

Abstract
Purpose
The transcription factor Sp1 controls number of cellular processes by regulating the expression of critical cell cycle, differentiation and apoptosis-related genes containing proximal GC/GT-rich promoter elements. We here provide both experimental and clinical evidence that Sp1 plays an important regulatory role in MM cell growth and survival.
Experimental design
We have investigated the functional Sp1 activity in MM cells using a plasmid with renilla luciferase reporter gene driven by Sp1-responsive promoter. We have also used both SiRNA and ShRNA-mediated Sp1 knock-down to investigate the growth and survival effects of Sp1 on MM cells, and further investigated the anti-MM activity of Terameprocol (TMP), a small molecule which specifically competes with Sp1-DNA binding in vitro and in vivo.
Results
We have confirmed high Sp1 activity in MM cells which is further induced by adhesion to bone marrow stromal cells (BMSC). Sp1 knock down decreases MM cell proliferation and induces apoptosis. Sp1-DNA binding inhibition by TMP inhibits MM cell growth both in vitro and in vivo, inducing caspase 9-dependent apoptosis and overcoming the protective effects of BMSCs.
Conclusions
Our results demonstrate Sp1 as an important transcription factor in myeloma that can be therapeutically targeted for clinical application by TMP.
Introduction
Specificity Protein 1 (Sp1) and other Sp and Kruppel-like factor (KLF) proteins are members of a family of transcription factors which bind GC/GT-rich promoter elements through three C2H2-type zinc fingers that are present at their C-terminal domains (1).
Sp1 regulates gene expression both by direct interaction with promoter elements as well as via protein-protein interactions or interplay with other transcription factors, such as Ets-1, c-myc, c-Jun, Stat1, and Egr-1, and/or components of the basal transcriptional machinery (2). Sp1 has been also linked to chromatin remodeling through interactions with chromatin-modifying factors such as p300 (3) and histone deacetylases (HDACs) (4).
Although Sp1 has been considered as a ubiquitous transcription factor, increasing evidence suggests that it plays a major role in regulating expression of cell differentiation, cell cycle and apoptosis related genes affecting cellular growth (5). Sp1 levels and/or activity are increased in multiple cancers including breast (6), colon (7), gastric (8), pancreatic (9-12), and thyroid cancer (13) as compared with normal tissues. Elevated Sp1 expression is inversely correlated with the survival of patients with gastric cancer (14) and identifies advanced stage tumors and predicts a poor clinical outcome in primary pancreatic adenocarcinoma (12). On the other hand, interference of Sp1 activity has been shown to suppress tumor cell growth (15, 16) as well as tumor formation in athymic mice (6, 17) suggesting that Sp1 plays a central regulatory role in controlling number of pathways of tumor development and progression and thus may be an attractive therapeutic target.
Sp1 could contribute to transformation via regulation of the expression of Sp1-responsive genes including those supporting cell growth (c-jun, Raf, cyclins, E2F1, TGF-β, IEX-1 and TCL1)(18, 19) and apoptosis (Bcl-2, Survivin)(5, 20). Sp1 also regulates genes involved in angiogenesis and metastasis, including VEGF, uPA (21), PSA, and MT1-MMP (22). Enhanced Sp1 activity is related to both increased Sp1 gene expression and its posttranslational modification; for example, Sp1 phosphorylation regulates target genes in both positive and negative directions (23, 24).
Although Sp1 has been described to modulate autocrine IL-6 secretion by MM cells affecting its growth (25), its role in MM pathobiology remains unexplored. The promoter of several genes such as NF-kB p65, IGF-IR, VEGF, hTERT and IL-6 that regulate MM cell growth, cell cycle progression, survival and apoptosis contains proximal GC-rich promoter sequences that interact with Sp1 protein for their optimal expression(25-28). In addition, the same kinase pathways that have been shown to increase Sp1 phosphorylation and the transactivation of target genes (21, 22) are also known to mediate proliferation, survival, drug resistance, and migration (ERK, Jak/STAT, PI3K/Akt, and PKC signaling cascades, respectively) in MM.
Here we report significant role of Sp1 in myeloma cell growth and survival. Our results suggest that Sp1 activity can be therapeutically targeted for clinical application in MM.
Materials and Methods
Cells
Bone marrow mononuclear cells (BMMNCs) and primary MM cells from BM aspirates from MM patients following informed consent and Dana-Farber Cancer Institute IRB approval were isolated using Ficoll-Hypaque density gradient sedimentation. MM patient cells were separated from BM samples by antibody-mediated positive selection using anti-CD138 magnetic activated cell separation microbeads (Miltenyi Biotech, Gladbach, Germany). BMSC were established as previously described (29). MM cell lines were cultured in RPMI 1640 (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS). The IL-6 dependent MM cell lines INA-6 (kindly provided from Dr R. Burger, University of Kiel, Germany) were cultured presence of 2.5 ng/mL rhIL-6 (R&D Systems, Minneapolis, MN).
Reagents
TMP was synthesized and dissolved in CPE as previously described (30).
Cell proliferation assay
MM cell proliferation was measured by [3H]-thymidine (Perkin-Elmer, Boston, MA) incorporation assay as previously described (29).
Bromodesoxyuridine (BrdU) staining
The proportion of myeloma cells in S-phase was determined by incorporation of BrdU. 1 × 106 MM cells were exposed to 10 μg/ml of BrdU for 30 min. The cells were then harvested and stained with FITC anti-BrdU Ab and 7-AAD using a BrdU Flow Kit (BD Bioscience Pharmingen) according to the manufacturer’s directions. Cells were analyzed by flow cytometric analysis with a Becton-Dickinson FACScan flow cytometer.
Apoptosis assay
Apoptosis was evaluated by flow cytometric analysis following Annexin-V and propidium iodide (PI) staining.
Sp1 binding activity
The Sp1 binding activity was analyzed using the Transcription Factor ELISA kit, a DNA-binding enzyme-linked immunosorbent assay (ELISA)–based assay (Panomics). Sp1 transcription factor binding to its consensus sequence on the plate-bound oligonucleotide was studied from nuclear extracts, following the manufacturer’s procedure. Briefly, nuclear proteins were extracted with a Nuclear Extraction kit (Panomics) and quantified using the Bio-Rad Protein Assay kit (Bio-Rad). A total of 15 μg of nuclear protein from each treatment were analyzed. Sp1 antibody was used as primary antibody and anti-rabbit IgG horseradish peroxidase was used as secondary antibody. The absorbance was measured at a wavelength of 450 nm on a spectrophotometer.
Promoter activity assay
The Sp1 promoter reporter constructs was purchased from Sabiosciences. To examine transcriptional regulation of the Sp1 promoter by TMP, MM cells were transiently transfected with 1 μg of Sp1 reporter plasmid or empty vector control by electroporation using AMAXA technology according to the manufacturer’s instructions. Luciferase assays were performed with a Luminoskan Ascent 2.4 luminometer and the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase values were normalized to Renilla luciferase activity and were either expressed as relative luciferase units (RLU) or as mean ‘fold induction’.
Sp1 knock down
siRNA
RNA interference was performed using the TranSilent Human Sp1 siRNA (Panomics, Inc., Redwood, CA) following the manufacturer’s instructions. Nontargeting scrambled negative control siRNA (Panomics, Inc., Redwood, CA) was used as negative control. Briefly, U266 and OPM2 cells were seeded to 80% confluence in six-well plates in triplicate and transiently transfected with 2 μM of Sp1 siRNA by electroporation using AMAXA technology.
shRNA
Lentiviral shRNA were used to knockdown Sp1 expression in MM cells. Scrambled and Sp1 pLKO shRNA vectors were provided by Dr. William Hahn (Dana-Farber Cancer Institute). Recombinant lentivirus was produced by transient infection of 293T cells following a standard protocol, as previously described (31).
Immunoblotting
Whole cell lysates, nuclear extracts or cytsolic fractions of lysates (30 μg) were subjected to SDS-PAGE using “Precast Gel” (Bio-Rad Laboratores, Melville, NY), transferred to a nitrocellulose membrane (Bio-Rad), and immunoblotted with anti -Sp1 (Abcam) -Survivin, -Caspase-3,-7,-8,-9, -PARP (Cell Signaling Technology, Danvers, MA) antibodies (Abs). After incubating with secondary Ab, membranes were developed by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ).
In vivo study
s.c. model
The in vivo efficacy of TMP was tested in a murine xenograft model of MM using U266, MM1S or OPM2 MM cell lines injected s.c. in SCID mice. Following detection of tumor, mice were treated with either vehicle or TMP (50 mg/Kg) s.c. for 5 consecutive days/week for 2 weeks. Tumor growth was measured as previously described (32). Excised tumors from mice were immediately fixed and stored in 10% formalin. The fixed tissue was then dehydrated through a series of graded alcohols and xylene and embedded in paraffin. The paraffin tissue blocks were thin sectioned and stained for microscopy with H&E or analyzed by immunocytochemical methods for Ki67, tunel and caspase-3. The survivin level analysis was performed following staining with a combination of anti-CD138 and anti-survivin antibody. Appropriate immunofluorescent secondary antibodies were applied along with the nuclear dye DAPI. The tissue sections were imaged and the relative amount of survivin localized within tumor-cell nuclear compartment was determined using an automatated quantitation of antigen expression (AQUA) analysis. The data reflects the results of 12 fields imaged at 200x magnification per tumor.
SCID-hu model
Six- to eight week old male CB-17 severe combined immunodeficient (SCID) mice (Taconic, Germantown, NY) were housed and monitored in our Animal Research Facility. All experimental procedures and protocols had been approved by the Institutional Animal Care and Use Committee (VA Boston Healthcare System). Human fetal bone grafts were s.c. implanted into SCID mice (SCID-hu) as previously described. Four weeks following bone implantation, 3 × 106 INA-6 MM cells were injected directly into the human bone implant. Mouse sera were serially monitored for shuIL-6R by ELISA (R&D Systems, Inc., Minneapolis, MN).
Statistical analysis
The statistical significance of differences was analyzed using the t test; differences were considered significant when P≤ 0.05. Tumor growth inhibition and Kaplan–Meier survival analysis were determined using the Graphpad analysis software.
Results
High Sp1 protein expression and activity in MM
Sp1 is ubiquitously expressed in cells; however, its nuclear localization is important for functional activity. We first evaluated nuclear Sp1 protein levels in MM cell lines and normal PBMC. All MM cell lines had high nuclear Sp1 expression while normal PBMCs had predominantly cytoplasmic Sp1 with relatively small amount of nuclear localization (Fig. 1A-B). We further confirmed increased Sp1 nuclear levels and binding activity in MM cells compared to PBMC and BMSC using an ELISA-based Sp1 binding assay (Fig. 1C).
Figure 1. High Sp1 protein expression and activity in MM cells.
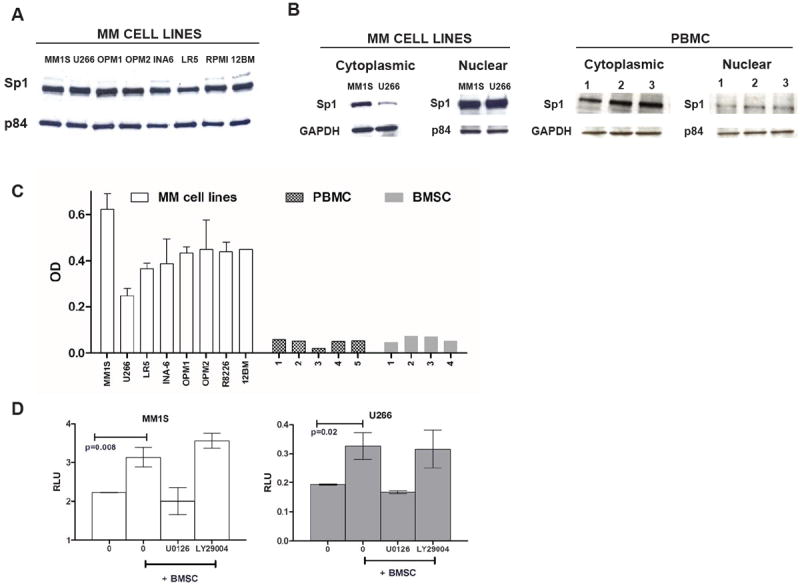
(A) Nuclear extracts from 8 MM cell lines were subjected to WB analysis using anti-Sp1 and p84 Abs. (B) Nuclear and cytoplasmic extracts from MM1S, U266 and PBMCs from 3 healthy donors were subjected to WB analysis using anti-Sp1 and GAPDH or p84 Abs. (C) 15 μg of nuclear proteins were analyzed for Sp1 activity using the Sp1 TF ELISA kit which measures Sp1 DNA binding activity. Absorbance was obtained with a spectrophotometer at 450 nm and presented as OD. (D) MM1S and U266 cells were transiently transfected with either negative control (NC)-luc or Sp1-promoter driven -Luc. After 48 h from transfection, MM cells were treated with 10 μM of U0126 or LY29004 or control for 30 minutes, washed and then cultured in absence (-) or presence (+) of BMSC for additional 6 h. The firefly luciferase activity was measured in cell lysate and normalized according to Renilla luciferase activity and expressed as relative luciferase units (RLU) to reflect the Sp1 promoter activity in the absence or presence of BMSC. The graph shows one representative experiment of two performed in triplicate. Results are shown as mean ± standard deviation.
As activation of Akt and Erk signaling pathways induces nuclear translocation and activation of Sp1 (21) and these pathways are activated in MM cells by their interaction with BM microenvironment, we investigated whether the presence of BMSC could modulate Sp1 activity in MM cells. Using a plasmid containing renilla luciferase reporter gene driven by Sp1-responsive promoter, we observed that the interaction between MM cells and BMSC significantly induced the transcriptional activity of Sp1 in MM cells (Fig. 1D). This increase was completely abrogated by the ERK pathway inhibitor U0126 but not by the AKT inhibitor LY29004.
Sp1 knock-down decreases MM cell proliferation
We have further evaluated the role of Sp1 in MM by analyzing the effect of Sp1 knockdown on MM cell growth and survival. We first knocked down endogenous Sp1 by RNA interference in U266 MM cells. Western blot and ELISA-based analysis confirmed reduction in both cytoplasmic and nuclear levels of Sp1 protein following transient transfection of MM cells with Sp1 siRNA compared with cells transfected with control scrambled siRNA (Fig. 2A-B). Interestingly, we also found that reduction of Sp1 levels was associated with inhibition of MM cell proliferation (Fig. 2C) and significant changes in the cell cycle with increase in G2/M and decrease in S phases (Fig. 2D). We have confirmed these data using OPM2 cells (Fig. 2E). Additionally, using 5 different Sp1-specific shRNA constructs, we have confirmed the inhibitory effect of Sp1 knockdown on MM1S cell proliferation and Sp1 activity (Fig. 2F). The cell populations with the largest reduction in Sp1 protein (shRNA #2, #3, #5) showed the greatest cell growth inhibition, compared to the vector control cell lines. Two of the Sp1 shRNAs that do not efficiently knock down Sp1 has less or no effect on phenotype, indirectly but reliably confirming the specificity of the effect. We used these 3 constructs to confirm ShRNA-related results with U266 cell line (Fig. 2G). Together, these results demonstrate the role of Sp1 in MM cell growth and survival.
Figure 2. Sp1 know-down decreases MM cell proliferation.
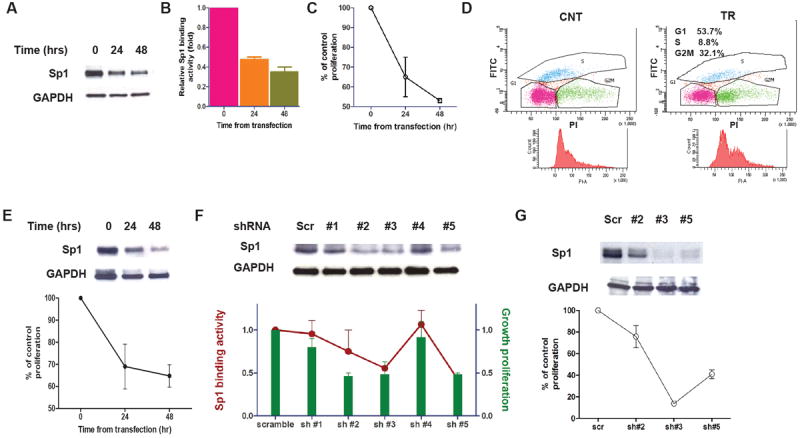
(A) U266 cells were transfected with TranSilent Human Sp1 siRNA. Cell lysates were obtained at indicated time and subjected to WB analysis to assess decrease in the Sp1 protein expression post-transfection using anti-Sp1 and GAPDH Abs. (B) The effect of Sp1 knockdown on Sp1 binding activity in MM cells transfected with Sp1 or control siRNA were was assessed by Sp1 TF ELISA and presented as proportional change from control cells. (C) The effect of Sp1 knockdown on cell proliferation in MM cells transfected with Sp1 or control siRNA was assessed by [3H]thymidine uptake, and presented as percentage of control cells. Data represent mean +/- SD of 3 independent experiments performed in triplicate. (D) 48 hours post-transfection with Sp1 or control siRNA, the measurement of cell incorporated BrdU (with FITC anti-BrdU) and total DNA content (with 7-AAD) in U266 cells allowed for the discrimination of cell subsets that resided in G0/G1, S, or G2/M phases of the cell cycle. (E) OPM2 cells were transfected with TranSilent Human Sp1 siRNA. Cell lysates were obtained at indicated time and subjected to WB analysis (upper panel), and cell proliferation was assessed by [3H]thymidine uptake at the indicated post-transfection time. (F) MM1S cells were infected with either scrambled (sc) or 5 different Sp1 shRNA (sh#1, sh#2, sh#3, sh#4, sh#5). Cell lysates were subjected to WB with anti-Sp1 and GAPDH Abs (upper panel). The transfected cells were analyzed for Sp1 binding activity (line) and cell growth (columns) 24 h after the second transfection by Sp1 TF ELISA and [3H]thymidine uptake respectively. The results are presented as change from cells infected with scramble shRNA (lower panel). (G) U266 cells were infected with either scrambled (sc) or 3 different Sp1 shRNA (sh#2, sh#3, sh#5). Cell lysates were subjected to WB with anti-Sp1 and GAPDH Abs (upper panel), and cell proliferation was assessed 24 h after the second transfection by [3H]thymidine uptake. The results are presented as change from cells infected with scramble shRNA (lower panel).
Inhibition of Sp1 activity by a chemical inhibitor induces MM cell growth arrest and apoptosis via caspases activation and reduction in survivin protein level
Our next approach was based on selective interference of Sp1- mediated transactivation of genes with TMP, a lignan tetra-O-methyl nordihydroguaiaretic acid derivative, which has been shown to specifically bind to Sp1-specific DNA binding domains within the responsive gene promoter regions and interfere with the transcription of these Sp1-controlled genes (33-36). We have confirmed decreased DNA binding activity of Sp1 by TMP as assessed by ELISA-based Sp1 binding assay (Fig. 3A), along with decreased basal and BMSC-induced Sp1 transcriptional activity (Fig. 3B). Importantly, Sp1 protein level in MM cells was not affected by 24 hours TMP treatment (Fig. 3C), suggesting that the modulation of Sp1 binding activity by TMP was not due to change of Sp1 protein level. We have confirmed these data using an additional MM cell line, observing that longer exposure to TMP led to decrease Sp1 protein expression (supplemental Figure 1).
Figure 3. Inhibition of Sp1 binding and transcriptional activity correlates with MM cell growth arrest.
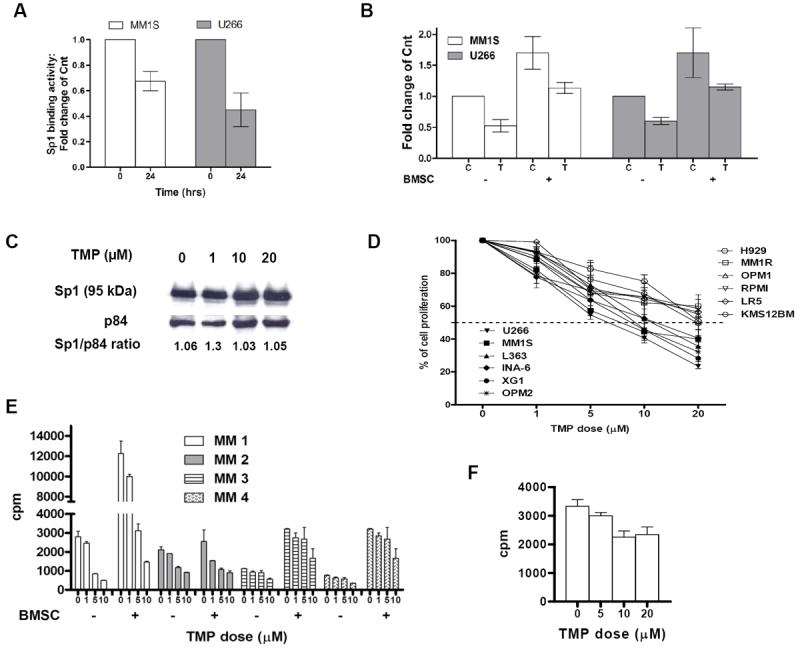
(A) U266 cells were cultured in the presence of 20 μM TMP for 24 hours (hrs) and nuclear extracts were subjected to Sp1 ELISA assay to assess Sp1 binding to its consensus sequence on the plate-bound oligonucleotide. (B) MM cells were transiently transfected with Sp1-Luc plasmid and 48 h post-transfection MM cell were cultured in absence (-) or presence (+) of BMSC and treated with placebo (C) or 20 μM of TMP (T) for additional 6 h. Luciferase activity was measured. Results are reported as mean of fold change from control (untreated cells). Mean values were calculated from five independent experiments and are shown as mean ± standard deviation. (C) U266 cells were treated with placebo or different concentrations of TMP (1-20 μM) for 24 h. Nuclear extracts were subjected to WB analysis using anti-Sp1 and p84 Abs to assess Sp1 protein levels. The ratio of Sp1 to p84 for each sample as assessed by densitometric quantitation of band intensity from WB is denoted. (D) Several MM cell lines were treated with various concentrations of TMP (1-20 μM) for 24 h and MM cell growth was assessed by [3H]thymidine uptake. Data are presented as % of vehicle-treated cell proliferation. (E) Primary CD138+ MM cells were cultured in the absence (-) or presence (+) of BMSC at different concentration of TMP for 24 hours. Cell proliferation was assessed by [3H]thymidine uptake, and expressed as cpm (count per minute). (F) BMSC from MM patients were treated with different concentration of TMP for 48 hours and cell proliferation was assessed by [3H]thymidine uptake, and expressed as cpm.
We next assessed the effect of the TMP-mediated inhibition of Sp1 binding activity on MM cell growth. We have examined the effect of the Sp1 inhibitor in MM cell lines with constitutive activation of the canonical and/or non-canonical NFkB pathways. TMP significantly inhibited DNA synthesis in all MM cells lines tested in a dose-dependent fashion (Fig 3D). In the most sensitive cell line the IC50 is in the range of 1-10 uM, while in the less sensitive cell lines the IC50 is in the range of 10-20 uM for a 24 hours period of treatment. Importantly, TMP inhibited proliferation of primary patient MM cells overcoming the MM growth promoting effect of BMSC (Fig. 3E) without affecting the viability of the normal BMSC (Fig. 3F).
Following exposure to TMP, we have also observed increase in G2/M and decrease in S phases of the cell cycle (Fig. 4A), as well as late induction of apoptotic cell death (Fig. 4B) in MM cells. We have further observed activation of the mitochondrial apoptotic pathway by TMP via activation of caspase-9, -3 and -7 and PARP cleavage while caspase-8 was not activated (Fig. 4C). Change in the protein expression of survivin, a known anti-apoptotic gene transcriptionally regulated by Sp1 (37), was confirmed following TMP treatment (Fig. 4C). Since caspase activation is a relatively late event, the reduction in survivin protein level during TMP-induced apoptosis appears to be an early event not mediated by caspase-dependent pathway. We have also observed decrease in cyclin-dependent kinase 1 (cdk1), a Sp1-regulated and cell cycle-controlling gene following exposure to TMP (data not shown). Finally, since Sp1 is an important regulator of the expression of important angiogenic factors, including VEGF which is believed to play a critical role in myeloma angiogenesis, we aimed to evaluate if inhibition of Sp1 binding activity by TMP impairs VEGF production by MM cells, As shown in supplemental Fig 2, six hours exposure to TMP were sufficient to decrease VEGF levels in the culture supernatant of both MM1S and U266.
Figure 4. TMP induces apoptosis via caspase activation.
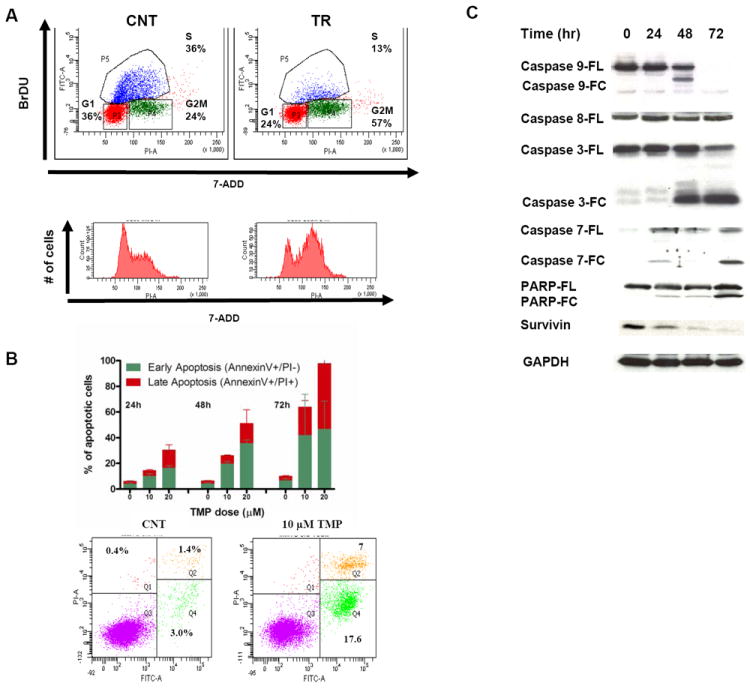
(A) Flow cytometric cell cycle analysis of BrdU incorporation was performed after treatment of cells with the inhibitor for 24 h. Data shown are percentage of cells in the different phases of the cell cycle. (B) MM1S were cultured in the absence or presence of TMP and apoptotic cell death was assessed by flow cytometric analysis following AnnexinV and propidium iodide staining. In the upper panel it is shown the % of AnnexinV+/PI- (early apoptosis) and AnnexinV+/PI+ (late apoptosis) cells at the indicated time, while a representative experiment (48 hours post treatment) is shown in the lower panel. (C) Whole cell lysate from MM1S cells treated with TMP (10μM) for the indicated time periods was subjected to Western blot analysis and probed with antibodies against caspases 3, 8, 9, 7, PARP, Survivin, cdc2 with GAPDH as loading control.
Thus, the antitumor activity of TMP may result, at least in part, from suppression of Sp1 activity and the consequent down-regulation of downstream targets that are key to cell growth, apoptosis and angiogenesis.
TMP inhibits MM cell growth and prolongs survival in vivo in a xenograft murine model of MM
Next, we investigated the anti-MM effect of Sp1 inhibition by TMP in vivo in 3 different murine xenograft models of human myeloma as well as in the SCID-hu model of human myeloma. In the xenograft models, we injected subcutaneously (s.c.) 3 different MM cell lines, U266, OPM2 and MM1S in SCID mice. Following detection of tumor, mice were treated with either 50 mg/kg TMP or placebo s.c. daily for 3 weeks. Tumors were measured in two perpendicular dimensions once every 3 days. Treatment with TMP, compared to vehicle alone, significantly inhibited MM tumor growth in all 3 murine models of MM (Fig. 5A). As seen in Fig. 5B, treatment with TMP also significantly prolonged survival in treated animals compared to control (p=0.017); the median overall survival was 18 days in the control group and 28 days in the TMP-treated group. TMP-related toxicity was not observed in mice, as determined by daily evaluation of activity and overall body weight change during the course of treatment. Histological examinations of tumor retrieved from MM1S-bearing mice confirmed decreased proliferation (as highlighted by Ki-67 staining), significant tumor cell apoptosis (caspase-3 and TUNEL staining) (Fig. 5C) as well as decreased expression of Survivin (Fig. 5D) following TMP treatment in vivo.
Figure 5. TMP inhibits MM cell growth and prolongs survival in vivo.
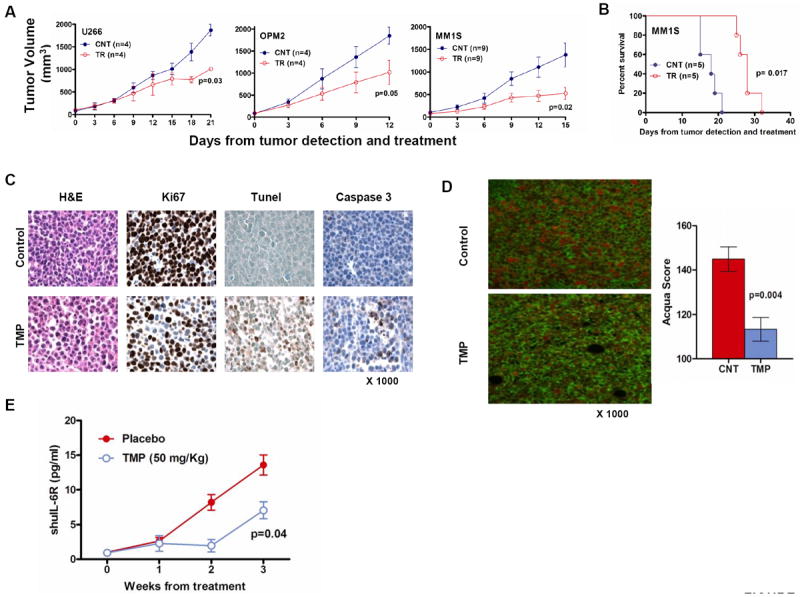
(A) U266, OPM2 ansd MM1s cells were injected s.c. in 3 different cohorts of SCID mice. Following detection of tumor, mice were treated with either 2 mg TMP or placebo s.c. daily for 3 weeks. Tumors were measured in two perpendicular dimensions once every 3 days. (B) Survival was evaluated from the first day of treatment until death using the Graphpad analysis software (C) Tumors were isolated from TMP-treated and control mice and sections were evaluated by histological examinations following Ki67 staining showing decreased proliferation; caspase3 and TUNEL stains showing significant tumor cell apoptosis. (D) Evaluation of survivin levels was performed in the paraffin embedded tumor sections stained with a combination of primary antibodies specific for CD138 and survivin. The tissue sections were imaged and the relative amount of survivin localized within tumor-cell nuclear compartment was determined using Automatated quantitation of antigen expression (AQUA) analysis. The data reflects the results of 12 fields imaged at 200x magnification per tumor. (E) In the SCID-hu model, mice, at the first detection of tumor, were treated with vehicle (n=3) or TMP (n=3). Serum samples were collected weekly and level of shuIL-6R was measured by ELISA as a marker of tumor growth. Baseline values before treatment were not significantly different among groups.
In the SCID-hu model, 4 weeks after implanting human fetal bone in mice, human myeloma cells are injected in the bone and sIL-6R levels are measured in murine blood as a marker of myeloma tumor growth. SCID-hu mice were injected with TMP five times a week for 3 weeks after first detection of shuIL-6R in mice. We observed significant antitumor activity of TMP as measured by shuIL-6R levels in murine blood, suggesting MM cell growth inhibitory effects of TMP (Fig. 5E).
Discussion
Alteration in expression and function of transcription factors has been frequently associated with neoplastic transformation. In this study, we provide experimental evidence that Sp1, a transcription factor that controls number of cellular processes, plays an important regulatory role in MM cell growth and survival.
Although Sp1 is ubiquitously expressed, its nuclear localization, observed in MM is functionally important. We have confirmed high Sp1 activity in MM cells both by demonstrating increased DNA binding as well as increased Sp1-responsive promoter activity measured by luciferase reporter assay. We here further confirmed effect of Sp1 on MM cell growth by using both SiRNA and ShRNA-mediated Sp1 knock-down using multiple constructs.
MM cell-BMSC interaction induces transcription and secretion of cytokines and growth factors that in turn confer proliferation and survival of MM cells. We here observed that this interaction leads to Sp1 activation; and inhibition of Sp1 activity by TMP led to suppression of MM-BMSC interaction-mediated growth of MM cells. Moreover, MM-BMSC interaction induces the activation of several signaling pathways, which in turn lead to Sp1 phosphorylation and transactivation of target genes. In line with these observations we report that the MM-BMSC-induced increase in both DNA binding and Sp1 transcriptional activity in MM cells was completely abrogated by inhibition of ERK pathway.
Compounds, such as TMP, able to disrupt the interaction between Sp1 and GC-rich motifs inhibit Sp1 activity. We have confirmed specific inhibition of both Sp1 binding and transcriptional activity in MM cells by TMP, including in the context of MM-BMSC interaction, without direct effect on Sp1 protein expression. Along with inhibition of Sp1 activity, we observed both in vitro and in vivo anti-myeloma effect of TMP. Importantly, there was no significant synergistic effect when MM cells transfected with Sp1 siRNA were treated with TMP (data not shown), confirming specificity of TMP’s mechanism of action. These results provide the rationale to evaluate efficacy of TMP in MM. TMP is currently in phase I/II clinical development for the treatment of glioma, treatment-refractory solid tumors and cervical dysplasia (38).
Altered survivin expression may be one of the mechanisms by which Sp1 may affect MM cell survival. Survivin is an inhibitor of apoptosis and a possible modulator of the terminal effector phase of cell death/survival and is highly expressed in number of human cancers including MM, but not in normal adult human tissue. Transcription of survivin is modulated by Sp1 (39-43) and in pancreatic cancer cells, inhibition of Sp1 activity has been shown to decrease survivin expression and subsequently sensitize the cells to radiotherapy (43, 44).
The importance of Sp1 in myeloma is also supported by the recent observation that conventional and novel anti-MM drugs have direct effect on Sp1 activity. For example, it has been shown that HDAC1 could interact with Sp1 to regulate its activity (45) and HDAC inhibitors induce Sp1 activity (46) suggesting potential for synergism by using HDAC and Sp1 inhibitors together. Interestingly, Bortezomib has been shown to inhibit DNA–binding activity of Sp1and disrupt the physical interaction of Sp1/NF-kB (47, 48). In MM, Bortezomib specifically down-regulates the expression of class I HDACs through caspase-8-dependent degradation of Sp1 protein (49). Having defined the cellular, signaling and the molecular mechanisms of sensitivity of MM to TMP, rationally designed combinations of conventional and novel agents to enhance cytotoxicity, to avoid or overcome drug resistance and to minimize adverse side effect profiles could be developed. More recently, it has been reported that both lenalidomide and pomalidomide upregulate Sp1 providing a rationale for their preclinical evaluation to increase cytotoxicity and overcome drug resistance (50).
In conclusion, we report significant role of Sp1 in myeloma cell growth and survival with its influence on clinical outcome in MM. Our preclinical in vitro and in vivo results suggest that specific inhibition of Sp1 activity may be an interesting potential therapeutic target alone and in combination with other agents in MM.
Supplementary Material
(A) MM1S cells were treated with placebo or different concentrations of TMP (1-20 μM) for 24 h. Nuclear extracts were subjected to WB analysis using anti-Sp1 and p84 Abs to assess Sp1 protein levels. The ratio of Sp1 to p84 for each sample as assessed by densitometric quantitation of band intensity from WB is denoted. (B) MM1S cells were treated with placebo or 20 μM TMP for the indicated time periods and nuclear extracts subjected to WB analysis using indicated Abs.
MM1S and U266 cells were incubated in the presence or absence of TMP for 6 h. After the culture period, the conditioned media were collected and VEGF levels were measured by ELISA assay. Data are presented as fold change inhibition of VEGF production as compared with the control culture. Values are means +/- SD from 3 independent experiments.
Statement of translational relevance.
The transcription factor Sp1 affects growth and metastatic potential of tumor cells. In MM, key genes such as NF-k p65, IGF-IR, VEGF, and IL-6 contain proximal GC-rich promoter sequences, and their interactions with Sp proteins are critical for their expression. Here we show that Sp1 plays an important role in myeloma cell growth and survival. Importantly, Sp1-specific inhibitor is able to induce tumor apoptosis in murine models of MM. Thus inhibition of Sp1 may be an attractive therapeutic modality in MM, alone or in combination with other agents.
Acknowledgments
This work was supported in part by DF/HCC myeloma SPORE Career Development Award to MF, grants from the Dept. of Veterans Affairs Merit Review Awards and from the National Institutes of Health Grants RO1-124929, and PO1-155249 to N.C.M., P50-100007 and PO1-78378 to NCM and KCA.
Footnotes
Author contributions: M.F. and N.C.M. designed research; M.F., P.N., and S.A. performed research; S.R. performed pathology; Y.T., R.P., S.M., P.T., T.H., C.L. and H.A. contributed reagents/analytic tools; M.F., K.C.A. and N.C.M analyzed data; M.F. and N.C.M wrote the manuscript.
References
- 1.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 2.Naar AM, Ryu S, Tjian R. Cofactor requirements for transcriptional activation by Sp1. Cold Spring Harb Symp Quant Biol. 1998;63:189–99. doi: 10.1101/sqb.1998.63.189. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki T, Kimura A, Nagai R, Horikoshi M. Regulation of interaction of the acetyltransferase region of p300 and the DNA-binding domain of Sp1 on and through DNA binding. Genes Cells. 2000;5:29–41. doi: 10.1046/j.1365-2443.2000.00302.x. [DOI] [PubMed] [Google Scholar]
- 4.Zhao S, Venkatasubbarao K, Li S, Freeman JW. Requirement of a specific Sp1 site for histone deacetylase-mediated repression of transforming growth factor beta Type II receptor expression in human pancreatic cancer cells. Cancer Res. 2003;63:2624–30. [PubMed] [Google Scholar]
- 5.Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–60. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 6.Abdelrahim M, Samudio I, Smith R, 3rd, Burghardt R, Safe S. Small inhibitory RNA duplexes for Sp1 mRNA block basal and estrogen-induced gene expression and cell cycle progression in MCF-7 breast cancer cells. J Biol Chem. 2002;277:28815–22. doi: 10.1074/jbc.M203828200. [DOI] [PubMed] [Google Scholar]
- 7.Abdelrahim M, Safe S. Cyclooxygenase-2 inhibitors decrease vascular endothelial growth factor expression in colon cancer cells by enhanced degradation of Sp1 and Sp4 proteins. Mol Pharmacol. 2005;68:317–29. doi: 10.1124/mol.105.011825. [DOI] [PubMed] [Google Scholar]
- 8.Kanai M, Wei D, Li Q, Jia Z, Ajani J, Le X, et al. Loss of Kruppel-like factor 4 expression contributes to Sp1 overexpression and human gastric cancer development and progression. Clin Cancer Res. 2006;12:6395–402. doi: 10.1158/1078-0432.CCR-06-1034. [DOI] [PubMed] [Google Scholar]
- 9.Shi Q, Le X, Abbruzzese JL, Peng Z, Qian CN, Tang H, et al. Constitutive Sp1 activity is essential for differential constitutive expression of vascular endothelial growth factor in human pancreatic adenocarcinoma. Cancer Res. 2001;61:4143–54. [PubMed] [Google Scholar]
- 10.Abdelrahim M, Baker CH, Abbruzzese JL, Sheikh-Hamad D, Liu S, Cho SD, et al. Regulation of vascular endothelial growth factor receptor-1 expression by specificity proteins 1, 3, and 4 in pancreatic cancer cells. Cancer Res. 2007;67:3286–94. doi: 10.1158/0008-5472.CAN-06-3831. [DOI] [PubMed] [Google Scholar]
- 11.Jungert K, Buck A, von Wichert G, Adler G, Konig A, Buchholz M, et al. Sp1 is required for transforming growth factor-beta-induced mesenchymal transition and migration in pancreatic cancer cells. Cancer Res. 2007;67:1563–70. doi: 10.1158/0008-5472.CAN-06-1670. [DOI] [PubMed] [Google Scholar]
- 12.Jiang NY, Woda BA, Banner BF, Whalen GF, Dresser KA, Lu D. Sp1, a new biomarker that identifies a subset of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2008;17:1648–52. doi: 10.1158/1055-9965.EPI-07-2791. [DOI] [PubMed] [Google Scholar]
- 13.Chiefari E, Brunetti A, Arturi F, Bidart JM, Russo D, Schlumberger M, et al. Increased expression of AP2 and Sp1 transcription factors in human thyroid tumors: a role in NIS expression regulation? BMC Cancer. 2002;2:35. doi: 10.1186/1471-2407-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Wei D, Huang S, Peng Z, Le X, Wu TT, et al. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin Cancer Res. 2003;9:6371–80. [PubMed] [Google Scholar]
- 15.Yuan P, Wang L, Wei D, Zhang J, Jia Z, Li Q, et al. Therapeutic inhibition of Sp1 expression in growing tumors by mithramycin a correlates directly with potent antiangiogenic effects on human pancreatic cancer. Cancer. 2007;110:2682–90. doi: 10.1002/cncr.23092. [DOI] [PubMed] [Google Scholar]
- 16.Ishibashi H, Nakagawa K, Onimaru M, Castellanous EJ, Kaneda Y, Nakashima Y, et al. Sp1 decoy transfected to carcinoma cells suppresses the expression of vascular endothelial growth factor, transforming growth factor beta1, and tissue factor and also cell growth and invasion activities. Cancer Res. 2000;60:6531–6. [PubMed] [Google Scholar]
- 17.Lou Z, O’Reilly S, Liang H, Maher VM, Sleight SD, McCormick JJ. Down-regulation of overexpressed sp1 protein in human fibrosarcoma cell lines inhibits tumor formation. Cancer Res. 2005;65:1007–17. [PubMed] [Google Scholar]
- 18.Abdelrahim M, Liu S, Safe S. Induction of endoplasmic reticulum-induced stress genes in Panc-1 pancreatic cancer cells is dependent on Sp proteins. J Biol Chem. 2005;280:16508–13. doi: 10.1074/jbc.C500030200. [DOI] [PubMed] [Google Scholar]
- 19.Chuang JY, Wang YT, Yeh SH, Liu YW, Chang WC, Hung JJ. Phosphorylation by c-Jun NH2-terminal kinase 1 regulates the stability of transcription factor Sp1 during mitosis. Mol Biol Cell. 2008;19:1139–51. doi: 10.1091/mbc.E07-09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verrecchia F, Rossert J, Mauviel A. Blocking sp1 transcription factor broadly inhibits extracellular matrix gene expression in vitro and in vivo: implications for the treatment of tissue fibrosis. J Invest Dermatol. 2001;116:755–63. doi: 10.1046/j.1523-1747.2001.01326.x. [DOI] [PubMed] [Google Scholar]
- 21.Benasciutti E, Pages G, Kenzior O, Folk W, Blasi F, Crippa MP. MAPK and JNK transduction pathways can phosphorylate Sp1 to activate the uPA minimal promoter element and endogenous gene transcription. Blood. 2004;104:256–62. doi: 10.1182/blood-2003-08-2661. [DOI] [PubMed] [Google Scholar]
- 22.Sroka IC, Nagle RB, Bowden GT. Membrane-type 1 matrix metalloproteinase is regulated by sp1 through the differential activation of AKT, JNK, and ERK pathways in human prostate tumor cells. Neoplasia. 2007;9:406–17. doi: 10.1593/neo.07193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan NY, Khachigian LM. Sp1 phosphorylation and its regulation of gene transcription. Mol Cell Biol. 2009;29:2483–8. doi: 10.1128/MCB.01828-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu S, Ferro TJ. Sp1: regulation of gene expression by phosphorylation. Gene. 2005;348:1–11. doi: 10.1016/j.gene.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Gerlo S, Haegeman G, Vanden Berghe W. Transcriptional regulation of autocrine IL-6 expression in multiple myeloma cells. Cell Signal. 2008;20:1489–96. doi: 10.1016/j.cellsig.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Cooke DW, Bankert LA, Roberts CT, Jr, LeRoith D, Casella SJ. Analysis of the human type I insulin-like growth factor receptor promoter region. Biochem Biophys Res Commun. 1991;177:1113–20. doi: 10.1016/0006-291x(91)90654-p. [DOI] [PubMed] [Google Scholar]
- 27.Takakura M, Kyo S, Kanaya T, Hirano H, Takeda J, Yutsudo M, et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999;59:551–7. [PubMed] [Google Scholar]
- 28.Ueberla K, Lu Y, Chung E, Haseltine WA. The NF-kappa B p65 promoter. J Acquir Immune Defic Syndr. 1993;6:227–30. [PubMed] [Google Scholar]
- 29.Fulciniti M, Tassone P, Hideshima T, Vallet S, Nanjappa P, Ettenberg SA, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood. 2009;114:371–9. doi: 10.1182/blood-2008-11-191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez RA, Goodman AB, Rhodes M, Blomberg JA, Heller J. The anticancer activity of the transcription inhibitor terameprocol (meso-tetra-O-methyl nordihydroguaiaretic acid) formulated for systemic administration. Anticancer Drugs. 2007;18:933–9. doi: 10.1097/CAD.0b013e32813148e0. [DOI] [PubMed] [Google Scholar]
- 31.Hideshima T, Mitsiades C, Ikeda H, Chauhan D, Raje N, Gorgun G, et al. A proto-oncogene BCL6 is up-regulated in the bone marrow microenvironment in multiple myeloma cells. Blood. 115:3772–5. doi: 10.1182/blood-2010-02-270082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tai YT, Fulciniti M, Hideshima T, Song W, Leiba M, Li XF, et al. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood. 2007;110:1656–63. doi: 10.1182/blood-2007-03-081240. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Hwu JR, Tseng WN, Gnabre J, Giza P, Huang RC. Antiviral activities of methylated nordihydroguaiaretic acids. 1. Synthesis, structure identification, and inhibition of tat-regulated HIV transactivation. J Med Chem. 1998;41:2994–3000. doi: 10.1021/jm970819w. [DOI] [PubMed] [Google Scholar]
- 34.Chen H, Teng L, Li JN, Park R, Mold DE, Gnabre J, et al. Antiviral activities of methylated nordihydroguaiaretic acids. 2. Targeting herpes simplex virus replication by the mutation insensitive transcription inhibitor tetra-O-methyl-NDGA. J Med Chem. 1998;41:3001–7. doi: 10.1021/jm980182w. [DOI] [PubMed] [Google Scholar]
- 35.Gnabre JN, Brady JN, Clanton DJ, Ito Y, Dittmer J, Bates RB, et al. Inhibition of human immunodeficiency virus type 1 transcription and replication by DNA sequence-selective plant lignans. Proc Natl Acad Sci U S A. 1995;92:11239–43. doi: 10.1073/pnas.92.24.11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dohm JA, Hsu MH, Hwu JR, Huang RC, Moudrianakis EN, Lattman EE, et al. Influence of ions, hydration, and the transcriptional inhibitor P4N on the conformations of the Sp1 binding site. J Mol Biol. 2005;349:731–44. doi: 10.1016/j.jmb.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Li F, Altieri DC. The cancer antiapoptosis mouse survivin gene: characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res. 1999;59:3143–51. [PubMed] [Google Scholar]
- 38.Smolewski P. Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. IDrugs. 2008;11:204–14. [PubMed] [Google Scholar]
- 39.Chun JY, Hu Y, Pinder E, Wu J, Li F, Gao AC. Selenium inhibition of survivin expression by preventing Sp1 binding to its promoter. Mol Cancer Ther. 2007;6:2572–80. doi: 10.1158/1535-7163.MCT-07-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mityaev MV, Kopantzev EP, Buzdin AA, Vinogradova TV, Sverdlov ED. Functional significance of a putative sp1 transcription factor binding site in the survivin gene promoter. Biochemistry (Mosc) 2008;73:1183–91. doi: 10.1134/s0006297908110035. [DOI] [PubMed] [Google Scholar]
- 41.Xu R, Zhang P, Huang J, Ge S, Lu J, Qian G. Sp1 and Sp3 regulate basal transcription of the survivin gene. Biochem Biophys Res Commun. 2007;356:286–92. doi: 10.1016/j.bbrc.2007.02.140. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Xie M, Yang J, Yang D, Deng R, Wan Y, et al. The expression of antiapoptotic protein survivin is transcriptionally upregulated by DEC1 primarily through multiple sp1 binding sites in the proximal promoter. Oncogene. 2006;25:3296–306. doi: 10.1038/sj.onc.1209363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konduri S, Colon J, Baker CH, Safe S, Abbruzzese JL, Abudayyeh A, et al. Tolfenamic acid enhances pancreatic cancer cell and tumor response to radiation therapy by inhibiting survivin protein expression. Mol Cancer Ther. 2009;8:533–42. doi: 10.1158/1535-7163.MCT-08-0405. [DOI] [PubMed] [Google Scholar]
- 44.Abdelrahim M, Baker CH, Abbruzzese JL, Safe S. Tolfenamic acid and pancreatic cancer growth, angiogenesis, and Sp protein degradation. J Natl Cancer Inst. 2006;98:855–68. doi: 10.1093/jnci/djj232. [DOI] [PubMed] [Google Scholar]
- 45.Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, et al. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol. 1999;19:5504–11. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J, Zhou JY, Wei WZ, Philipsen S, Wu GS. Sp1-mediated TRAIL induction in chemosensitization. Cancer Res. 2008;68:6718–26. doi: 10.1158/0008-5472.CAN-08-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann E, et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-kappaB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood. 2008;111:2364–73. doi: 10.1182/blood-2007-08-110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goffin L, Seguin-Estevez Q, Alvarez M, Reith W, Chizzolini C. Transcriptional regulation of matrix metalloproteinase-1 and collagen 1A2 explains the anti-fibrotic effect exerted by proteasome inhibition in human dermal fibroblasts. Arthritis Res Ther. 12:R73. doi: 10.1186/ar2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kikuchi J, Wada T, Shimizu R, Izumi T, Akutsu M, Mitsunaga K, et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 116:406–17. doi: 10.1182/blood-2009-07-235663. [DOI] [PubMed] [Google Scholar]
- 50.Escoubet-Lozach L, Lin IL, Jensen-Pergakes K, Brady HA, Gandhi AK, Schafer PH, et al. Pomalidomide and lenalidomide induce p21 WAF-1 expression in both lymphoma and multiple myeloma through a LSD1-mediated epigenetic mechanism. Cancer Res. 2009;69:7347–56. doi: 10.1158/0008-5472.CAN-08-4898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) MM1S cells were treated with placebo or different concentrations of TMP (1-20 μM) for 24 h. Nuclear extracts were subjected to WB analysis using anti-Sp1 and p84 Abs to assess Sp1 protein levels. The ratio of Sp1 to p84 for each sample as assessed by densitometric quantitation of band intensity from WB is denoted. (B) MM1S cells were treated with placebo or 20 μM TMP for the indicated time periods and nuclear extracts subjected to WB analysis using indicated Abs.
MM1S and U266 cells were incubated in the presence or absence of TMP for 6 h. After the culture period, the conditioned media were collected and VEGF levels were measured by ELISA assay. Data are presented as fold change inhibition of VEGF production as compared with the control culture. Values are means +/- SD from 3 independent experiments.