogenic dye CFSE.
As described in the Materials and Methods section, a mixture containing equal amounts of SIINFEKL peptide-pulsed CFSEhigh and non-pulsed CFSElow cells from naive mice was intravenously injected into mice on day 24 post-vaccination. The cell-permeant and relatively non-fluorescent carboxyfluorescein diacetate succinimidyl ester passively diffuses into cells where cytosolic esterases remove the acetate groups to produce the cell-impermeant and fluorescent carboxyfluorescein succinimidyl ester, while the succinimidyl moiety covalently attaches to the primary amines of proteins [31]. As a result, the CFSE dye becomes trapped intracellularly unless the plasma membrane integrity is compromised, as in the case of antigen-specific CTL-mediated lysis, thereby allowing soluble proteins to diffuse out of the cell. The specific lysis of SIINFEKL-pulsed cells in vivo in vaccinated and control mice was analyzed by flow cytometry 16 hr after adoptive transfer of the labeled cells. As shown in Fig. 3A, SIINFEKL-pulsed (CFSEhigh) and non-pulsed spleen cells (CFSElow) were present at similar levels in the non-immunized animals. The activated splenocytes from the LLO-LPDII-primed mice were able to lyse 89.5% of the SIINFEKL-pulsed (CFSEhigh) target cells, which was significantly higher than that of PBS- or HI-LPDII-primed mice (Fig. 3B, p < 0.05). Meanwhile, no significant cytolytic activity differences were observed between LLO-LPDII-primed mice and the protein formulation-primed mice (p = 0.415).
Fig. 3.
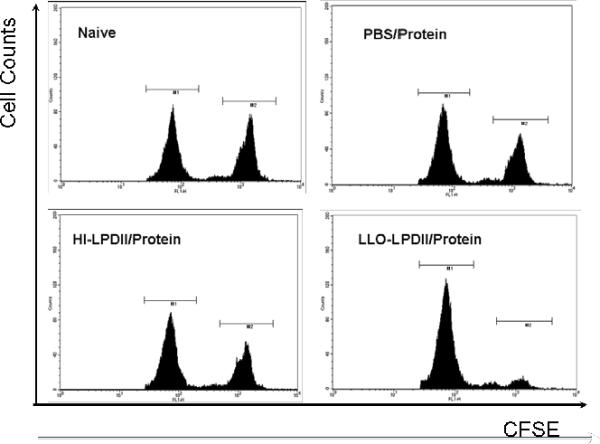

Enhanced functional cytolytic CD8+ T cells were detected in LLO-LPDII-primed mice by an in vivo CTL assay. (A) C57BL/6 mice were primed subcutaneously with PBS, 50 μg of OVA protein encapsulated in LLO-containing liposomes, 50 μg of pNGVL3.OVA in LLO-LPDII or in HI-LPDII, respectively. 12 days post-prime, all of the mice were boosted with 50 μg of OVA protein encapsulated in LLO-containing liposomes. 12 days post-boost, mice were i.v. injected with an equivalent amount of SIINFEKL-pulsed (labeled with 4 μM CFSE; CFSEhigh) and non-pulsed (labeled with 0.4 μM CFSE; CFSElow) splenocytes obtained from syngeneic naive donor mice. 16 hr after adoptive transfer of CFSEhigh and CFSElow cells, spleen cells from the recipient mice were harvested, and the proportions of the CFSEhigh and CFSElow cells were analyzed by flow cytometry. Representative flow cytometry data show counts of remaining non-pulsed CFSElow splenocytes (left peak) versus remaining SIINFEKL-pulsed CFSEhigh splenocytes (right peak) in histograms for naive, PBS prime/ protein boost, LLO-LPDII DNA prime/ protein boost, and HI-LLO-LPDII DNA prime/ protein boost mice. (B) Percentage of antigen peptide (SIINFEKL)-specific cell lysis is shown. The mean of the percentage from each group was compared to that of the PBS-primed group and was statistically analyzed (n = 4; * p < 0.05)
3.4. Increased antigen-specific T cell frequency and cytokine production by LLO-LPDII
The induction of IFN-γ-producing CD8+ T cells was evaluated by the ex vivo ELISPOT assay. This assay has the advantage of detecting only activated/memory T cells in addition to the sensitive detection of cytokine release at the single cell level. In good correlation with the in vivo CTL data (see the above section, Fig. 3), the antigen-specific IFN-γ secreting CTLs were twice as numerous in the LLO-LPDII-primed group as those in the HI-LLO-LPDII-primed group (Fig. 4A). Compared with the PBS-primed group, the LLO-LPDII-primed group exhibited a 2.5-fold increase in the SIINFEKL-specific IFN-γ producing CD8+ T cell frequencies. The difference between the LLO-LPDII-primed and protein-primed groups was not significant (p = 0.400). These results, along with the in vivo CTL data, demonstrate that the LLO-LPDII-prime was clearly superior to the HI-LLO-LPDII-prime, and as efficient as protein-LLO-liposome-based priming.
Fig. 4.
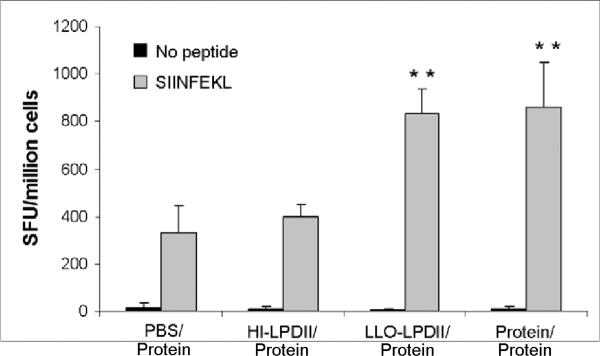
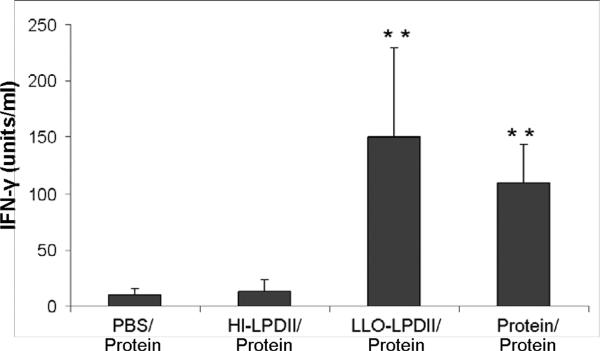
Higher antigen-specific IFN-γ-secreting T cell frequency (A) and enhanced IFN-γ secretion (B) generated by LLO-LPDII immunization. (A) Groups of four C57BL/6 mice were s.c. primed by either PBS, 50 μg of OVA protein encapsulated in LLO-containing liposomes, 75 μg of pNGVL3.OVA in HI-LPDII or LLO-LPDII, respectively. 12 days later, all the mice were boosted with 50 μg of OVA protein encapsulated in LLO-containing liposomes. OVA peptide-specific IFN-γ-secreting T cell frequency was monitored by ELISPOT assay at day 12 post-boost. Results are shown as IFN-γ-specific spot-forming units (SFU) per 106 cells (mean ± SD). The mean of SFU from each group was compared to that of the PBS-primed group and was statistically analyzed (** p < 0.01). (B) Spleen cells were harvested from the immunized mice and stimulated in vitro with OVA peptide (SIINFEKL) at 5 μM or with media only. IFN-γ in the culture supernatant after 72 hours was measured by ELISA. Results are shown as IFN-γ units/ml (mean ± SD). The mean IFN-γ concentration from each group was compared to that of the PBS primed-group and was statistically analyzed (** p < 0.01).
The antigen-specific cytokine (IFN-γ) response was monitored as an additional measure of cellular immunity generated by different vaccination modalities. Splenocytes from the mice primed with LLO-LPDII secreted significantly higher (p < 0.01) levels of IFN-γ (150.3 ± 78.6 units/ml) than those of the PBS-primed (10.2 ± 5.4 units/ml) and HI-LLO-LPD-primed groups (13.2 ± 9.9 units/ml) (Fig. 4B). The level of IFN-γ generated by splenocytes without the addition of peptide was not detectable in all of the groups. Splenocytes from all four groups exhibited a strong cytokine response when treated with the T cell mitogen ConA (data not shown). Similarly to the ELISPOT data, no significant difference was observed between the LLO-LPDII-primed and protein-LLO-liposome-primed groups.
4. Discussion
The development of plasmid DNA delivery vehicles that can effectively protect DNA from degradation in a physiological environment while increasing the efficiency of transport of DNA through multiple cellular barriers to the cytosol and nucleus is critical for the successful realization of gene therapy including DNA-based vaccination strategies. In this study, an LLO-incorporated LPDII platform was tested for the first time in vivo for enhanced efficiency over a non-LLO LPDII system, and further evaluated for its potential utility in a DNA prime/ protein boost vaccination scenario using a mouse model. Advantages of this delivery system include: (i) protection of DNA from degradation by DNases in the physiological milieu due to the extensive condensation by cationic protamine as well as complexed liposomes, (ii) greater compatibility of the LPDII anionic delivery system with physiological environments, and (iii) increased efficiency of endosomal escape of the delivered DNA, facilitated by the incorporated LLO, resulting in enhanced transfection efficiency. While their relative transfection efficiency may in general be lower than that of cationic delivery vehicles, an important attribute of anionic liposome-based LPDII delivery systems is their potential ability to function in the presence of serum. Interestingly, the LLO-incorporated LPDII appears to maintain the advantages of anionic delivery systems while concomitantly enhancing the transfection efficiency both in vitro and in vivo. We postulate that this improvement in overall efficiency of gene delivery results from the highly efficient LLO-mediated endosomal release of DNA. This conclusion is partly supported by our in vitro data in which LLO-LPDII particles showed transfection levels comparable to cationic lipid formulations in the presence of 10% FBS in the transfection media. Since biological fluids are unavoidable in vivo, such compatibility is essential to developing safe and effective gene delivery vehicles.
After intravenous injection into mice, the highest gene expression of LLO-LPDII was observed in spleen and liver. The underlying reason may be that upon injection into the circulation, LLO-LPDII particles, like other particulate drug carriers, are recognized as foreign by the reticuloendothelial system [32]. The fenestrated endothelia lining the capillaries of the liver and spleen would allow particles such as LLO-LPDII to diffuse into these tissues where they would encounter Kupffer cells and resident macrophages, respectively, resulting in their rapid removal from the general circulation following intravenous administration [33]. The in vivo gene expression profile observed upon i.v. injection would certainly be different from that of s.c. administered LLO-LPDII. However, it demonstrates that the effect of LLO incorporation in the LPDII is clearly reflected in animals, especially in comparison with the LPDII with heat-inactivated LLO. The in vivo gene expression profile of the anionic LLO-LPDII is quite different from those of previous reports of cationic LPD delivery systems [34, 35]. In the case of cationic LPD, the highest gene expression was observed in the lungs after intravenous injection, presumably because of agglutinates formed by electrostatic interaction between positively charged LPD and negatively charged RBCs [36]. The gene expression profile of LLO-LPDII after s.c. injection, which was used for DNA prime, was not examined as it was beyond the scope of this study; this remains to be addressed with a careful mechanistic investigation of the cell types and tissues that are transfected after s.c. injection in comparison with i.v. injection.
A consistent body of literature indicates that DNA immunization induces immune responses via professional antigen-presenting cells such as macrophages and dendritic cells [37]. Our in vitro transfection data demonstrate that LLO is capable of enhancing the efficiency of LPDII-mediated transfection in the macrophage-like cell line P388D1; this LLO-mediated effect on LPDII-mediated plasmid DNA was further confirmed in our in vivo immunization experiments.
A strong antigen-specific CTL response is an essential component of the protection from, and clearance of, intracellular infections and tumors; it follows that CD8+ CTL-mediated antigen-specific lysis of target cells (e.g., tumor cells, virus-infected cells) is desirable in many vaccine applications [38, 39]. Accordingly, the induction of antigen-specific cytotoxic effector cells, along with direct quantification and characterization of the response, is considered one of the most important criteria for evaluating DNA vaccination efficacy. We have employed two sensitive and specific methods, the in vivo CTL assay combined with an IFN-γ ELISPOT assay, to evaluate the relative efficiency of the LLO-LPDII delivery system in facilitating an antigen-specific CTL response. These results, obtained using two specific and distinct methods, complement each other in that the ELISPOT assay detects the ability of the peptide/ major histocompatibility complex I (MHC I) to activate CD8+ T cells via the detection of IFN-γ secretion by activated T cells, whereas the in vivo CTL assay detects the ability of the specific effector cells to kill the antigen peptide-bearing target cells [40]. Our results clearly demonstrate that the LLO-LPDII-primed group generates a significantly stronger antigen-specific CTL response than the PBS-primed or heat-inactivated LLO-LPDII-primed groups.
Several studies have recently suggested that heterologous prime-boost strategies incorporating two distinct forms of antigens and vectors (DNA and protein) are far superior at inducing immune responses compared with homologous boosting [26, 41]. The advantages of this approach include potential synergistic effects on the induction of an immune response and the generation of a robust T cell-mediated immune response [42]. In the present study, we have used a DNA prime and protein boost protocol to investigate the immunogenicity of a model antigen expressed from a DNA-based vaccine delivered via LLO-LPDII. The question of what would be the best boost after DNA prime was not the major focus of this study although it would certainly be important in moving the proposed strategy toward a successful application in vaccines. Therefore the boost was kept constant, i.e., protein in LLO-liposomes, and did not include DNA in the formulation. The protein formulation for boost is prepared by co-encapsulation of LLO and OVA into pH-sensitive liposomes, as previously reported by us [18]. Exogenous soluble protein antigens in general, typical for subunit protein-based vaccines, do not gain efficient entry into the cytosolic compartment to generate MHC I-dependent antigen presentation. However, it has been demonstrated that pH-sensitive LLO-liposomes carrying OVA antigen, the protein boost formulation utilized in the current study, are capable of inducing robust OVA-specific CTL responses [18]; all of the immunized mice were boosted using this protein formulation in the investigation reported here. As a positive control, one group of mice was both primed and boosted by the protein formulation. Our data suggest that both the LLO-LPDII-primed group and the protein-primed group could elicit equally strong antigen-specific CTL activities.
In the present study, E. coli was employed to produce both plasmid DNA and recombinant LLO. As a gram-negative bacterium, E. coli contains lipopolysaccharide (LPS) in the outer membrane of its cell wall, which may have co-purified with plasmid DNA and the recombinant LLO. As one of the best studied immunostimulatory components of bacteria, LPS has been shown to interact with toll-like receptor 4 to elicit innate and adaptive immune responses [43, 44]. However, as shown in Figures 3 and 4, the CTL activity generated by HI-LPDII-primed group is much lower than that of the LLO-LPDII-primed group, and no significantly different CTL activity was observed between the PBS-primed and the HI-LPDII-primed groups. As mentioned before, the HI-LPDII formulation was prepared by heating LLO-containing liposomes at 75 °C for 10 min prior to complex formation, a condition that inactivates LLO but not LPS. Thus, our data suggest that the enhanced CTL activity of LLO-LPDII was generated by DNA immunization, and rules out the effect of residual LPS, if any, in the formulations.
In conclusion, LLO is capable of facilitating in vitro and in vivo gene delivery of the anionic LPDII platform. The LLO-containing LPDII system is a potent delivery vehicle for DNA-based vaccines and can induce enhanced cellular responses, thus shown as a potent DNA prime formulation in the DNA prime/ protein boost regimen of vaccination. The LLO-containing LPDII system has enormous potential as a candidate carrier for DNA-based vaccine delivery and possibly for general gene therapy.
Acknowledgements
This research was supported by National Institutes of Health grant numbers AI047173 and AI058080. We would like to thank Dr. Suna Choi, Na Hyung Kim, Chasity Andrews and Stefanie Goodell for their knowledge and support in preparing this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kawakami S, Higuchi Y, Hashida M. Nonviral approaches for targeted delivery of plasmid DNA and oligonucleotide. Journal of Pharmaceutical Sciences. 2008;97(2):726–745. doi: 10.1002/jps.21024. [DOI] [PubMed] [Google Scholar]
- [2].Mintzer MA, Simanek EE. Nonviral Vectors for Gene Delivery. Chemical Reviews. 2008;109(2):259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- [3].Shedlock DJ, Weiner DB. DNA vaccination: antigen presentation and the induction of immunity. J Leukoc Biol. 2000;68(6):793–806. [PubMed] [Google Scholar]
- [4].Bivas-Benita M, Ottenhoff TH, Junginger HE, Borchard G. Pulmonary DNA vaccination: concepts, possibilities and perspectives. J Control Release. 2005;107(1):1–29. doi: 10.1016/j.jconrel.2005.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Huygen K. Plasmid DNA vaccination. Microbes Infect. 2005;7(5–6):932–938. doi: 10.1016/j.micinf.2005.03.010. [DOI] [PubMed] [Google Scholar]
- [6].Wang R, Doolan DL, Le TP, Hedstrom RC, Coonan KM, Charoenvit Y, Jones TR, Hobart P, Margalith M, Ng J, Weiss WR, Sedegah M, de Taisne C, Norman JA, Hoffman SL. Induction of antigen-specific cytotoxic T lymphocytes in humans by a malaria DNA vaccine. Science. 1998;282(5388):476–480. doi: 10.1126/science.282.5388.476. [DOI] [PubMed] [Google Scholar]
- [7].Frelin L, Sallberg M. A small step closer to the Holy Grail of DNA vaccines: undisputed clinical benefit in humans. Genome Med. 2009;1(1):15. doi: 10.1186/gm15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9(10):776–788. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Khan AS, Draghia-Akli R, Shypailo RJ, Ellis KI, Mersmann H, Fiorotto ML. A Comparison of the Growth Responses Following Intramuscular GHRH Plasmid Administration Versus Daily Growth Hormone Injections in Young Pigs. Mol Ther. 2009;18(2):327–333. doi: 10.1038/mt.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bergman PJ, Camps-Palau MA, McKnight JA, Leibman NF, Craft DM, Leung C, Liao J, Riviere I, Sadelain M, Hohenhaus AE, Gregor P, Houghton AN, Perales MA, Wolchok JD. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the Animal Medical Center. Vaccine. 2006;24(21):4582–4585. doi: 10.1016/j.vaccine.2005.08.027. [DOI] [PubMed] [Google Scholar]
- [11].Yoshikawa T, Imazu S, Gao JQ, Hayashi K, Tsuda Y, Shimokawa M, Sugita T, Niwa T, Oda A, Akashi M, Tsutsumi Y, Mayumi T, Nakagawa S. Augmentation of antigen-specific immune responses using DNA-fusogenic liposome vaccine. Biochem Biophys Res Commun. 2004;325(2):500–505. doi: 10.1016/j.bbrc.2004.10.056. [DOI] [PubMed] [Google Scholar]
- [12].Lee RJ, Huang L. Folate-targeted, anionic liposome-entrapped polylysine-condensed DNA for tumor cell-specific gene transfer. J Biol Chem. 1996;271(14):8481–8487. doi: 10.1074/jbc.271.14.8481. [DOI] [PubMed] [Google Scholar]
- [13].Shi G, Guo W, Stephenson SM, Lee RJ. Efficient intracellular drug and gene delivery using folate receptor-targeted pH-sensitive liposomes composed of cationic/anionic lipid combinations. Journal of Controlled Release. 2002;80(1–3):309–319. doi: 10.1016/s0168-3659(02)00017-2. [DOI] [PubMed] [Google Scholar]
- [14].Banerjee R. Liposomes: applications in medicine. J Biomater Appl. 2001;16(1):3–21. doi: 10.1106/RA7U-1V9C-RV7C-8QXL. [DOI] [PubMed] [Google Scholar]
- [15].Portnoy DA, Auerbuch V, Glomski IJ. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol. 2002;158(3):409–414. doi: 10.1083/jcb.200205009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Choi S, Lee KD. Enhanced gene delivery using disulfide-crosslinked low molecular weight polyethylenimine with listeriolysin o-polyethylenimine disulfide conjugate. J Control Release. 2008;131(1):70–76. doi: 10.1016/j.jconrel.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee KD, Oh YK, Portnoy DA, Swanson JA. Delivery of macromolecules into cytosol using liposomes containing hemolysin from Listeria monocytogenes. J Biol Chem. 1996;271(13):7249–7252. [PubMed] [Google Scholar]
- [18].Mandal M, Lee KD. Listeriolysin O-liposome-mediated cytosolic delivery of macromolecule antigen in vivo: enhancement of antigen-specific cytotoxic T lymphocyte frequency, activity, and tumor protection. Biochim Biophys Acta. 2002;1563(1–2):7–17. doi: 10.1016/s0005-2736(02)00368-1. [DOI] [PubMed] [Google Scholar]
- [19].Mathew E, Hardee GE, Bennett CF, Lee KD. Cytosolic delivery of antisense oligonucleotides by listeriolysin O-containing liposomes. Gene Ther. 2003;10(13):1105–1115. doi: 10.1038/sj.gt.3301966. [DOI] [PubMed] [Google Scholar]
- [20].Saito G, Amidon GL, Lee KD. Enhanced cytosolic delivery of plasmid DNA by a sulfhydryl-activatable listeriolysin O/protamine conjugate utilizing cellular reducing potential. Gene Ther. 2003;10(1):72–83. doi: 10.1038/sj.gt.3301859. [DOI] [PubMed] [Google Scholar]
- [21].Portnoy DA, Jones S. The cell biology of Listeria monocytogenes infection (escape from a vacuole) Ann N Y Acad Sci. 1994;730:15–25. doi: 10.1111/j.1749-6632.1994.tb44235.x. [DOI] [PubMed] [Google Scholar]
- [22].Beauregard KE, Lee KD, Collier RJ, Swanson JA. pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J Exp Med. 1997;186(7):1159–1163. doi: 10.1084/jem.186.7.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lorenzi GL, Lee KD. Enhanced plasmid DNA delivery using anionic LPDII by listeriolysin O incorporation. J Gene Med. 2005;7(8):1077–1085. doi: 10.1002/jgm.750. [DOI] [PubMed] [Google Scholar]
- [24].Ramshaw IA, Ramsay AJ. The prime-boost strategy: exciting prospects for improved vaccination. Immunol Today. 2000;21(4):163–165. doi: 10.1016/s0167-5699(00)01612-1. [DOI] [PubMed] [Google Scholar]
- [25].Li Y, Jin J, Yang Y, Bian Z, Chen Z, Fan M. Enhanced immunogenicity of an anti-caries vaccine encoding a cell-surface protein antigen of Streptococcus mutans by intranasal DNA prime-protein boost immunization. J Gene Med. 2009;11(11):1039–1047. doi: 10.1002/jgm.1386. [DOI] [PubMed] [Google Scholar]
- [26].Tanghe A, D'Souza S, Rosseels V, Denis O, Ottenhoff TH, Dalemans W, Wheeler C, Huygen K. Improved immunogenicity and protective efficacy of a tuberculosis DNA vaccine encoding Ag85 by protein boosting. Infect Immun. 2001;69(5):3041–3047. doi: 10.1128/IAI.69.5.3041-3047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yu S, Feng X, Shu T, Matano T, Hasegawa M, Wang X, Ma H, Li H, Li Z, Zeng Y. Potent specific immune responses induced by prime-boost-boost strategies based on DNA, adenovirus, and Sendai virus vectors expressing gag gene of Chinese HIV-1 subtype B. Vaccine. 2008;26(48):6124–6131. doi: 10.1016/j.vaccine.2008.09.017. [DOI] [PubMed] [Google Scholar]
- [28].Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234(3):466–468. [PubMed] [Google Scholar]
- [29].Byers AM, Kemball CC, Moser JM, Lukacher AE. Cutting edge: rapid in vivo CTL activity by polyoma virus-specific effector and memory CD8+ T cells. J Immunol. 2003;171(1):17–21. doi: 10.4049/jimmunol.171.1.17. [DOI] [PubMed] [Google Scholar]
- [30].Chen W, Huang L. Induction of cytotoxic T-lymphocytes and antitumor activity by a liposomal lipopeptide vaccine. Mol Pharm. 2008;5(3):464–471. doi: 10.1021/mp700126c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Weston SA, Parish CR. New fluorescent dyes for lymphocyte migration studies: Analysis by flow cytometry and fluorescence microscopy. Journal of Immunological Methods. 1990;133(1):87–97. doi: 10.1016/0022-1759(90)90322-m. [DOI] [PubMed] [Google Scholar]
- [32].Juliano RL. Factors affecting the clearance kinetics and tissue distribution of liposomes, microspheres and emulsions. Advanced Drug Delivery Reviews. 1988;2(1):31–54. [Google Scholar]
- [33].Mumtaz S, Ghosh PC, Bachhawat BK. Design of liposomes for circumventing the reticuloendothelial cells. Glycobiology. 1991;1(5):505–510. doi: 10.1093/glycob/1.5.505. [DOI] [PubMed] [Google Scholar]
- [34].Li S, Huang L. In vivo gene transfer via intravenous administration of cationic lipidprotamine-DNA (LPD) complexes. Gene Ther. 1997;4(9):891–900. doi: 10.1038/sj.gt.3300482. [DOI] [PubMed] [Google Scholar]
- [35].Li S, Rizzo MA, Bhattacharya S, Huang L. Characterization of cationic lipidprotamine-DNA (LPD) complexes for intravenous gene delivery. Gene Ther. 1998;5(7):930–937. doi: 10.1038/sj.gt.3300683. [DOI] [PubMed] [Google Scholar]
- [36].Eliyahu H, Servel N, Domb AJ, Barenholz Y. Lipoplex-induced hemagglutination: potential involvement in intravenous gene delivery. Gene Ther. 2002;9(13):850–858. doi: 10.1038/sj.gt.3301705. [DOI] [PubMed] [Google Scholar]
- [37].Heinzerling L, Basch V, Maloy K, Johansen P, Senti G, Wuthrich B, Storni T, Kundig TM. Critical role for DNA vaccination frequency in induction of antigen-specific cytotoxic responses. Vaccine. 2006;24(9):1389–1394. doi: 10.1016/j.vaccine.2005.09.018. [DOI] [PubMed] [Google Scholar]
- [38].Oldstone MB. The role of cytotoxic T lymphocytes in infectious disease: history, criteria, and state of the art. Curr Top Microbiol Immunol. 1994;189:1–8. doi: 10.1007/978-3-642-78530-6_1. [DOI] [PubMed] [Google Scholar]
- [39].Shimizu K, Thomas EK, Giedlin M, Mule JJ. Enhancement of tumor lysate- and peptide-pulsed dendritic cell-based vaccines by the addition of foreign helper protein. Cancer Res. 2001;61(6):2618–2624. [PubMed] [Google Scholar]
- [40].Chen J, Wu Q, Yang P, Hsu HC, Mountz JD. Determination of specific CD4 and CD8 T cell epitopes after AAV2- and AAV8-hF.IX gene therapy. Mol Ther. 2006;13(2):260–269. doi: 10.1016/j.ymthe.2005.10.006. [DOI] [PubMed] [Google Scholar]
- [41].Xiao-wen H, Shu-han S, Zhen-lin H, Jun L, Lei J, Feng-juan Z, Ya-nan Z, Ying-jun G. Augmented humoral and cellular immune responses of a hepatitis B DNA vaccine encoding HBsAg by protein boosting. Vaccine. 2005;23(14):1649–1656. doi: 10.1016/j.vaccine.2004.10.013. [DOI] [PubMed] [Google Scholar]
- [42].Yang K, Whalen BJ, Tirabassi RS, Selin LK, Levchenko TS, Torchilin VP, Kislauskis EH, Guberski DL. A DNA vaccine prime followed by a liposome-encapsulated protein boost confers enhanced mucosal immune responses and protection. J Immunol. 2008;180(9):6159–6167. doi: 10.4049/jimmunol.180.9.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zacharowski K, Zacharowski PA, Koch A, Baban A, Tran N, Berkels R, Papewalis C, Schulze-Osthoff K, Knuefermann P, Zahringer U, Schumann RR, Rettori V, McCann SM, Bornstein SR. Toll-like receptor 4 plays a crucial role in the immune-adrenal response to systemic inflammatory response syndrome. Proc Natl Acad Sci U S A. 2006;103(16):6392–6397. doi: 10.1073/pnas.0601527103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lu Y-C, Yeh W-C, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]