

Abstract
Mitochondrial fitness is central to heart health. In many cell types mitochondria are dynamic, interconnected filamentous networks. By comparison, mitochondria of healthy post-mitotic adult cardiomyocytes are shortened, round, hypo-dynamic organelles. Mitochondrial networks are absent in cardiomyocytes; fission, fusion, and organelle mobility are not normally observed. Nevertheless, mitochondrial fission factor Drp1 and fusion factors Mfn1, Mfn2, and Opa1 are abundant and indispensable in adult hearts. Here, we review recent insights into roles for mitochondrial dynamics factors not strictly related to morphometric remodeling, advancing the argument that fission and fusion of cardiomyocyte mitochondria supports surveillance, sequestration, and mitophagic removal of damaged organelles.
Keywords: Mitochondrial dynamics, heart, mitochondrial fission and fusion, Drp1, mitofusins, mitochondrial quality control, mitophagy, mitochondrial morphology
Overview
The past decades have witnessed an explosion in our understanding of the lives of mitochondria. What likely began as a commensal relationship between endosymbiotic protobacteria and host unicellular organisms has, over eons, evolved into the most breathtakingly successful and expansive example of synergistic mutualism in the animal kingdom. As mitochondria and their hosts (i.e. us) co-evolved, we have become almost completely co-dependent. Thus, mitochondria exported approximately 99% of their genome to cell nuclei, and consequently rely upon host cells to transcribe DNA and translate mRNA encoding all but 13 (of ~1,000) mitochondrial proteins; the cells do the work of making mitochondrial proteins, and the mitochondria import them through a complex and highly regulated sorting machinery (Schmidt et al., 2010). In exchange, host cells have come to rely on highly efficient mitochondrial oxidative phosphorylation, rather than glycolysis, as the major source of ATP to fuel normal cell functions. While we continue to unravel the mechanisms underlying mitochondrial metabolism, research into the newer areas of mitochondrial dynamics and quality control has revealed unanticipated behaviors that mediate interactions between individual mitochondria within the greater cellular collective, and that determine how these organelles communicate and collaborate with their host cells to maintain overall fitness of the mitochondrial pool. Far from being static transmutators of hydrocarbons and oxygen to ATP, mitochondria are dynamic entities with an ability to homeostatically orchestrate (or pathologically disrupt) cell specification/differentiation during embryonic development, programmed cell elimination during organ/organism senescence, and virtually every other cellular function in the intervening period.
It has become axiomatic that mitochondria constantly undergo fission, fusion, and migration within cells (Liesa and Shirihai, 2013), collectively termed “mitochondrial dynamism”. The supporting evidence for this conclusion is overwhelming; one needs only to look at movies of living cultured fibroblasts after their mitochondria have been stained with, or are genetically expressing, one or more fluorescent markers (Archer, 2013). The filamentous mitochondrial networks are constantly remodeling, with sections of the network periodically breaking away and then re-establishing new interconnections elsewhere. Mitochondrial morphometry seems especially plastic during cell mitosis (Taguchi et al., 2007), perhaps in part to accommodate massive changes in the cell’s internal architecture, size, and shape, and to facilitate distribution of mitochondria to both daughter cells. Dissolution of the mitochondrial network is also frequently observed during programmed apoptotic cell death (Frank et al., 2001). While many of the molecular mediators have been identified, the underlying mechanisms for mitochondrial remodeling are not yet fully understood (Youle and van der Bliek, 2012).
The general characterization of mitochondria as “highly dynamic” does not apply to adult cardiac myocytes. Compared to prototypical cultured fibroblasts, normal cardiomyocyte mitochondria exhibit a smaller, rounder, so-called “fragmented” morphometry, and do not routinely travel within the cell. For this reason, the widely accepted notion that mitochondrial fragmentation is detrimental (and mitochondrial fusion is beneficial) to mitochondrial and cellular functioning likely does not apply to adult cardiomyocytes, at least under normal conditions. Here, we review recent findings supporting the view that mitochondrial dynamism plays different roles according to biological context. We examine accumulating evidence that adult cardiomyocyte mitochondria represent a “special case” in which lessons learned from studying cultured non-cardiomyocytes should be applied with caution as our understanding of the processes that govern, and the cellular effects that are evoked by, mitochondrial fission and fusion continues to evolve.
Wherefore art thou mitochondria?
Mitochondria serve the same function in cells as does the kitchen of a busy restaurant: both structures provide a discrete space in which imported raw materials undergo potentially dangerous manipulations to produce “food” for consumption. In both instances compartmentalization is not only central to functional efficiency, but helps to sequester unpleasant or deleterious by-products of the primary process. Mitochondrial architecture (and that of related chloroplasts) is therefore defined by its principal purpose, oxidative phosphorylation. Enzymes of the electron transport system reside on a highly convoluted inner mitochondrial membrane (IMM) (or in chloroplasts, on the thylakoids) encompassing a protein-rich mitochondrial matrix that also contains mitochondrial DNA and ribosomes (Figure 1). The matrix and IMM are surrounded by the outer mitochondrial membrane (OMM), creating a small intermembrane space; the proton gradient that drives IMM ATP synthase (a.k.a. F1/Fo ATPase) exists between this mitochondrial intermembrane space and the matrix. The dual-membrane mitochondrial structure has some intriguing implications. First, the matrix, IMM, and intermembrane space are physically sequestered by the OMM from the rest of the host cell; access is limited by passive diffusion or active translocation across the OMM. Second, as mitochondria replicate and communicate via fission and fusion, these processes have to navigate the complicated internal organelle architecture (the molecular mechanisms by which this is accomplished are described below). Finally, mitochondrial sequestration inside the OMM insulates the rest of the cell from potent biochemical reactions taking place within the organelle. The insular physical relationship between mitochondrion and cell is essential both to promote mitochondrial respiratory function and for cytoprotection in the face of mitochondria-generated reactive oxygen species (ROS) (Figure 1). But this degree of compartmentalization introduces problems for organelle-cell and organelle-organelle communications. The biological solution was for the OMM not simply to passively contain mitochondrial oxidation-reduction reactions, but for it to actively engage both the mitochondrion and the cell as the physical and functional interface for information exchange (i.e. signaling). Thus, two additional functions of the OMM are to regulate mitochondrial morphometry and network morphology in accordance with changing minute-by-minute needs of the host cell, and to mediate cell-based mitochondrial removal mechanisms for organelles that are damaged beyond repair. Mitochondrial dynamism is central to both of these functions.
Figure 1. Compartmentalization of mitochondrial processes and signaling interactions with host cell.

OMM is outer mitochondrial membrane; IMM is inner mitochondrial membrane; IMS is intermembrane space between the outer and inner membranes; the matrix is the space within the inner membrane. Lower: Electron transport chain resides on the IMM and ATP is synthesized by F1/Fo ATPase (also known as complex V). Excessive ROS can damage DNA and proteins within and outside the mitochondria, and activate regulatory pathways. Upper: Endoplasmic/sarcoplasmic reticulum (ER/SR) membrane tethers to the OMM, facilitating inter-organelle calcium signaling. Right: Calcium can trigger mitochondrial permeability transition pore (MPTP) opening, leading to electron efflux and water influx that causes mitochondrial swelling and even cell death.
What does mitochondrial dynamism accomplish?
Mitochondria are gregarious. In cultured fibroblasts they are literally conjoined, forming a continuous (albeit constantly changing) filamentous network through which signaling events can be transmitted as propagating waves (Szabadkai et al., 2004). The changing network morphology is the net consequence of multiple simultaneous organelle fusion and fission events orchestrated by local factors such as mitochondrial polarization status, proximity of endoplasmic reticulum, and activity of the Parkin mitophagy signaling pathway (Friedman et al., 2011; Liu et al., 2009; Twig et al., 2008; Yang and Yang, 2013). Even in neurons, in which long axonal structures (a meter or more of axon between cell body and distal synapse) are not conducive to maintenance of interconnected mitochondrial networks, individual mitochondria migrate bidirectionally along the axons, intermittently fusing with and transferring contents into other mitochondria (Cagalinec et al., 2013). These types of observations engendered the notion that mitochondria are “highly dynamic, interconnected organelles” (DuBoff et al., 2013).
In most cells mitochondrial networks are dynamic. It seems obvious therefore that a major determinant of mitochondrial morphology is the relative frequency of mitochondrial fusion and fission. Mitochondrial shape is typically reported as the aspect ratio, i.e. length/width (normal value is ~6 in fibroblasts) (Song et al., 2014b). When fusion and fission are balanced, mitochondrial aspect ratio will be stable. However, when the balance shifts in favor of more fusion (and/or less fission), mitochondrial elongation and network connectivity will increase. And when an imbalance produces less fusion (and/or more fission), this will provoke accumulation of smaller, shorter “fragmented” organelles. Unless it is established that the underlying mechanism for organelle dysmorphology is increased mitochondrial fission, we prefer the use of “shorter” to describe smaller, rounder mitochondria; this neutral term does not imply that active mitochondrial “fragmentation” (rather than passive loss of fusion) induced the structural change.
Dissolution of mitochondrial networks and mitochondrial shortening (in this case, actual fragmentation) are frequently observed at the beginning and end of cellular life, i.e. during mitotic cell division and apoptotic cell death (Frank et al., 2001; Taguchi et al., 2007). However, genetically ablating mitochondrial fusion factors (Chen et al., 2005; Song, 2014b) or driving pro-fission factors (Szabadkai et al., 2004) provokes mitochondrial shortening without killing the cells (although mitochondrial fitness is impaired). Indeed, mitochondrial network fragmentation can be protective (Szabadkai et al., 2004), and as described below, adult cardiomyocytes that lack interconnected mitochondrial networks (their mitochondria appear constitutively “fragmented”) have both the greatest mitochondrial density and highest respiratory capacity of any mammalian cell. Thus, at least in cardiomyocytes, mitochondrial networking is dispensable to normal cell functioning. Nevertheless, mitochondrial fusion clearly plays important and incompletely understood roles in organism development, as revealed by embryonic lethality in mice lacking either Mfn1 or Mfn2 in the germ line (Chen et al., 2003) or both Mfn1 and Mfn2 in developing embryonic cardiac myocytes (Kasahara et al., 2013).
Although not universally accepted, conventional wisdom holds that mitochondrial fusion/fission and the resulting intermixing of matrix components between two mitochondria are important for repairing mitochondrial damage by functional complementation (Chen et al., 2010a; Nakada et al., 2001; Youle and van der Bliek, 2012). For example, mitochondrial “kiss and run” describes transient fusion, and then immediate fission, of two mitochondria that exchange biological components without changing organelle morphology (Liu et al., 2009). Although this may be reparative, it also may simply be a mechanism for inter-organelle information exchange. Further support for a reparative function of mitochondrial fusion is the classic observation that fusion-defective murine embryonic fibroblasts (MEFs) exhibit widespread heterogeneity in mitochondrial membrane potential (Chen et al., 2005; Olichon et al., 2003). Partial mitochondrial depolarization in this context was widely interpreted as the consequence of loss of fusion-mediated complementation, but recent identification of non-canonical functions for mitochondrial fusion proteins in mitophagy raise the possibility that mitochondrial degeneration in fusion-impaired cells reflects interrupted organelle quality surveillance and culling (Chen and Dorn, 2013; Lee et al., 2012; Pham et al., 2012; Sebastian et al., 2012).
Mitofusins and Drp1 – the OMM dynamic duo
Before examining the atypical functions of mitochondrial dynamics factors in cardiomyocytes, it is worth reviewing the molecular mechanism of their conventional roles in cells having mitochondrial networks. Consider that a mitochondrial network is a bit like a balloon animal. It is malleable and the overall structure can be readily altered by fairly simple manipulations within or between individual organelles/balloons. The process of mitochondrial constriction that precedes actual organelle fission is similar for mitochondria and party balloons: the structure is pinched and constricted to form a separate entity. In mitochondria this is accomplished by the cytosolic dynamin family GTPase, Dynamin-related protein 1 (Drp1) (Figure 2). Normally, ~97% of Drp1 is cytosolic (Smirnova et al., 2001), but its recruitment to one or more binding proteins on OMMs (Otera et al., 2010; Palmer et al., 2011; Tieu and Nunnari, 2000; Zhao et al., 2011), self-assembly into spirals, and GTP-mediated constriction, severs the organelle (Friedman et al., 2011; Ingerman et al., 2005). Mitochondrial constriction sites are often marked by endoplasmic reticulum (ER), and the ER-mitochondria contact is prior to and independent of Drp1 recruitment (Friedman et al., 2011). As one would predict, mitochondria in fibroblasts lacking Drp1 exhibit elongated/filamentous morphologies and are resistant (but not immune) to mitochondrial fragmentation normally induced by mitochondrial uncoupling agents or during mitosis (Figure 2). Fragmentation of Drp1-deficient mitochondria during cytokinesis reveals, however, that mitochondrial fission can also be induced by factors other than Drp1 (Ishihara et al., 2009), perhaps including Parkin that evokes mitochondrial fragmentation during mitophagy (Yang and Yang, 2013).
Figure 2. Mitochondrial dynamics factors and mitochondrial fission and fusion.
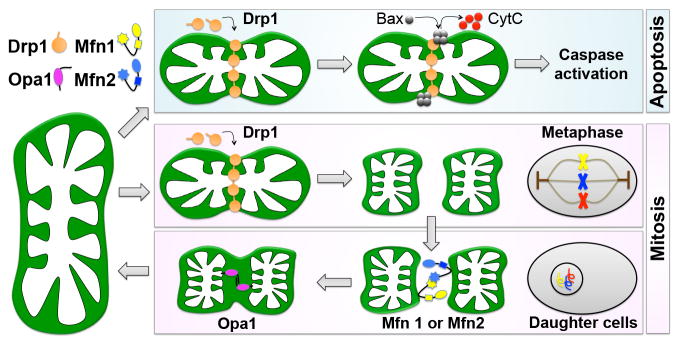
Left: Drp1 is dynamin-related protein 1; Opa1 is optic atrophy 1; Mfn1 and Mfn2 are mitofusins. Upper right: Mitochondrial fission before and during apoptosis. Lower right: Mitochondrial morphology during mitosis. Mitochondria undergo fission during the mitotic phase and after cytokinesis, fragmented mitochondria restore the filamentous morphology through mitochondrial fusion in daughter cells.
Mitochondrial fragmentation is almost always observed before or during apoptosis (Figure 2), and Drp1 has therefore been mechanistically implicated in programmed cell death. Drp1 stimulates Bax oligomerization and cytochrome c release by promoting tethering and hemifusion of membranes during mitochondrial fission (Frank et al., 2001). In some studies, genetic or chemical inhibition of Drp1 decreased cytochrome c release and blocked cell death, suggesting a contributory effect of mitochondrial fission in apoptosis (Cassidy-Stone et al., 2008; Frank et al., 2001). In others however, inhibition of Drp1-mediated fission did not prevent release of proapoptotic Smac/DIABLO or reduce cell death (Estaquier and Arnoult, 2007; Parone et al., 2006), and in at least some instances enhanced Drp1-mediated fission can protect against apoptosis (Szabadkai et al., 2004). Thus, it is probably most correct to conclude that Drp1-mediated mitochondrial fission can be a contributory factor in cell apoptosis, and likely helps in the cell-wide disassembly that characterizes this form of “clean” cell suicide, but is not essential to actual apoptotic cell demise.
When conceptualizing mitochondrial fusion the balloon analogy fails because balloons have only one wall, and fusion of the two mitochondrial membranes (OMM and IMM) requires separate steps, working from outside in. In mammals, the evolutionarily conserved OMM fusion proteins (which are also dynamin family GTPases) are the mitofusins (Mfn1 and Mfn2) (Chen et al., 2003; Santel and Fuller, 2001) (Figure 2). Mitofusins are constitutively localized at the OMM where they have several physiologically distinct functions. Their titular function is fusion of the OMM, which requires GTPase activity and is irreversible. Mfn1 and Mfn2 are largely redundant for this function, and can interact either homotypically (Mfn1-Mfn1, Mfn2-Mfn2) or heterotypically (Mfn1-Mfn2), although differences in their GTPase activity (Ishihara et al., 2004) likely underlie the modest differences in mitochondrial morphology exhibited by MEFs lacking one or the other Mfn (Chen et al., 2005). By comparison, MEFs lacking both Mfn1 and Mfn2 have markedly shortened and partially depolarized mitochondria (Chen et al., 2003; Koshiba et al., 2004; Olichon et al., 2003).
A mechanistically distinct but functionally related effect of mitofusins is organelle tethering. This is distinct from membrane fusion because it is fully reversible and does not require hydrolysis of GTP. Tethering of two mitochondria both precedes and enables OMM fusion (Koshiba et al., 2004). Mfn2 (but not Mfn1) can, however, also localize to endoplasmic reticulum (and sarcoplasmic reticulum in cardiomyocytes), thereby tethering ER/SR to mitochondria and facilitating inter-organelle calcium signaling (Chen et al., 2012b; de Brito and Scorrano, 2008). The consequences of Mfn2-facilitated calcium cross-talk are: 1. Enhanced bioenergetic responsiveness to temporary increases in metabolic demand; and 2. Increased calcium-mediated opening of mitochondrial permeability transition pores (MPTP) under conditions of ischemic stress (Dorn et al., 2014a). Finally, after post-translational modification, Mfn2 can act as a mitochondrial receptor protein for Parkin translocation during mitophagic organelle culling (Chen and Dorn, 2013); the importance of this in hearts is discussed below.
As a matter of completeness, Optic atrophy 1 (Opa1) is the fourth dynamin family GTPase involved in mitochondrial dynamism. Opa1 is localized on, and mediates fusion of, the IMM (Figure 2). It is also essential to maintaining proper IMM architecture in the form of cristae (Frezza et al., 2006). Depletion of Opa1 induces heterogeneity in mitochondrial morphometry because IMM fusion is interrupted, but OMM fusion still occurs (Song et al., 2009).
At first glance, a broad biological imperative for mitochondrial dynamism seems to be supported by the embryonic lethality provoked by germ-line ablation of these core fission/fusion factors in mice (Chen et al., 2003; Davies et al., 2007; Ishihara et al., 2009). However, most of the genetic human syndromes attributed to mutations in these factors show remarkably restricted effects. For example, loss of function mutations of Mfn2 are linked to Charcot-Marie-Tooth Disease (type 2A), which is an autosomal dominant syndrome characterized by neuropathy of the peripheral axons (Verhoeven et al., 2006). Likewise, Opa1 mutations are linked to autosomal-dominant optic atrophy, a neurodegenerative condition that specifically targets retinal ganglion cells (Delettre et al., 2000). Thus, multiple loss-of-function mutations in these two mitochondrial fusion proteins are clearly deleterious, but the major adverse effects are limited to distinct cellular subtypes within the neurological system. These experiments of Nature pose some intriguing questions: If mitochondrial fusion is generally important, why are the consequences of human Mfn2 and Opa1 mutations so limited? And given that GTPase activity of Mfn1 is more efficient than Mfn2 (Ishihara et al., 2004), why have there not been reports of human disease linked to damaging mutations of Mfn1 (which are approximately as abundant as damaging mutations of Mfn2 in the ~6,000 exomes currently reported by the NHLBI Exome Sequencing Project)? Finally, if Mfn1 and Mfn2 exhibit functional redundancy as mitochondrial fusion factors, and mitochondrial fusion is their most critical role, shouldn’t their selective ablation from different mouse tissues evoke similar phenotypes? They do not. In each instance with which we are familiar, loss of Mfn2 has induced dysfunction, but loss of Mfn1 has been well tolerated (Chen and Dorn, 2013; Lee et al., 2012; Pham et al., 2012; Sebastian et al., 2012). It is said that when your theory no longer fits the facts you should develop a new theory. Thus, one possible explanation is that mitochondrial fusion is not important, which we do not accept. Another possibility is that mitochondrial reliance on dynamism is cell type-dependent. Based in part on the above observations, and informed by studies in the heart, we postulate that non-canonical functions of mitochondrial fusion and fission factors contribute to cell and organ dysfunction evoked by genetic mutation or ablation.
Mitochondrial dynamism in the heart - NOT
The broad generalizations about mitochondrial dynamism introduced above simply do not apply to mitochondria in cardiac myocytes of normal adult hearts. Cardiomyocyte mitochondria are not “highly dynamic”, or “interconnected”, or “constantly undergoing fission and fusion”. Let’s examine each of these mischaracterizations individually: While it is almost certain that mitochondria within adult cardiomyocytes can eventually migrate to a different position within the cell, such organelle translocation is not directly observed (Dorn et al., 2011). Over periods of time when the cardiomyocyte would have been contracting thousands of times, cardiomyocyte mitochondria appear static. Nor are they connected into networks. It is apparent from ultrastructural examination of myocardium that individual cardiomyocyte mitochondria exist as relatively short organelles (aspect ratio ~1.5) collectively grouped together between myofibrillar elements in lanes that extend parallel to the long axis of the cell (see Figure 4). Even in the perinuclear region of cardiomyocytes, where mitochondria tend to be clustered in large numbers, filamentous organelles and interconnected networks are not observed. We have speculated that intrinsically fragmented mitochondria confer a biomechanical advantage to striated muscle because interconnected organelles would constitute an internal resistance element detrimental to myocyte contraction (Dorn, 2014b). Finally, cardiomyocyte mitochondria do not undergo fusion and fission at a rate that can be directly measured. Based on temporally defined cardiomyocyte-specific ablation of Mfn1 and Mfn2 in combination, or Drp1 alone, the mitochondrial fusion-fission cycle takes approximately 2 weeks in adult mice (Chen et al., 2011; Song et al., 2014b). Nevertheless, mitochondrial fusion and fission proteins are expressed at high abundance in adult hearts and can be regulated in heart disease (Chen et al., 2009; Chen et al., 2011; Fang et al., 2007; Givvimani et al., 2014; Li et al., 2014; Martin et al., 2014; Shen et al., 2007) and cardiac-specific ablation of either Mfn1 and Mfn2 in combination, or Mfn2 alone, or Drp1, (but not Mfn1 alone) evokes cardiac dysfunction, albeit with strikingly different characteristics (Chen and Dorn, 2013; Chen et al., 2011; Papanicolaou et al., 2011; Papanicolaou et al., 2012b; Song et al., 2014b). As discussed below, these mouse gene manipulations and similar studies in Drosophila fruit flies have helped to deconvolute the multifaceted functioning of mitochondrial dynamics factors in hearts.
Figure 4. Mitochondrial morphologies in different cell types.
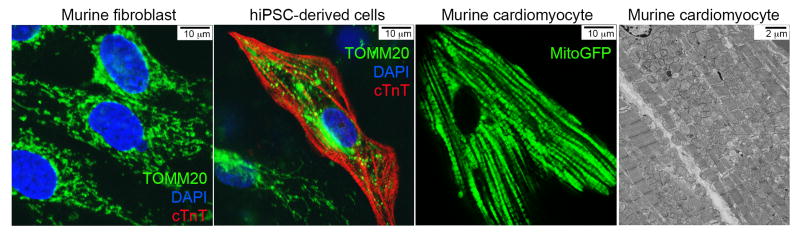
Murine fibroblasts and human induced pluripotent stem cells (hiPSC)-derived cells were co-stained with anti-TOMM20 (green) and anti-cardiac Troponin T (cTnT, red), and mounted in DAPI. In the image of hiPSC-derived cells, the cTnT positive cell (middle) is a spontaneously contracting cardiomyocyte with mostly fragmented mitochondria and the cTnT negative cell (lower left) is a fibroblast with filamentous mitochondria. The mouse cardiomyocyte was isolated from an adult mouse heart expressing a mitochondrial-targeted fluorescent green marker. The transmission electron microscopic image of adult mouse heart was taken at a magnification of 2,000x.
As noted, mitochondrial fusion in cardiomyocytes occurs infrequently (Chen et al., 2011), and therefore has not been directly quantified in cardiomyocytes. Because there are currently no useful pharmacological fusion inhibitors, its occurrence and importance have been inferred from the phenotypes provoked by cardiomyocyte-specific genetic ablation or suppression of OMM fusion proteins. In Drosophila, cardiomyocyte-specific expression of an RNAi specific to fly mitofusin (there is only one, called MARF) produced unusually small mitochondria in hypocontractile heart tubes; both the organ and organelle phenotypes were rescued by cardiomyocyte-specific expression of human Mfn1 or Mfn2 (Dorn et al., 2011). The functionally analogous genetic manipulation in adult mouse hearts, i.e. combined cardiomyocyte-specific ablation of Mfn1 and Mfn2, likewise produced abnormally small mitochondria and a cardiomyopathy (Chen et al., 2011; Papanicolaou et al., 2012a). Surprisingly, the initial description of mitofusin-deficient hearts did not uncover a functional defect in the cardiac mitochondria (Chen et al., 2011), but subsequent work revealed that the unusually small mitochondria that accumulate after suppression of mitochondrial fusion escaped isolation using standard protocols. Using appropriately modified protocols, the “fragmented” mitochondria in Mfn1/Mfn2 double deficient hearts were found to have impaired respiration and increased ROS production (Song et al., 2014b). This same report identified the underlying mechanism for the eccentric cardiomyopathy in mitochondrial fusion-defective Mfn1/Mfn2 deficient hearts as impaired mitophagy without cardiomyocyte loss (Song et al., 2014b). This finding is consistent with an important role for Mfn2 in the PINK1-Parkin mitophagy signaling pathway (Chen and Dorn, 2013), which is described in greater detail below.
Although less thoroughly characterized, interrupting fusion of IMM by suppressing Opa1 likewise evokes mitochondrial dysmorphology and cardiomyopathy in Drosophila (Dorn et al., 2011) and mice (Chen et al., 2012a; Piquereau et al., 2012). Conversely, Opa1 overexpression in cultured H9C2 cells produces mitochondrial elongation (which occurred without the anticipated reduction in apoptosis under conditions of simulated ischemia) (Chen et al., 2009). Together, these early results of Opa1 manipulation in cardiomyocytes suggest that (like Mfn1 and Mfn2) Opa1 is indispensable to normal cardiac functioning, but (like Mfn2) its most important role may not simply be to promote mitochondrial fusion. Indeed, the most important functions of Opa1 may derive from its orchestration of mitochondrial cristae architecture (Cipolat et al., 2006; Cogliati et al., 2013; Frezza et al., 2006).
It is standard practice to examine biological questions from opposing perspectives, in anticipation of obtaining reciprocal results. Accordingly, in addition to modulating fusion, when dissecting the consequences of mitochondrial dynamism in the heart one should determine what happens when fission is interrupted. Pharmacological inhibition can have less molecular specificity and less precise cellular targeting than genetic interruption, but investigations using small molecular inhibitors of Drp1-mediated mitochondrial fission have provided useful information. As introduced above, mitochondrial fragmentation is frequently observed during cell apoptosis. It seemed reasonable therefore to see if inhibition of mitochondrial fragmentation with the Drp1 inhibitor, Mdivi-1, could prevent apoptosis (Cassidy-Stone et al., 2008). The first attempt to use this approach in cardiomyocytes showed that Mdivi-1 pretreatment of cultured HL-1 cells improved cell survival and delayed MPTP opening after simulated ischemia-reperfusion; mice pretreated with Mdivi-1 developed smaller myocardial infarcts after coronary occlusion-reperfusion (Ong et al., 2010). Mdivi-1 also improved left ventricular function and decreased myocardial fibrosis in pressure overload-induced heart failure (Givvimani et al., 2012). Mochly-Rosen’s group has reported similar findings using a completely different pharmacological Drp1 inhibitor, a synthetic 7-amino-acid peptide that selectively disrupts the interaction between Drp1 and its mitochondrial receptor Fis1 (Qi et al., 2013). This peptide, designated P110, improved bioenergetics and functional recovery in primary cardiomyocytes, ex vivo perfused hearts, and in vivo ischemia-reperfused hearts (Disatnik et al., 2013). It has also slowed neurodegeneration in a transgenic mouse model of Huntington’s disease (Guo et al., 2013). These data strongly suggest that preventing mitochondrial fission through Drp1 inhibition can be beneficial in ischemically injured hearts and other disorders in which excessive mitochondrial fission has a pathophysiological role.
As noted, pharmacological inhibition and genetic ablation are not the same and do not provide the same information. There are limited data on the consequences of genetic loss of Drp1 function in vertebrate hearts. The potential for Drp1 to play a role was revealed in an N-ethyl-N-nitrosourea mutagenesis screen, wherein the “Python” mouse having a Drp1C452F mutation developed dilated cardiomyopathy associated with reduced levels of respiratory complexes and ATP depletion (Ashrafian et al., 2010). The recent availability of Drp1 floxed allele mice (Ishihara et al., 2009; Udagawa et al., 2014; Wakabayashi et al., 2009) has permitted organ-specific Drp1 deletion, including in the heart. Three groups independently combined Drp1 floxed allele mice with cardiomyocyte-specific Cre deletors to produce cardiac-specific Drp1 knockout mice (Ikeda et al., 2014; Kageyama et al., 2014; Song et al., 2014b). A detailed comparison of the data and conclusions of the three papers has been reported elsewhere (Dorn, 2015). Briefly, Drp1 deficiency induced mitochondrial enlargement in all three reports, as expected from unopposed organelle fusion. In either the perinatal period or in fully developed adult hearts, loss of cardiac Drp1 proved lethal. Likewise, in each instance of Drp1 deletion, abnormalities in mitochondrial quality and/or quantity were associated with disturbances in mitochondrial autophagy. Thus, all three studies linked mitochondrial fission to quality control, although the specific nature of the mitophagic dysfunction remains unclear.
Our group’s Drp1 heart knockout study was unique in that we directly compared the consequences of conditional cardiac interruption of mitochondrial fission to interruption of mitochondrial fusion. Cardiac-specific Drp1 ablation produced fission-defective cardiomyocyte mitochondria that were not only enlarged, but whose abundance progressively decreased. Biochemical markers of metabolic stress and mitochondrial autophagy increased, especially in the terminal stages of the progressive dilated cardiomyopathy. Drp1 ablation provoked programmed cardiomyocyte necrosis that ultimately caused myocardial fibrosis and lethal cardiac failure. Studies in Drp1-deficient MEFs revealed inappropriate mitochondrial clearance and MPTP-mediated cell death, and inhibition of MPTP opening by pharmacological or genetic suppression of cyclophilin D rescued cells and hearts, respectively (Song et al., 2014b). By comparison, conditional combined cardiac-specific Mfn1/Mfn2 ablation produced fusion-defective cardiomyocyte mitochondria whose size decreased, but abundance increased with time. Mitochondrial respiratory fitness was markedly impaired in these hearts and the mitochondrial uncoupled protein response was potently induced, but mitophagy was not induced. Furthermore, although Mfn1/Mfn2 deficient mitochondria progressively developed striking structural and functional abnormalities, cardiomyocyte viability was not compromised (Song et al., 2014b). Thus, deficiency of both mitochondrial fusion proteins interrupted cardiomyocyte mitophagy; we previously showed that cyclophilin D ablation does not rescue this type of mitophagy-suppressed heart phenotype (Song et al., 2014a).
It’s the mitophagy…
James Carville famously developed three focus points for Bill Clinton’s 1992 presidential campaign; each of these applies also to mitochondrial dynamism in the heart. The first is “Change vs. more of the same”. The morphology of cardiomyocyte mitochondria is just “more of the same”. These cells are ~40% full of monotonously similar rounded-rectangular mitochondria. There are no mitochondrial networks to remodel, and fission and fusion occur rarely. Even genetic ablation of both mitofusin proteins or the Drp1 fission protein provokes only a modest change in cardiomyocyte mitochondria size (~two-fold decrease or increase, respectively), compared to complete mitochondrial fragmentation or hyper-fusion in cultured MEFs undergoing the same conditional gene manipulations (Song et al., 2014b).
Carville’s second focus point was “Don’t forget health care”. The cumulative data seem to indicate that, rather than shape change, mitochondrial dynamism in hearts is central to maintaining mitochondrial fitness, i.e. “health care”. Notwithstanding that the absolute rates of mitochondrial fission and fusion are slow in mammalian hearts, creating an imbalance between these two processes through their genetic interruption disrupts mitochondrial quality control in opposite directions (Song et al., 2014b).
This leads us to the third and most celebrated of Carville’s focus points, “It’s the economy, stupid”; for cardiomyocyte mitochondria this translates to “It’s the mitophagy…..” Mitophagy, literally the cell eating its mitochondria using the autophagy apparatus, is a protective mechanism against cell damage by oxygen radicals released from senescent or functionally impaired mitochondria (Youle and van der Bliek, 2012). It may also be a mechanism for recycling mitochondrial components. The connection between mitochondrial dynamism and mitophagy was elegantly described by Shirihai’s group in studies of cultured INS1 and COS7 cells. They observed asymmetric fission of modestly damaged (i.e. partially depolarized) mitochondria producing one healthy (hyperpolarized) daughter that re-joined, by fusion, the functional mitochondrial pool, whereas the other depolarized daughter mitochondrion was specifically targeted for mitophagic elimination (Twig et al., 2008) (Figure 3). The critical involvement of mitochondrial fission in sequestering damaged mitochondrial components into one daughter targeted for removal, and of mitochondrial fusion in re-introducing the healthy daughter to the mitochondrial collective, reflects a central homeostatic function for mitochondrial dynamism independent of morphometric or network remodeling. Whereas cardiomyocyte mitochondria are severely hypo-dynamic, maintenance of mitochondrial fitness is clearly a physiological imperative in this most energy-demanding and mitochondrial rich organ (Anan et al., 1995; Doenst et al., 2013; Kolwicz et al., 2013). An immediate example is the aforementioned lethal phenotypes and the seemingly opposite effects on mitophagy in fission-deficient mouse hearts reported by Ikeda, et al (Ikeda et al., 2014) and our group (Song et al., 2014b): at an early stage, the absence of asymmetric fission prevents damaged mitochondrial components being segregated for mitophaghic elimination. Conservation of damaged components not only impairs mitochondrial function but also interrupts normal mitophagy. With the damage accumulates over time, absence of asymmetric fission eventually provokes a generalized widespread mitophagic response. Consequently, we next examine additional evidence for and against an important role for the mitochondrial dynamism-mitophagy interactome (Dorn, 2014b) in mammalian heart function.
Figure 3. Relationship of mitochondrial dynamism to mitophagy.

Mitochondria accumulate damage over time. Impaired (partially depolarized) mitochondrion divides into one depolarized daughter and one hyperpolarized daughter through asymmetric fission. The depolarized daughter is eliminated via Mfn2-Parkin-mediated mitophagy and the hyperpolarized daughter re-joins the functional mitochondrial pool through organelle fusion. Healthy mitochondria (hyperpolarized) are colored in green and damaged mitochondria (depolarized) in yellow.
Mitophagy is controlled by signaling through two Parkinson’s disease factors, the mitochondrial kinase PINK1 and the cytosolic E3 ubiquitin ligase, Parkin (Youle and van der Bliek, 2012). PINK1 and Parkin co-involvement in mitochondrial quality control was initially uncovered through their genetic epistasis in Drosophila (Clark et al., 2006; Park et al., 2006). It was only recently, however, that Youle and colleagues uncovered how the PINK1-Parkin system achieves selective mitophagy (Narendra et al., 2010). Both PINK1 and Parkin are nuclear-encoded. PINK1 is imported into mitochondria where it is almost immediately degraded by mitochondrial proteases (Deas et al., 2011; Jin et al., 2010). PINK1 proteolysis is impaired in damaged or senescent depolarized mitochondria, resulting in its accumulation in these damaged organelles. Stabilized PINK1 then recruits cytosolic Parkin to those depolarized mitochondria, provoking its ubiquitination of OMM proteins (Sarraf et al., 2013), and thereby targeting the damaged organelle for autophagosomal engulfment (Yang and Yang, 2013). In this manner PINK1 and Parkin constitute the central effectors of mitophagy (Figure 3). Their importance to heart function, and that of mitophagy in general, is supported by the observation that mice lacking PINK1 exhibit abnormalities of cardiac mitochondria associated with pathological cardiac hypertrophy and cardiac dysfunction linked to myocardial fibrosis and increased cardiomyocyte apoptosis (Billia et al., 2011). Likewise, mice lacking Parkin develop structurally and functionally abnormal mitochondria and associated age-dependent cardiac abnormalities resembling premature senescence (Hoshino et al., 2013; Kubli et al., 2013a; Kubli et al., 2013b). Furthermore, cardiomyocyte-specific Parkin overexpression protects against age-related mitochondrial dysfunction and cardiac senescence (Hoshino et al., 2013). However, whereas direct evidence exists for mitochondrial involvement in the skeletal myopathy of Parkinson’s disease (Ahlqvist et al., 1975; Blin et al., 1994; Shoffner et al., 1991), cardiac dysfunction is not common; the only evidence for cardiac involvement is inferential and derived from purely statistical associations (Zesiewicz et al., 2004). Likewise, cardiac phenotypes in Parkin knockout mice are modest, and develop only later in life (Hoshino et al., 2013; Kubli et al., 2013b), but this may be a consequence of compensatory upregulation of related ubiquitin ligases in the germ-line Parkin knockout mouse heart (Bhandari et al., 2014).
In an fascinating example of scientific convergence, emerging data have revealed essential roles for pro-apoptotic Bcl-2 family proteins Nix and Bnip3 in homeostatic mitochondrial signaling and quality control (reviewed in (Dorn, 2014b)). Thus, Bnip3 and Nix (also called Bnip3L) are induced and evoke apoptotic cardiomyocyte death in damaged or stressed mouse hearts (Chen et al., 2010b; Diwan et al., 2007; Diwan et al., 2008; Syed et al., 2004; Yussman et al., 2002), but their combined ablation in otherwise normal hearts provokes the accumulation of abnormal mitochondria and causes an apparent cardiomyopathy of mitophagic dysfunction (Dorn, 2010). The molecular underpinnings of Nix and Bnip3 involvement in mitochondrial quality control are diverse: Nix (and likely Bnip3) regulates calcium signaling between cardiomyocyte mitochondria and endo/sarcoplasmic reticulum (Chen et al., 2010b; Diwan et al., 2009). Nix and Bnip3 also promote the physical association of depolarized mitochondria with autophagosomes (Ding et al., 2010; Glick et al., 2012; Hanna et al., 2012; Schwarten et al., 2009). The involvement of Nix, Bnip3, and other Bcl2 proteins in mitochondrial quality control led us to postulate that programmed elimination of individual dysfunctional mitochondria (i.e. mitophagy) and programmed elimination of individual dysfunctional cells (i.e. apoptosis) is more a matter of scale rather than distinct molecular mechanism (Dorn, 2014b).
Recent work from our group further supported the importance of mitophagic mitochondrial quality control to mammalian hearts, and described an unsuspected interaction between mitochondrial dynamism and mitophagy. During the course of investigating the mitochondria-SR tethering function of Mfn2 in mouse hearts (Chen et al., 2012b) we observed that postnatal cardiac-specific deletion of Mfn2 provoked the gradual accumulation of abnormal cardiomyocyte mitochondria, and hypothesized that absence of Mfn2 induced a defect in mitochondrial quality control. Consistent with this notion we demonstrated that Mfn2 functionally links mitochondrial PINK1 activity to translocation and activation of cytosolic Parkin, and identified PINK1-phosphorylated Mfn2 as the first known mitochondrial Parkin receptor (Chen and Dorn, 2013). More recently, we demonstrated two different roles for mitochondrial-derived ROS in this model of defective cardiac mitophagy. First, in accordance with conventional wisdom, mitochondrial ROS were increased in Mfn2-deficient hearts, and their normalization by transgenic expression of mitochondrial-targeted catalase (mCAT) delayed and improved the cardiomyopathy (Song et al., 2014a). This uncovered ROS cardiotoxicity linked to retention of abnormal mitochondria after normal mitophagic culling mechanisms were interrupted, similar to aged Parkin knockout mouse hearts (Hoshino et al., 2013). However, we also uncovered evidence that mitochondrial ROS can induce compensatory secondary mitochondrial autophagy mechanisms: suppressing mitochondrial ROS to levels far below normal both exacerbated the accumulation of damaged mitochondria and accelerated the associated cardiomyopathy in the same mitophagically impaired hearts (Song et al., 2014a). This demonstrates that increased ROS in damaged or senescent mitochondria can stimulate mitochondrial culling independent of the classic PINK1-Mfn2-Parkin pathway. Taken together, these studies mechanistically link mitochondrial fusion and mitophagy through Mfn2, and mitotoxicity and quality control through ROS. Tantalizingly, the dual roles of Mfn2 as an initiator of mitochondrial fusion and a critical effector of PINK1-Parkin mediated mitophagy position it at the pivot point in the cellular decision pathway for managing damaged mitochondria via repair vs. removal.
What the future may hold…
As much as it might simplify biomedical research, a man will never be a mouse (or a fly), and a cardiomyocyte is not a fibroblast. If one accepts our premise that the most basic lessons of mitochondrial dynamism gleaned from studies of cultured fibroblasts and similar cells can be misleading when applied to the unique characteristics of adult cardiac myocytes, then it is obvious that we need better research platforms and experimental systems. This is not news. Researchers have long used immortalized cells having some characteristics of cardiac myocytes (e.g. H9C2, AT-1 and HL-1 cells) or neonatal rat and mouse cardiac myocytes to better understand the cell biology of cardiac hypertrophy and failure; frequently such studies were used to supplement and complement data from in vivo studies of genetically modified mice (Dorn and Molkentin, 2004). This approach has been applied with some success to studies of mitochondrial dynamism and quality control (Ong et al., 2010; Siddall et al., 2013). However, the mitochondrial architecture of these cells more resembles fibroblasts than adult cardiomyocytes. We have performed studies in Drosophila in which cardiomyocyte mitochondria have many of the same morphometric and functional characteristics as in mammals, but mechanistic investigations are limited by the small size and minimal protein content of fruit fly heart tubes (Bhandari et al., 2014; Chen et al., 2012b; Chen and Dorn, 2013; Chen et al., 2013; Dorn et al., 2011).
Recently, we and others have taken advantage of improved techniques for differentiating cardiomyocytes from human inducible pluripotent stem cells (hiPSC-CM) (Lian et al., 2013; Takahashi et al., 2007) to manipulate and examine the effects of mitochondrial fusion factors in functional cultured human cardiomyocytes. In theory, hiPSC-CM can be a nearly unlimited resource for studying the cell biology of cardiac disorders (Priori et al., 2013). Joseph Wu and colleagues have demonstrated the utility of this system for studying familial dilated cardiomyopathy (Sun et al., 2012) and viral myocarditis (Sharma et al., 2014). Wu and Mochly-Rosen recently used hiPSC-CM to understand mitochondrial and cellular dysfunction conferred by a human mutation in aldehyde dehydrogenase (Ebert et al., 2014). In our hands, like mitochondria of adult cardiac myocytes, mitochondria of hiPSC-CM tend to be short and localized between myofibrillar elements (Figure 4). By comparison, mitochondria of those hiPSC-derived cells that fail to differentiate into cardiomyocytes retain the filamentous morphometry and network architecture characteristic of fibroblasts (Figure 4). Since only 60–80% of the hiPSC-derived cells actually differentiate into spontaneously contracting cardiac myocytes (unpublished results), this provides a unique side-by-side system perfect for comparing and contrasting the effects of different genetic and environmental manipulations on cardiomyocyte and non-myocyte mitochondria. Together with new in vivo genetic models, such improved in vitro systems are likely to propel investigations toward more fully understanding the functional integration of mitochondrial dynamism, mitochondrial quality control, and other biological aspects of the ancient commensalism that exists between our mitochondria and us.
Acknowledgments
Supported by NIH HL59888 (GWD) and an American Heart Association predoctoral fellowship award (MS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlqvist G, Landin S, Wroblewski R. Ultrastructure of skeletal muscle in patients with Parkinson’s disease and upper motor lesions. Lab Invest. 1975;32:673–679. [PubMed] [Google Scholar]
- Anan R, Nakagawa M, Miyata M, Higuchi I, Nakao S, Suehara M, Osame M, Tanaka H. Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation. 1995;91:955–961. doi: 10.1161/01.cir.91.4.955. [DOI] [PubMed] [Google Scholar]
- Archer SL. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. New Engl J Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, Lygate CA, Hough T, Townsend S, Williams D, et al. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. doi: 10.1371/journal.pgen.1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW., 2nd Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res. 2014;114:257–265. doi: 10.1161/CIRCRESAHA.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. 2011;108:9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin O, Desnuelle C, Rascol O, Borg M, Peyro Saint Paul H, Azulay JP, Bille F, Figarella D, Coulom F, Pellissier JF, et al. Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J Neurol Sci. 1994;125:95–101. doi: 10.1016/0022-510x(94)90248-8. [DOI] [PubMed] [Google Scholar]
- Cagalinec M, Safiulina D, Liiv M, Liiv J, Choubey V, Wareski P, Veksler V, Kaasik A. Principles of the mitochondrial fusion and fission cycle in neurons. J Cell Sci. 2013;126:2187–2197. doi: 10.1242/jcs.118844. [DOI] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010a;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91–99. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, Cortopassi G, Chiamvimonvat N, Bers DM, Votruba M, et al. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc. 2012a;1:e003012. doi: 10.1161/JAHA.112.003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ Res. 2012b;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW., 2nd Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA. 2010b;107:9035–9042. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu Y, Dorn GW., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sparks M, Bhandari P, Matkovich SJ, Dorn GW., 2nd Mitochondrial Genome Linearization Is a Causative Factor for Cardiomyopathy in Mice and Drosophila. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5432. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, Votruba M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. 2007;16:1307–1318. doi: 10.1093/hmg/ddm079. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011;20:867–879. doi: 10.1093/hmg/ddq526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW., 2nd Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009;119:203–212. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW., 2nd Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008;117:396–404. doi: 10.1161/CIRCULATIONAHA.107.727073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW., 2nd Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardio Transl Res. 2010;3:374–383. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Clark CF, Eschenbacher WH, Kang MY, Engelhard JT, Warner SJ, Matkovich SJ, Jowdy CC. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Molkentin JD. Manipulating cardiac contractility in heart failure: data from mice and men. Circulation. 2004;109:150–158. doi: 10.1161/01.CIR.0000111581.15521.F5. [DOI] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Song M, Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardio. 2014a doi: 10.1016/j.yjmcc.2014.09.015. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Kitsis RN. The mitochondrial dynamism-mitophagy-cell death interactome: Multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res. 2014b doi: 10.1161/CIRCRESAHA.116.303554. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW., 2nd Gone fission…Diverse consequences of cardiac Drp1 deficiency. Circ Res. 2015 doi: 10.1161/CIRCRESAHA.114.305672. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBoff B, Feany M, Gotz J. Why size matters - balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci. 2013;36:325–335. doi: 10.1016/j.tins.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Kodo K, Liang P, Wu H, Huber BC, Riegler J, Churko J, Lee J, de Almeida P, Lan F, et al. Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci Transl Med. 2014;6:255ra130. doi: 10.1126/scitranslmed.3009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- Fang L, Moore XL, Gao XM, Dart AM, Lim YL, Du XJ. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. 2007;80:2154–2160. doi: 10.1016/j.lfs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PloS one. 2012;7:e32388. doi: 10.1371/journal.pone.0032388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givvimani S, Pushpakumar S, Veeranki S, Tyagi SC. Dysregulation of Mfn2 and Drp-1 proteins in heart failure. Can J Physiol Pharmacol. 2014;92:583–591. doi: 10.1139/cjpp-2014-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW, 2nd, Brady MJ, Macleod KF. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32:2570–2584. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest. 2013;123:5371–5388. doi: 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Comm. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart Against Energy Stress. Circ Res. 2014 doi: 10.1161/CIRCRESAHA.116.303356. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara A, Cipolat S, Chen Y, Dorn GW, 2nd, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science. 2013;342:734–737. doi: 10.1126/science.1241359. [DOI] [PubMed] [Google Scholar]
- Kolwicz SC, Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Kubli DA, Quinsay MN, Gustafsson AB. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol. 2013a;6:e24511. doi: 10.4161/cib.24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013b;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet. 2012;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- Li J, Li Y, Jiao J, Wang J, Li Y, Qin D, Li P. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol Cell Biol. 2014;34:1788–1799. doi: 10.1128/MCB.00774-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weaver D, Shirihai O, Hajnoczky G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009;28:3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM, Kelly DP. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res. 2014;114:626–636. doi: 10.1161/CIRCRESAHA.114.302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. 2012a;111:1012–1026. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF, Jr, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012b;302:H167–179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham AH, Meng S, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet. 2012;21:4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, Huynh le H, Nicolas V, Alavi MV, et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. 2012;94:408–417. doi: 10.1093/cvr/cvs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Di Pasquale E, Condorelli G. Induced pluripotent stem cell-derived cardiomyocytes in studies of inherited arrhythmias. J Clin Invest. 2013;123:84–91. doi: 10.1172/JCI62838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802. doi: 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–698. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Marceau C, Hamaguchi R, Burridge PW, Rajarajan K, Churko JM, Wu H, Sallam KI, Matsa E, Sturzu AC, et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus b3-induced myocarditis and antiviral drug screening platform. Circ Res. 2014;115:556–566. doi: 10.1161/CIRCRESAHA.115.303810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–23361. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Watts RL, Juncos JL, Torroni A, Wallace DC. Mitochondrial oxidative phosphorylation defects in Parkinson’s disease. Ann Neurol. 1991;30:332–339. doi: 10.1002/ana.410300304. [DOI] [PubMed] [Google Scholar]
- Siddall HK, Yellon DM, Ong SB, Mukherjee UA, Burke N, Hall AR, Angelova PR, Ludtmann MH, Deas E, Davidson SM, et al. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PloS one. 2013;8:e62400. doi: 10.1371/journal.pone.0062400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW., 2nd Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res. 2014a;115:348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fusion and fission factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2014b doi: 10.1016/j.cmet.2014.12.011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–3532. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra147. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, Sanbe A, Robbins J, Dorn GW., 2nd Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res. 2004;95:1200–1206. doi: 10.1161/01.RES.0000150366.08972.7f. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa O, Ishihara T, Maeda M, Matsunaga Y, Tsukamoto S, Kawano N, Miyado K, Shitara H, Yokota S, Nomura M, et al. Mitochondrial Fission Factor Drp1 Maintains Oocyte Quality via Dynamic Rearrangement of Multiple Organelles. Curr Biol. 2014 doi: 10.1016/j.cub.2014.08.060. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Verhoeven K, Claeys KG, Zuchner S, Schroder JM, Weis J, Ceuterick C, Jordanova A, Nelis E, De Vriendt E, Van Hul M, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 2006;129:2093–2102. doi: 10.1093/brain/awl126. [DOI] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Yang WY. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat Comm. 2013;4:2428. doi: 10.1038/ncomms3428. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW., 2nd Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. 2002;8:725–730. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- Zesiewicz TA, Strom JA, Borenstein AR, Hauser RA, Cimino CR, Fontanet HL, Cintron GB, Staffetti JF, Dunne PB, Sullivan KL. Heart failure in Parkinson’s disease: analysis of the United States medicare current beneficiary survey. Parkinsonism Relat Disord. 2004;10:417–420. doi: 10.1016/j.parkreldis.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]