

Abstract
The pedunculopontine nucleus (PPN) is a new deep brain stimulation (DBS) target for Parkinson's disease (PD), but little is known about PPN firing pattern alterations in PD. The anesthetized rat is a useful model for investigating the effects of dopamine loss on the transmission of oscillatory cortical activity through basal ganglia structures. After dopamine loss, synchronous oscillatory activity emerges in the subthalamic nucleus and substantia nigra pars reticulata in phase with cortical slow oscillations. To investigate the impact of dopamine cell lesion-induced changes in basal ganglia output on activity in the PPN, this study examines PPN spike timing with reference to motor cortex (MCx) local field potential (LFP) activity in urethane- and ketamine-anesthetized rats. Seven – ten days after unilateral 6-hydroxydopamine lesion of the medial forebrain bundle, spectral power in PPN spike trains and coherence between PPN spiking and PPN LFP activity increased in the ∼1 Hz range in urethane-anesthetized rats. PPN spike timing also changed from firing predominantly in-phase with MCx slow oscillations in the intact urethane-anesthetized rat to firing predominantly antiphase to MCx oscillations in the hemi-parkinsonian rat. These changes were not observed in the ketamine-anesthetized preparation. These observations suggest that dopamine loss alters PPN spike timing by increasing inhibitory oscillatory input to the PPN from basal ganglia output nuclei, a phenomenon that may be relevant to motor dysfunction and PPN DBS efficacy in PD patients.
Keywords: pedunculopontine nucleus, Parkinson's disease, motor cortex, oscillations, local field potential, dopamine, deep brain stimulation, basal ganglia, urethane, ketamine
Introduction
The pedunculopontine nucleus (PPN) has robust connections with the basal ganglia (Spann and Grofova, 1989, 1991; Lavoie and Parent, 1994a, 1994c; Shink et al., 1996, 1997), thalamus (Lavoie and Parent, 1994b), cerebellum (Hazrati and Parent, 1992), and spinal cord (Spann and Grofova, 1989) and has been suggested to play a key role in the control of posture and locomotion (Masdeu et al., 1994; Lee et al., 2000; Pahapill and Lozano, 2000). The PPN has also recently emerged as a target for deep brain stimulation (DBS) in Parkinson's disease (PD) and early DBS results show amelioration of medically intractable akinesia and gait abnormalities (Mazzone et al., 2005; Plaha and Gill, 2005; Stefani et al., 2007).
However, there is conflicting information about the effects of dopamine depletion on PPN neuronal activity in PD. Non-human primate and patient data suggest that the PPN is over-inhibited in PD. While excitotoxic lesions of the PPN in normal primates cause akinesia (Kojima et al., 1997; Munro-Davies et al., 1999; Matsumura and Kojima, 2001), low-frequency (2.5-10 Hz) stimulation (Jenkinson et al., 2004) or pharmacological disinhibition (Nandi et al., 2002) of the PPN can dramatically improve motor behavior in parkinsonian MPTP-lesioned primates. Stimulation in the 10-25 Hz range, generally thought to drive local neuronal activity, is also effectively used in the PPN to ameliorate motor symptoms in PD patients (Mazzone et al., 2005; Plaha and Gill, 2005; Stefani et al., 2007). However, in contrast to the non-human primate and patient results, rodent studies suggest either excitation of the PPN (Breit et al., 2001, 2005; Jeon et al., 2003), inhibition of the PPN (Florio et al., 2007; Gomez-Gallego et al., 2007), or no change in PPN neuronal activity following dopamine cell lesion (Mena-Segovia et al., 2005; Heise and Mitrofanis, 2006).
The urethane-anesthetized rat has been shown to be a useful model for investigating the effects of dopamine loss on the transmission of oscillatory cortical activity through basal ganglia structures. Cortical activity in the urethane-anesthetised rat is highly synchronized, characteristically oscillates at ∼1Hz, and is relatively stable over time (Steriade et al., 1993; Contreras and Steriade, 1995, 1997; Amzica and Steriade, 1998). Many studies have shown changes in firing pattern and increased oscillatory activity in the basal ganglia following dopamine cell lesion in the anesthetized rat (Sanderson et al., 1986; MacLeod et al., 1990; Hollerman and Grace, 1992; Burbaud et al., 1995; Hassani et al., 1996; Murer et al., 1997; Rohlfs et al., 1997; Tseng et al., 2000, 2001a, 2001b; Perier et al., 2000; Vila et al., 2000; Magill et al., 2001; Ni et al., 2001; Belluscio et al., 2003; Tai et al., 2003; Walters et al., 2005, 2007; Parr-Brownlie et al., 2007; Zold et al., 2007). Recently, this cortical oscillatory activity has been used as a probe signal in the urethane-anesthetized rat to investigate the effects of dopamine loss on the phase relationships between oscillatory activity in the external segment of the globus pallidus (GPe), the substantia nigra pars reticulata (SNpr), the subthalamic nucleus (STN), and the striatum (Walters et al., 2007). Increases in slow oscillations (0.3–2.5 Hz) in firing rate emerged in all structures following dopamine cell lesion (Walters et al., 2007). Phase relationships of oscillatory activity in the GPe, STN, and SNpr suggest that the increased incidence of oscillatory activity in the SNpr is supported by the convergence of antiphase inhibitory oscillatory input from the GPe and excitatory oscillatory input from the STN (Walters et al., 2007).
It is unknown whether the oscillatory cortical activity that is transmitted through basal ganglia structures following dopamine loss is also transmitted to the PPN. Evidence of a connection between the PPN and the motor cortex (MCx) in non-human primates (Matsumura et al., 2000) and the recent evidence of this connection in humans (Aravamuthan et al., 2007; Muthusamy et al., 2007) suggest that this direct transmission of cortical oscillations is possible. Cortical oscillatory activity could also be transmitted to the PPN indirectly via the basal ganglia. Most input to the PPN comes from three basal ganglia regions. The internal segment of the globus pallidus (GPi) and SNpr send inhibitory efferents to the PPN (Shink et al., 1996, 1997) while excitatory input comes predominantly from the STN (Spann and Grofova, 1991). Therefore, convergent excitatory oscillatory input from the STN and inhibitory oscillatory input from the GPi/SNpr (Walters et al., 2007) may shape oscillatory activity in the PPN in PD.
The aim of the present study was to investigate whether loss of dopamine affects the relationship between neuronal activity in the PPN and MCx in rats under urethane or ketamine-xylazine anesthesia. Effects of dopamine cell lesion on spike timing relationships between PPN and MCx were assessed by simultaneously recording spike trains in the PPN and local field potential (LFP) activity in the MCx and PPN in anesthetized intact rats and in the lesioned hemisphere of hemi-parkinsonian rats 7 – 10 days after a unilateral injection of 6-OHDA into the medial forebrain bundle.
Methods
All experiments were conducted in accordance with the NIH Guide for Care and Use of Laboratory Animals and approved by the NINDS Animal Care and Use Committee. Every effort was made to minimize the number of animals used and their discomfort.
Nigrostriatal lesions and behavioral testing
Male Sprague–Dawley rats (Taconic Farms, Rockville, MD, USA) weighing 275–325 g at the time of surgery were anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and placed in a stereotaxic apparatus with the incisor bar at −3.5 mm. Ocular lubricant Lacrilube was applied to prevent corneal drying. A hole was drilled in the skull above the appropriate coordinates and a 27-gauge injection cannula lowered to the left medial forebrain bundle: 4.4 mm anterior to the lambdoid suture, 1.2 mm lateral to the sagittal suture and 8.3 mm ventral to the surface of the skull. Six μg of 6-OHDA HBr in 3 μl of 0.9% saline containing 0.01% ascorbic acid were infused via the cannula over 3 min. The cannula was left in place for 3 min after the infusion. Rats were injected with desmethylimipramine (15 mg/kg, i.p.) 30 min prior to the intracerebral infusion to protect noradrenergic neurons. The post-operative diet of lesioned rats was supplemented with fruit and enriched gelatin to maintain weight. Five to seven days after surgery, rats were screened for lesion efficacy by step testing (Olsson et al., 1995). Only rats that demonstrated a strong lesion effect in behavior (number of steps by contralateral limb/number of steps by ipsilateral limb<5%, previously shown to correlate with 99% loss of dopamine in the striatum ipsilateral to the lesioned hemisphere (Parr-Brownlie et al., 2007)) were used for electrophysiology.
Surgical and recording procedures
Recordings were made in intact rats and in rats with unilateral 6-OHDA-induced dopamine cell lesions 7 to 10 days after the lesioning surgery. Extracellular unit activity and LFPs of PPN neurons were recorded through the same electrode in intact rats or in lesioned rats ipsilateral to the dopamine cell lesion. PPN activity was also simultaneously recorded with LFP activity in the ipsilateral MCx. Recordings were conducted in rats under either urethane anesthesia (1.6 g/kg i.p. initially, with additional supplements as needed) or ketamine-xylazine anesthesia (100 mg/kg ketamine and 10 mg/kg xylazine i.p., with supplemental doses of ketamine as needed). LFP activity also provided an indication of depth of anesthesia. Anesthesia was maintained at levels sufficient to eliminate reflexive responses and whisking movements but not induce burst suppression patterning (where LFP activity would be mainly oscillating at frequencies <0.3 Hz).
Rats were placed into a stereotaxic apparatus (David Kopf, Tujunga, CA, USA), the scalp reflected, and holes drilled in the skull over the target areas. Body temperature was maintained at 36–38 °C using a heating pad. With micromanipulators (MO-8, Narishige, East Meadow, NY, USA), single barrel glass recording electrodes filled with 2% Pontamine Sky Blue dye in 2 M NaCl were lowered through craniotomies to the PPN or MCx. Spike trains and LFPs were recorded with microelectrodes with 3-4 MΩ resistances (measured at 135 Hz) with tip diameter of 1–2 μm. Electrode location varied in the PPN without consistent placement in a specific subregion. MCx LFP recording sites were targeted to layer V (1.6 mm anterior and 2.2 mm lateral from bregma and 2.0 mm below the surface of the cortex). Recording sites were similarly distributed in the lesioned and intact hemispheres (Fig. 1A).
Figure 1.
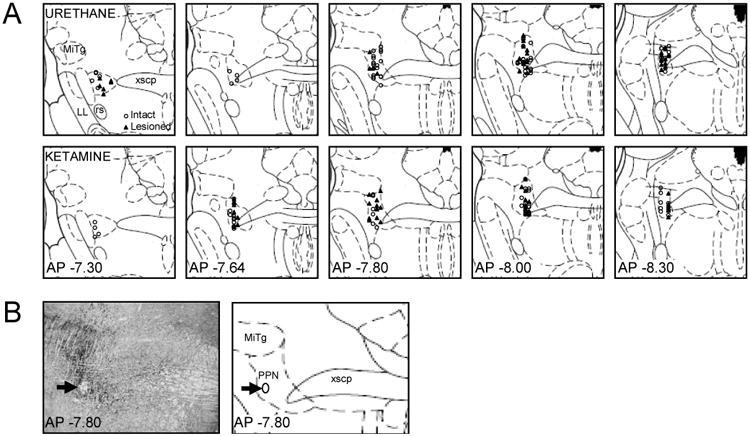
Location of Recorded PPN Cells in Intact and 6-OHDA Lesioned Urethane and Ketamine-Anesthetized Rats. Recordings in intact rats are shown as open circles and recordings in lesioned rats are shown as closed triangles (A). An example of histological confirmation of recording location in the PPN is shown on a 20 μm coronal brain section stained for acetylcholinesterase (B). Arrows indicate the site of a dye deposit on an image (left) and schematic (right) of the stained section. The dye deposit itself is washed off during acetylcholinesterase staining to leave a hole in the slice that is visible on the image (left) and indicated as an ellipse on the schematic (right). Distance from bregma is indicated for each coronal slice in mm. Abbreviations: PPN: pedunculopontine nucleus, PAG: periaqueductal gray, xscp: decussation of the superior cerebellar peduncle, MiTg: microcellular tegmental nucleus, LL: lateral lemniscus, rs: rubrospinal tract. Schematics of coronal slices are adapted from The Rat Brain in Stereotaxic Coordinates, 2nd Ed. (Paxinos and Watson, 1986).
Extracellularly recorded action potentials were amplified (World Precision Instruments Duo 773, World Precision Instruments, Sarasota, FL, USA) and monitored on digital oscilloscopes (Hewlett-Packard, Palo Alto, CA, USA) and audio monitors (Grass, West Warwick, RI, USA). Spikes and LFPs were sampled at 25 kHz and 1000 Hz and band pass filtered at 250–5000 Hz and 0.1–100 Hz, respectively. Discriminated signals were collected on a PC with a CED interface and Spike2 data acquisition and analysis software (Cambridge Electronic Design, Cambridge, UK). All waveforms were biphasic. Final recording sites were marked by iontophoresis of dye with 40 min of constant current injection (approximately -20 μA). Rats were sacrificed, brains were removed and frozen, and 20 μm brain sections were mounted on subbed slides for histological examination. Only units confirmed as being within the target nucleus by location of dye deposit and/or electrolytic lesion were analyzed. When multiple recording sites were investigated, prior recording sites were reconstructed from the coordinates of the blue dye deposit or electrolytic lesion. The location of recording sites within the PPN was confirmed by staining the slices for acetylcholinesterase (Fig. 1B, Paxinos and Watson, 1986). Slides were incubated overnight in a 4 mM CuSO4, 16 mM glycine, and 0.4 mM acetylthiocholine-iodide solution in 50 mM Na+CH3COO- buffer (pH 5.0). The solution was also supersaturated with ethopropazine, a butyrcholinesterase inhibitor. Slides were rinsed in water and the tissue fixed in formaldehyde overnight. The tissue was then dehydrated in ethanol, rinsed in Histo-Clear, and coverslipped with Permount.
Data analysis
For data analyses, 300 s of spike train and LFP data were isolated from baseline recordings. These epochs were selected from the 5–10 min of baseline activity, were representative of neuronal activity, and free from artifacts. In addition to determining firing rate, spiking and LFP activity were analyzed for significant oscillatory activity maintained over time within the frequency range of 0.3–2.5 Hz. LFPs were smoothed to 20 Hz and high pass filtered at 0.2 Hz. Spike channels were converted from event channels to waveform channels with a 20 Hz sampling rate using a Spike2 script. Fast Fourier transform (FFT) analyses were used to determine the dominant frequency in the spike train and LFP for each 300 s data segment using a block size of 256 (yielding a frequency resolution of 0.078 Hz). Coherence between spiking and LFP activity and coherence between LFP activity in the two recording locations were examined as well using a script in Spike2. Each 300s data segment was binned at 50 ms, using a block size of 256 yielding 23 non-overlapping 12.8 s windows. For these parameters, coherence >0.127 was considered significant (P<0.05), as determined by the equation 1-(1-α)1/(L-1), where α is 0.95 and L is the number of windows used (Rosenberg et al., 1989). In order to determine whether there were differences in amplitude between LFPs, the root mean square (RMS) of the amplitude of the LFP for each 300 s epoch was calculated. As all signals are corrected for any DC offset, the mean of the LFP signal is ∼0 mV. Therefore, the RMS about the mean LFP amplitude (equal to the standard deviation of LFP amplitude) serves as a good measure of neuronal activity (Goldberg et al., 2004; Moran et al., 2006; Zaidel et al., 2008).
LFP power spectra, LFP total power between 0.3-2.5 Hz, the main peak frequencies in LFP power spectra, the RMS of LFP amplitude, and LFP-LFP coherence spectra were averaged by rat while spike power spectra, spike total power, and spike-to-LFP coherence spectra were averaged across all cells. In order to ensure that LFP power spectra and LFP total power comparisons were evaluated between rats at similar levels of anesthesia, recordings with total power values outside of a 2 standard deviation (SD) range from the mean total power (between 0.3-2.5 Hz, 0.3-1.2 Hz, or 1.2-2.5 Hz, averaged across rats) were excluded from analysis. All LFP recordings excluded using the above criterion were also excluded from LFP-LFP coherence analyses.
Phase relationships between spikes and LFPs were assessed using spike-triggered waveform averages (STWAs) and plotted as polar histograms. LFP channels were smoothed to 20 Hz and band pass filtered at 0.3–2.5 Hz. Spike-triggered waveform averages were calculated for 4 s before and after the spike trigger over a 300 s epoch. Spiking at the peaks and troughs of LFP oscillations was considered to be at 0° and 180° phase, respectively. STWAs were determined to be significant if the peak-to-trough amplitude of the STWA around the time of spiking was greater than 4 SDs of the amplitude of an STWA generated from the same data using randomly distributed spikes.
Data are presented as the mean ± standard error of the mean. Data were statistically evaluated with grouped t-test. Criterion of significance was P<0.05. Statistics were generated with Microsoft Excel (Microsoft Corporation, 2002). Polar data are presented as the mean phase angle of the distribution±the angular deviation (analogous to SD) and as polar histograms. Distribution of phase angles was tested against the null hypothesis of a random distribution with the Rayleigh test (Batschelet, 1981). The P-value for the Rayleigh test indicates the degree of grouping of the polar data and depends on the strength of the phase concentration and the number of data points.
Drugs and Staining Chemicals
Glycine, acetylthiocholine-iodide, CuSO4, Na+CH3COO-, ethopropazine, ethanol, formaldehyde, desmethylimipramine HCl, urethane, and 6-OHDA HBr were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Histo-Clear was obtained from Raymond A Lamb Inc. (Durham, NC, USA), Permount from Biomeda Corp. (Foster City, CA, USA), Pontamine sky blue from BDH Laboratory Supplies (Poole, UK), ketamine HCl from Fort Dodge Animal Health (Fort Dodge, IA, USA), and xylazine from Phoenix Pharmaceutical, Inc. (St. Joseph, MO, USA). The ocular lubricant Lacrilube was purchased from Allergan Pharmaceuticals (Irvine, CA, USA).
Results
PPN and MCx spike and LFP relationships in the urethane-anesthetized rat
Slow ∼1 Hz oscillations are dominant in cortical output in the urethane-anesthetized state (Steriade et al., 1993) and loss of dopamine is thought to facilitate the transmission of these oscillations through the basal ganglia network (Murer et al., 2002). This cortical oscillatory activity could also be transmitted to the PPN directly via a connection between the motor cortex and PPN (demonstrated in non-human primates and humans (Matsumura et al., 2000; Aravamuthan et al., 2007; Muthusamy et al., 2007) but not verified in rodents) or indirectly via the basal ganglia (Inglis and Winn, 1995; Lee et al., 2000; Pahapill and Lozano, 2000). To examine relationships between oscillatory activity in the PPN and MCx after dopamine cell lesion, PPN spike and PPN LFP activity were recorded simultaneously with MCx LFP activity in intact rats and in lesioned rats 7-10 days after injection of 6-OHDA into the medial forebrain bundle.
The effect of dopamine cell lesion on PPN and MCx LFP activity
Slow ∼1 Hz oscillations that emerge in LFP and spike trains in the basal ganglia of urethane-anesthetized rats after dopamine cell lesion have been shown to be highly coherent with slow oscillations in cortical activity (Magill et al., 2001; Tseng et al., 2001b; Belluscio et al., 2003; Parr-Brownlie et al., 2007; Walters et al., 2007; Zold et al., 2007). In the present study, PPN LFPs were significantly coherent with MCx LFPs in the ∼1 Hz range (0.3-2.5 Hz) in intact rats (0.65 ± 0.03, n=10 rats) and lesioned rats (0.52 ± 0.06, n=11 rats) (Fig. 2A). Dopamine cell lesion had no effect on the coherence between PPN and MCx LFPs.
Figure 2.

Characteristics of PPN LFP and MCx LFP activity in the urethane-anesthetized preparation. Coherence between PPN and MCx LFP activity (A, n=10 intact rats, 11 lesioned rats) does not change significantly in the ∼1 Hz range (left) following dopamine cell lesion nor does mean coherence in the 0.3-2.5 Hz range (right). Dashed horizontal lines indicate the P=0.05 level of significance for coherence. LFP activity recorded in the PPN (B, n=15 intact rats, 11 lesioned rats) and MCx (C, n=11 intact rats, 11 lesioned rats) is described by comparing LFP power spectra in the 0.3-2.5 Hz range (left), total LFP power in the 0.3-2.5 Hz range (middle), and the RMS of LFP amplitude (right) between intact and lesioned rats. Both PPN and MCx LFP activity exhibit decreases in power in the ∼1 Hz range (left), decreases in total power between 0.3-2.5 Hz (middle), and decreases in LFP amplitude (right) following dopamine cell lesion. *P<0.05 compared with intact.
However, dopamine cell lesion did result in significant decreases in LFP power in both the MCx and PPN. PPN LFP power in the lesioned rat (n=11 rats) was significantly less than PPN LFP power in the intact rat (n=15 rats) in the ∼1 Hz range. Total PPN LFP power between 0.3-2.5 Hz showed a 44% decrease with dopamine cell lesion (0.59 ± 0.05 mV2·Hz·10-3 in the intact rat vs. 0.33 ± 0.05 mV2·Hz·10-3 in the lesioned rat). In addition, PPN LFP amplitude was significantly decreased following dopamine cell lesion. The RMS of PPN LFP amplitude in the lesioned rat (0.08 ± 0.01 mV) was 31% less than that in the intact rat (0.11 ± 0.01 mV) (Fig. 2B).
Similarly, MCx LFP power in the lesioned rat (n=11 rats) was significantly less than MCx LFP power in the intact rat (n=11 rats) in the ∼1 Hz range, though the peak spectral frequency was not significantly different between lesioned (0.77 ± 0.03 Hz) and intact (0.67 ± 0.05 Hz) animals. Total MCx LFP power was decreased by 46% (15.2 ± 1.7 mV2·Hz·10-3 in the intact rat vs. 8.2 ± 2.0 mV2·Hz·10-3 in the lesioned rat) and the RMS of MCx LFP amplitude was decreased by 33% with dopamine cell lesion as well (0.36 ± 0.05 mV in the lesioned rat vs. 0.53 ± 0.03 mV in the intact rat) (Fig. 2C).
The effect of dopamine cell lesion on PPN neuronal activity
To gain further insight into processes underlying decreases in PPN LFP amplitude and examine the potential impact of oscillatory output from the basal ganglia on the PPN, rate and temporal dynamics of PPN spiking were examined. Oscillatory activity in PPN spiking was examined by converting PPN spike trains to Gaussian waveforms with a 20 Hz sampling rate and then determining the power of this waveform signal using FFT analysis. In contrast to the decreased power and amplitude observed in PPN LFP following dopamine cell lesion, increased oscillatory activity was observed in PPN spike trains. A comparison of the power in PPN spike trains revealed that spike train power in the lesioned rat was significantly greater than spike train power in the intact rat in the ∼1 Hz range (Fig. 3A). Total spike power between 0.3-2.5 Hz in the lesioned rat (0.52 ± 0.12 Hz3, n=38 cells, 11 rats) was double the total spike power in the intact rat (0.26 ± 0.03 Hz3, n=59 cells, 17 rats).
Figure 3.
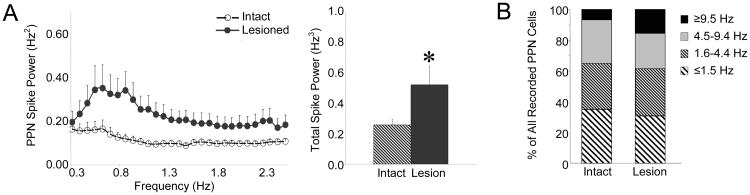
Characteristics of PPN spike trains in the urethane-anesthetized preparation. Oscillatory activity in PPN spike trains (A, n=59 PPN cells in 17 intact rats, 38 cells in 11 lesioned rats) was determined by converting PPN spike trains to Gaussian waveforms with a 20 Hz sampling rate and then using FFT to determine power in the 0.3-2.5 Hz range. There is an increase in power in the ∼1 Hz range in PPN spike trains following dopamine cell lesion (left) with a significant increase in total power between 0.3-2.5 Hz following dopamine cell lesion (right). * P<0.05 compared with intact. Firing rate distributions (B, n=59 cells in 17 intact rats, 38 cells in 11 lesioned rats) are indicated for the intact and 6-OHDA lesioned animals. There was no significant difference in firing rate between intact and lesioned rats.
This increase in PPN spike train oscillatory activity was not associated with significant changes in PPN firing rate. Dopamine cell lesion had no significant effect on PPN firing rate and the percentages of PPN neurons categorized as very slow (≤1.5 Hz), slow (1.6-4.4 Hz), medium (4.5-9.4 Hz), or fast (≥9.5 Hz) firing were comparable between the intact and lesioned preparations. In the intact animal, the mean firing rate in the PPN (n=59 cells, 17 rats) was 3.8 ± 0.5 Hz and ranged from 0.01 – 13.8 Hz with 93% of all cells firing at rates less than 9.5 Hz. The mean PPN firing rate in the lesioned rat (n=38 cells, 11 rats) was 5.8 ± 1.3 Hz and ranged from 0.1 – 39.8 Hz with 85% of all cells firing at rates less than 9.5 Hz (Fig. 3B).
The relationship between PPN spiking and LFP activity in the PPN and MCx
Consistent with increased oscillatory activity in PPN spike trains, coherence between PPN spiking and LFP oscillatory activity in the PPN and MCx increased in the ∼1 Hz range following dopamine cell lesion; PPN spiking and LFP oscillatory activity, which were not significantly coherent in the intact rat, became significantly coherent following dopamine cell lesion. Mean coherence between PPN spiking and PPN LFP increased by 50% with dopamine cell lesion from 0.10 ± 0.01 in the intact animals (n=59 cells, 17 rats) to 0.15 ± 0.02 in the lesioned animals (n=38 cells, 11 rats) (Fig. 4A). In addition, coherence between PPN spiking and MCx LFP activity was significantly greater in the lesioned rat in the ∼1 Hz range and mean coherence between PPN spiking and MCx LFP activity increased by 73% from 0.11 ± 0.09 in the intact rat (n=42 cells, 12 rats) to 0.19 ± 0.02 in the lesioned rat (n=38 cells, 11 rats). This increased coherence between spiking and LFP activity, in addition to the increased oscillatory activity in PPN spike trains in the ∼1 Hz range, predict increased phase locking between PPN spiking and LFP activity following dopamine cell lesion (Fig. 4B).
Figure 4.

Relationships between oscillatory activity in PPN spiking and LFP activity in the urethane-anesthetized preparation. Coherence between PPN spike trains and PPN LFP (A, n=59 cells in 17 intact rats, 38 cells in 11 lesioned rats) and coherence between PPN spike trains and MCx LFP (B, n=42 cells in 12 intact rats, 38 cells in 11 lesioned rats) LFP activity increase following dopamine cell lesion in the ∼1 Hz range (left). Mean coherence between PPN spiking and PPN LFP activity and mean coherence between PPN spiking and MCx LFP activity also increase across the 0.3-2.5 Hz range (right). * P<0.05 compared with intact.
Consistent with this prediction, significant phase locking between PPN spiking and LFP oscillations was evident in both the intact and lesioned rats. In the intact rat, PPN spiking occurred predominantly at the troughs (periods of peak negativity or greatest depolarization) of PPN and MCx LFP oscillations. The phase angles between PPN spiking and PPN LFP oscillations were significantly unimodally clustered around 180 ± 62° (n=54 cells, 16 rats) with 76% of PPN cells firing in the troughs of PPN LFP oscillations (exhibiting phase angles between 90 and 270°). Similarly, the phase angles between PPN spiking and MCx LFP activity were significantly unimodally clustered around 180 ± 69° (n=37 cells, 12 rats) with 71% of all cells firing in the troughs of MCx LFP oscillations. This suggests that PPN firing occurs in phase with firing in the MCx in the intact rat (Fig. 5A) since cortical firing also tends to occur at the troughs of cortical LFPs (Murthy and Fetz, 1996a, 1996b; Donoghue et al., 1998; Destexhe et al., 1999; Parr-Brownlie et al., 2007; Rasch et al., 2008).
Figure 5.

PPN spike timing relative to LFP oscillatory activity in the urethane-anesthetized preparation. Typical PPN spike trains in intact (A) and lesioned (B, n=37 significantly oscillatory cells in lesioned rats) rats are shown at the top of each panel overlaid on simultaneously recorded PPN LFP (left, n=54 cells with significant peaks in their STWAs in 16 intact rats, 37 cells in 11 lesioned rats) or MCx LFP (right, n=37 cells in 12 intact rats, 39 cells in 11 lesioned rats) activity. PPN spike-triggered LFP waveform averages (lower left of each panel) illustrate the time of PPN spiking relative to the phase of LFP oscillatory activity in the example spike train. Polar histogram plots (lower right of each panel) summarize the distribution of phases of PPN spikes with respect to PPN LFP (left) and MCx LFP (right) oscillations. In the intact rat, spiking occurs at or near the trough (∼180°) of PPN and MCx LFP activity. Dopamine cell lesion significantly changes this phase relationship as PPN spiking occurs primarily at the peaks (∼0°) of LFP oscillations in lesioned rats. *Significantly (P<0.05) unimodal distributions of phase relationships between PPN spiking and LFP activity.
Dopamine cell lesion resulted in significantly different spike-timing relationships between PPN spiking and LFP oscillatory activity. In contrast to the trough-locked firing observed in intact animals, PPN firing tended to occur at the peaks (periods of least depolarization) of LFP oscillations in the lesioned rats. The phase angles between PPN spiking and PPN LFP oscillations were significantly unimodally clustered around 0 ± 65° (n=37 cells, 11 rats) with 68% of all cells firing in the peaks of PPN LFP oscillations (exhibiting phase angles that are less than 90° and greater than 270°). The phase angles between PPN spiking and MCx LFP were also significantly unimodally clustered around 1 ± 68° (n=39 cells, 11 rats) with 62% of all cells firing in the peaks of MCx LFP oscillations. This demonstrates a predominantly antiphase relationship between spiking in the PPN and MCx following dopamine cell lesion (Fig. 5B).
PPN and MCx spike and LFP relationships in the ketamine-anesthetized rat
Ketamine, a common anesthetic and non-competitive NMDA antagonist, is often used in conjunction with xylazine as an anesthetic or as an anesthetic supplement to urethane during rat electrophysiological recordings (Rudolph and Antkowiak, 2004; Wolff and Winstock, 2006). Ketamine differs from urethane in its mechanism of action and has also been shown to induce slow oscillations in the cortex but at somewhat faster frequencies (Magill et al., 2000; Fontanini et al., 2003; Musizza et al., 2007). To study the effects of dopamine cell lesion in this different commonly-used anesthetic preparation, PPN spiking, PPN LFP, and MCx LFP were examined in intact and hemi-parkinsonian ketamine-xylazine anesthetized rats.
Similar to results observed with urethane anesthesia, coherence between PPN and MCx LFPs in the 0.3-2.5 Hz range was significant in both intact and lesioned ketamine-anesthetized rats with mean coherence at 0.41 ± 0.03 in the intact animals (n=9 rats) and at 0.44 ± 0.02 in the lesioned animals (n=10 rats) (Fig. 6A). However, in contrast to the urethane-anesthetized preparation, PPN LFP power (0.50 ± 0.08 mV2·Hz·10-3 in the intact rat and 0.40 ± 0.03 mV2·Hz·10-3 in the lesioned rat) and the RMS of LFP amplitude (0.11 ± 0.01 mV in the intact rat and 0.10 ± 0.01 mV in the lesioned rat) did not differ significantly between the intact (n=9 rats) and lesioned ketamine-anesthetized animals (n=10 rats) (Fig. 6B). MCx LFP power (9.1 ± 1.5 mV2·Hz·10-3 in the intact rat and 8.7 ± 1.5 mV2·Hz·10-3 in the lesioned rat) and the RMS of LFP amplitude (0.42 ± 0.05 mV in the intact rat and 0.44 ± 0.04 mV in the lesioned rat) also did not differ significantly between the intact (n=9 rats) and lesioned ketamine-anesthetized animals (n=12 rats) (Fig. 6C). As expected, ketamine anesthesia induced higher frequency cortical oscillations than did urethane, though there was no significant difference in peak spectral frequency between intact (1.66 ± 0.07 Hz) and lesioned (1.64 ± 0.07 Hz) animals.
Figure 6.
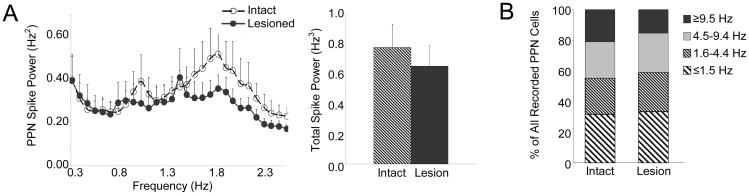
Characteristics of PPN LFP and MCx LFP activity in the ketamine-anesthetized preparation. Coherence between PPN LFP and MCx LFP activity (A, n=9 intact rats, 10 lesioned rats) also does not change significantly in the 0.8-2.5 Hz range (left) following dopamine cell lesion nor does mean coherence in the 0.3-2.5 Hz range (right). LFP activity recorded in the PPN (B n=9 intact rats, 10 lesioned rats) and MCx (C n=9 intact rats, 12 lesioned rats) is described by comparing LFP power spectra in the 0.3-2.5 Hz range (left), total LFP power in the 0.3-2.5 Hz range (middle), and the RMS of LFP amplitude (right) between intact and lesioned rats. Dopamine cell lesion does not change PPN LFP or MCx LFP power or amplitude in the 0.3-2.5 range.
Also in contrast to the results obtained in the urethane-anesthetized preparation, dopamine cell lesion had no effect on oscillatory activity in PPN spike trains in the ketamine-anesthetized rat. Mean total PPN spike power between 0.3-2.5 Hz was 0.77 ± 0.16 Hz3 in the intact rat (n=37 cells, 9 rats) and 0.65 ± 0.14 Hz3 in the lesioned rat (n=38 cells, 12 rats) (Fig. 7A). PPN firing rates were also comparable between the dopamine cell-lesioned and intact ketamine-anesthetized animals. In the intact rat, the mean firing rate in the PPN (n=37 cells, 9 rats) was 5.2 ± 0.9 Hz and ranged from 0.1 – 18.3 Hz with 93% of all cells firing at rates less than 9.5 Hz. The mean PPN firing rate in the lesioned rat (n=38 cells, 12 rats) was 5.1 ± 1.0 Hz and ranged from 0.1 – 31 Hz with 95% of all cells firing at rates less than 9.5 Hz (Fig. 7B).
Figure 7.
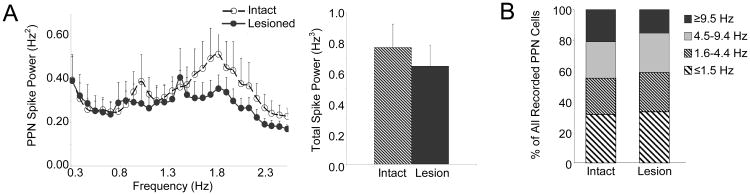
Characteristics of PPN spike trains in the ketamine-anesthetized preparation. Oscillatory activity in PPN spike trains (A, n=37 cells in 9 intact rats, 38 cells in 12 lesioned rats) was not significantly different between intact and lesioned rats in the ∼1 Hz range (left). Dopamine cell lesion had no significant effect on total power in PPN spike trains between 0.3-2.5 Hz (right). Firing rate distributions (B, n=37 cells in 9 intact rats, 38 cells in 12 lesioned rats) are indicated for the intact and dopamine cell lesioned animals. There was also no significant difference in firing rate between the intact and lesioned rat.
Finally, the spike-LFP relationships observed in the urethane-anesthetized preparation were also not observed in the ketamine-anesthetized preparation. There was no change in the coherence between PPN spiking and PPN LFP or MCx LFP oscillations following dopamine cell lesion in the ketamine-anesthetized animals. Mean coherence between PPN spiking and PPN LFP activity was 0.15 ± 0.02 in the intact rat (n=37 cells, 9 rats) and 0.17 ± 0.02 in the lesioned rat (n=38 cells, 12 rats) (Fig. 8A). Mean coherence between PPN spiking and MCx LFP activity was 0.18 ± 0.02 in the intact rat (n=37 cells, 9 rats) and 0.19 ± 0.02 in the lesioned rat (n=38 cells, 12 rats) (Fig. 8B). PPN spike timing relationships with PPN LFP or MCx LFP oscillations were also not evident in either the intact or lesioned animals; the distribution of phase relationships between PPN spiking and PPN LFP (n=38 significantly oscillatory cells in 9 intact rats, 32 cells in 12 lesioned rats) or MCx LFP (n=36 cells in 9 intact rats, 35 cells in 12 lesioned rats) was not significantly unimodally or bimodally clustered (Fig. 8C).
Figure 8.

Relationships between PPN spiking and LFP activity in the ketamine-anesthetized preparation. Coherence between PPN spike trains and PPN LFP (A, n=37 cells in 9 intact rats, 38 cells in 12 lesioned rats) and coherence between PPN spike trains and MCx LFP (B, n=37 cells in 9 intact rats, 38 cells in 12 lesioned rats) do not change following dopamine cell lesion. There is also no change in mean coherence between PPN spiking and PPN LFP or MCx LFP activity in the 0.3-2.5 Hz range (right). Polar histogram plots (C) summarize the distribution of phases of PPN spikes with respect to PPN LFP (top, n=38 significantly oscillatory cells in 9 intact rats, 32 cells in 12 lesioned rats) and MCx LFP (bottom, n=36 cells in 9 intact rats, 35 cells in 12 lesioned rats) oscillations. There is no consistent phase-locking between PPN spiking and LFP activity in the intact (left) or lesioned (right) rats.
Discussion
The robust connections between the PPN and basal ganglia indicate that the PPN will be affected by alterations in basal ganglia output after dopamine cell death. The aim of this study was to explore this issue by determining how loss of dopamine affects PPN activity and its relationship to slow oscillatory activity in the MCx in a rodent model of PD. Both urethane and ketamine-xylazine anesthetized preparations were used to examine this relationship 7-10 days after unilateral 6-OHDA-induced dopamine cell lesion. Oscillatory activity in PPN spike trains and coherence between PPN spiking and PPN LFP were increased after dopamine cell lesion in the urethane-anesthetized preparation. In addition, changes in PPN spike timing with respect to MCx LFP slow oscillations were observed. Viewed in the context of the oscillatory activity that emerges in basal ganglia nuclei following dopamine cell lesion, these results suggest that the increased oscillatory activity in basal ganglia output is transmitted to the PPN most directly via inhibitory oscillatory output from the SNpr or GPi. These changes in spike timing and oscillatory activity were not observed in rats anesthetized with ketamine/zylazine, suggesting that the NMDA antagonist properties of ketamine may attenuate some of the effects of dopamine cell lesion.
Predicted effect of basal ganglia output on PPN activity after dopamine cell lesion in the urethane-anesthetized rat
Dopamine depletion increases transmission of oscillatory activity from the cortex to the basal ganglia via the striatum (Murer et al., 2002). Consistent with this phenomenon, increases in burstiness and oscillatory activity have been shown in spike trains of several basal ganglia nuclei following dopamine cell lesion in the anesthetized rat model of PD (Sanderson et al., 1986; MacLeod et al., 1990; Hollerman and Grace, 1992; Burbaud et al., 1995; Hassani et al., 1996; Murer et al., 1997; Rohlfs et al., 1997; Magill et al., 2000; Tseng et al., 2000, 2001a; Perier et al., 2000; Vila et al., 2000; Ni et al., 2001; Belluscio et al., 2003; Tai et al., 2003; Walters et al., 2005, 2007; Parr-Brownlie et al., 2007; Zold et al., 2007).
The effect of oscillatory output from the basal ganglia on spike timing in the PPN is an interesting issue with respect to mechanisms underlying motor dysfunction in PD as the PPN has been suggested to play a role in the control of gait (Garcia-Rill et al., 1985, 1987; Reese et al., 1995) and is in a position to be affected by DBS of the STN (Florio et al., 2007; Stefani et al., 2007). Since the PPN is strongly interconnected with both the STN (Spann and Grofova, 1991) and SNpr (Shink et al., 1996, 1997), the oscillatory outputs of these nuclei are likely to affect PPN spike timing. However, as increased oscillatory input to the PPN from the STN is excitatory and in phase with increased inhibitory oscillatory input from the SNpr (Walters et al., 2007), these temporally coincident oscillatory inputs might effectively cancel each other in the PPN at the level of individual neurons or at the level of the overall PPN neuronal population.
The present results show that PPN spiking activity becomes more oscillatory in the urethane-anesthetized rat. Mean power in PPN spike trains in the ∼1 Hz range significantly increased following dopamine cell lesion and total power in PPN spike trains in the 0.3-2.5 Hz range doubled. Furthermore, coherence between PPN spiking and PPN LFP and coherence between PPN spiking and MCx LFP activity also increased significantly following dopamine cell lesion indicating that PPN spiking becomes more entrained to the slow oscillations observed in the MCx following dopamine loss in the urethane-anesthetized rat. These results are consistent with the view that phase cancellation in PPN spiking activity is not the dominant result of the coincident excitatory and inhibitory oscillatory input from the STN and SNpr/GPi.
Inhibitory oscillatory output from the basal ganglia output nuclei dominates phase relationships between PPN spiking and LFP activity following dopamine cell lesion in the urethane-anesthetized rat
To further explore the relationship between oscillatory activity in the PPN and oscillatory activity in the basal ganglia and motor cortex, PPN spike-triggered MCx LFP averages were analyzed. The troughs of LFP oscillations represent the periods of greatest negative potential in the extracellular space and thus correspond to the periods of greatest membrane depolarization of recorded cells. Consistently, cortical spiking activity has been shown to predominantly occur at the troughs of cortical LFP oscillations in awake animals (Murthy and Fetz, 1996a, 1996b; Donoghue et al., 1998), in anesthetized animals (Parr-Brownlie et al., 2007; Rasch et al., 2008), and at the troughs of slow cortical LFP oscillations (<1 Hz) observed during sleep (Destexhe et al., 1999). Firing in the STN and SNpr is also locked to the troughs of MCx LFP oscillations after dopamine loss suggesting that firing in these structures is in phase with firing in the MCx (Magill et al., 2001; Tseng et al., 2001b; Murer et al., 2002; Belluscio et al., 2003; Sharott et al., 2005; Walters et al., 2007).
Changes in phase relationships observed between PPN spiking and MCx LFP activity in the urethane anesthetized preparation support the view that inhibitory oscillatory output from the basal ganglia, i.e. from the SNpr and GPi, dominates PPN spike timing following dopamine loss. In the intact urethane-anesthetized rat, PPN spiking most frequently coincided with the troughs of MCx LFP oscillations. These results suggest that input from the STN may influence PPN spike timing relationships resulting in the more frequent occurrence of trough-locked PPN firing in phase with firing in the MCx. Excitatory input to the PPN directly from the MCx could also result in PPN firing in phase with MCx firing in the intact rat. However, it is unknown whether a direct connection in the rat exists. In contrast, in the parkinsonian 6-OHDA lesioned rat, PPN spiking most frequently coincided with the peaks of MCx LFP oscillations. Since increases in bursty and oscillatory activity in the STN are associated with inphase increases in oscillatory activity in the SNpr in the lesioned rat (Walters et al., 2007), the results from this study suggest that PPN neurons are more influenced by trough-locked inhibitory input from the SNpr resulting in predominantly peak-locked PPN firing out of phase with firing in the MCx. These results also corroborate observations in PD patients and evidence from parkinsonian non-human primates that suggest that the PPN is over-inhibited in PD (Nandi et al., 2002; Jenkinson et al., 2004; Mazzone et al., 2005; Plaha and Gill, 2005; Stefani et al., 2007).
It should be pointed out that while the majority (62-76%) of PPN cells exhibited the phase locking described above (firing in phase with MCx firing in the intact rat, but antiphase to MCx firing in the lesioned rat), 24-38% of all PPN cells exhibited the opposite phase relationship (firing antiphase to MCx firing in the intact rat but in phase with MCx firing in the lesioned rat). This diversity in spike timing within the PPN, despite the transmission of a stable ∼1Hz cortical rhythm, is not surprising noting the numerous inputs to the PPN (Inglis and Winn, 1995; Lee et al., 2000; Pahapill and Lozano, 2000), the existence of at least three different electrophysiologically-defined PPN neuronal subtypes (Kang and Kitai, 1990; Florio et al., 2007), and the multiple neurochemically-defined PPN neuronal subtypes including cholinergic (Spann and Grofova, 1992), glutamatergic (Rye et al., 1987), dopaminergic (Rye et al., 1987), and GABAergic (Ford et al., 1995) neuronal populations.
Coincident antiphase oscillatory inputs to the MCx and PPN following dopamine cell lesion may result in coherent decreases in MCx and PPN LFP power in the urethane-anesthetized rat
Since the troughs of LFPs should correspond to the periods when cells closest to the recording site are most likely to fire, one might expect PPN spiking to be predominantly trough-locked to PPN LFP oscillations in both the intact and lesioned rat. However, discrepant relationships between PPN spike timing and PPN LFP were observed in the present study. The results show that PPN spiking is predominantly peak-locked to PPN LFP oscillations in the lesioned rat. This dissociation between PPN spike timing and PPN LFP phase after dopamine cell lesion raises questions about the extent to which PPN LFP reflects local net depolarization related solely to the observed PPN spiking activity. In this context, it is interesting to note that PPN LFP activity was significantly and highly coherent with MCx LFP activity in both intact and lesioned rats and that MCx LFP amplitude observed in this study is almost 5 times larger than PPN LFP amplitude. This may be a result of the more regular organization of MCx neuronal elements and suggests that PPN LFP may not be as robust a measure of neuronal activity as MCx LFP.
The present study also demonstrated that dopamine cell lesion induced significant decreases in MCx and PPN LFP total power between 0.3-2.5 Hz in conjunction with mean decreases in LFP amplitude. Decreases in PPN LFP power are incongruent with the observed increases in synchronization in PPN firing and the increased coherence between PPN spiking and PPN LFP activity discussed above, both of which suggest that PPN LFP power should increase with dopamine cell lesion. In addition, decreases in cortical LFP in the ∼1 Hz frequency range after dopamine cell lesion is inconsistent with the idea that basal ganglia-thalamo-cortical circuits may become more entrained to the slow oscillatory rhythm after loss of dopamine (Goldberg et al., 2002, 2004; Leblois et al., 2006).
A number of factors may contribute to the decrease in power and amplitude in the PPN and MCx LFPs. Inhibitory oscillatory input from the SNpr could compete with excitatory oscillatory input from the STN resulting in the decreased PPN LFP amplitude and power observed in this study. A factor that could contribute to decreases in MCx LFP amplitude and LFP power following dopamine cell lesion is a reduction in the number of regularly firing MCx cells. As basal ganglia output from the SNpr is inhibitory and in phase with cortical activity (Murer et al., 1997; Tseng et al., 2000, 2001a; Walters et al., 2007), this inhibitory basal ganglia output might modulate excitatory thalamocortical activity and thus reduce the number of MCx cells firing synchronously in the urethane-anesthetized rat following dopamine lesion. This hypothesis is supported by studies in animal models of PD demonstrating hypoactivity of glutamatergic thalamic projection neurons to the cortex (Palombo et al., 1988; Schwartzman et al., 1988; Mitchell et al., 1989; Gnanalingham et al., 1995; Brownell et al., 2003; Rolland et al., 2007). Evidence for attenuation of cortical output following dopamine cell lesion also supports this hypothesis. Decreased striatal dendritic length and a decrease in the number of striatal dendritic spines have been observed in 6-OHDA lesioned rats and transgenic mouse models of PD (Ingham et al., 1989, 1993, 1998; Day et al., 2006; Solis et al., 2007) and could be related to decreased connectivity and glutamatergic input to the striatum (McAllister, 2000; Day et al., 2006) from the cortex.
Comparison of the neurological effects of urethane and ketamine: ketamine attenuates the effects of dopamine cell lesion
Ketamine anesthesia is associated with a faster slow oscillation in MCx LFP (Magill et al., 2000; Fontanini et al., 2003; Musizza et al., 2007) and a different mechanism of action from urethane. Results showed that PPN spike timing with respect to MCx was not significantly modulated in ketamine-xylazine anesthetized rats. Unlike in the urethane-anesthetized preparation, 6-OHDA lesion in the ketamine-anesthetized rat had no significant effect on PPN spike power, PPN or MCx LFP power, coherence between PPN spike and LFP activity, coherence between PPN LFP and MCx LFP activity, or on the phase relationships between PPN spiking and LFP oscillations.
Urethane has been shown to have a modest effect on several neurotransmitter-gated ion channels at concentrations close to that used for maintaining anesthesia (Hara and Harris, 2002) but may preferentially affect Ba2+-sensitive K+ leak conductance (Sceniak and Maciver, 2006). These effects on ion channel physiology may confer urethane with its anesthetic properties. Ketamine's anesthetic effect, on the other hand, is thought to result from its action as a non-competitive NMDA-receptor antagonist (Rudolph and Antkowiak, 2004; Wolff and Winstock, 2006), which ultimately results in depression of neuronal responses by interfering with the excitatory effects of glutamate (Anis et al., 1983; Wolff and Winstock, 2006).
Ketamine may have effects similar to other NMDA antagonists such as amantadine which have effectively been used as adjuvant parkinsonian therapies (Papa et al., 1995; Papa and Chase, 1996; Blanchet et al., 1997, 2003; Verhagen et al., 1998; Luginger et al., 2000), potentially acting at the level of the STN to reduce glutamatergic output from this structure (Greenamyre and O'Brien, 1991; Allers et al., 2005).
Relevance to faster oscillations in PD patients
It is important to note that the anesthetized rodent 6-OHDA lesion model of PD is not a comprehensive representation of PD and oscillatory activity in the awake human PD patient. For example, oscillations in the θ (3-8 Hz), α (8-12 Hz), and β (12-30 Hz) frequencies tend to dominate LFP recordings in the cortex and basal ganglia of awake PD patients (Salinas and Sejnowski, 2001; Brown, 2003) while we examined the transmission of slow ∼1 Hz oscillatory activity from the MCx to the basal ganglia and PPN in the anesthetized rodent preparation. Our results in the anesthetized animal should be interpreted conservatively with regard to phase relationships during the transmission of faster frequency oscillations through basal-ganglia/PPN/thalamocortical networks as lags due to axonal conduction and synaptic transmission may become more relevant. However, the network dynamics observed in this study may be relevant to transmission of lower physiological frequencies such as the <30 Hz frequencies that have been associated with parkinsonian motor symptoms (Brown, 2003) and the <11 Hz frequencies that have been recently observed in PPN LFP recordings in PD patients (Androulidakis et al., 2008).
Implications for DBS in PD patients
DBS is thought to ameliorate parkinsonian symptoms by disrupting pathological oscillations that emerge in the basal ganglia in the parkinsonian brain (Benazzouz and Hallett, 2000; Obeso et al., 2000; Brown, 2003; Brown and Williams, 2005; Wichmann and Delong, 2006). The PPN exhibits robust connections with the basal ganglia and dopamine cell lesion has been shown to result in increased transmission of oscillatory activity from the cortex to the basal ganglia (Murer et al., 2002). Our results suggest that this increased oscillatory activity in the basal ganglia is also transmitted downstream to the PPN as evidenced by increased power in PPN spike trains following dopamine cell lesion. Therefore, similar to DBS of the STN, it is possible that DBS of the PPN works by optimally disrupting the transmission of pathological oscillations to the PPN.
DBS of the PPN could also drive PPN activity in the parkinsonian brain. Low frequency stimulation, generally thought to drive neuronal activity, is found to be most therapeutically effective when stimulating the PPN in PD patients (Mazzone et al., 2005; Plaha and Gill, 2005; Stefani et al., 2007) and studies in non-human primates suggest that the PPN is over-inhibited in PD (Kojima et al., 1997; Munro-Davies et al., 1999; Matsumura and Kojima, 2001; Nandi et al., 2002; Jenkinson et al., 2004). Our results also suggest that the PPN is dominated by inhibitory oscillatory input from the SNpr in the parkinsonian brain. Therefore, driving PPN neuronal activity via low frequency stimulation could attenuate the effects of excessive inhibitory input to the PPN in PD.
Alternatively, DBS of the PPN could also affect PPN spike timing relationships. The significant changes in PPN spike timing following dopamine cell lesion observed in this study suggest that PPN spike timing in the parkinsonian brain is potentially dysfunctional. Low frequency stimulation of the PPN, in addition to driving PPN neuronal activity, may disrupt this pathological spike timing relationship or properly synchronize PPN firing with excitatory input.
Acknowledgments
This research was supported by the Intramural Research Program of the NINDS, NIH. Additional support also came from the George C. Marshall Commission (BRA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allers KA, Bergstrom DA, Ghazi LJ, Kreiss DS, Walters JR. MK801 and amantadine exert different effects on subthalamic neuronal activity in a rodent model of Parkinson's disease. Exp Neurol. 2005;191:104–118. doi: 10.1016/j.expneurol.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Amzica F, Steriade M. Cellular substrates and laminar profile of sleep K-complex. Neuroscience. 1998;82:671–686. doi: 10.1016/s0306-4522(97)00319-9. [DOI] [PubMed] [Google Scholar]
- Androulidakis AG, Khan S, Litvak V, Pleydell-Pearce CW, Brown P, Gill SS. Local field potential recordings from the pedunculopontine nucleus in a Parkinsonian patient. NeuroReport. 2008;19:59–62. doi: 10.1097/WNR.0b013e3282f2e2d1. [DOI] [PubMed] [Google Scholar]
- Anis NA, Berry SC, Burton NR, Lodge D. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol. 1983;79:565–575. doi: 10.1111/j.1476-5381.1983.tb11031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravamuthan BR, Muthusamy KA, Stein JF, Aziz TZ, Johansen-Berg H. Topography of cortical and subcortical connections of the human pedunculopontine and subthalamic nuclei. NeuroImage. 2007;37:694–705. doi: 10.1016/j.neuroimage.2007.05.050. [DOI] [PubMed] [Google Scholar]
- Batschelet E. Circular Statistics in Biology. Academic Press; New York: 1981. [Google Scholar]
- Belluscio MA, Kasanetz F, Riquelme LA, Murer MG. Spreading of slow cortical rhythms to the basal ganglia output nuclei in rats with nigrostriatal lesions. Eur J Neurosci. 2003;17:1046–1052. doi: 10.1046/j.1460-9568.2003.02543.x. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Hallett M. Mechanism of action of deep brain stimulation. Neurology. 2000;55:S13–S16. [PubMed] [Google Scholar]
- Blanchet PJ, Metman LV, Chase TN. Renaissance of amantadine in the treatment of Parkinson's disease. Adv Neurol. 2003;91:251–257. [PubMed] [Google Scholar]
- Blanchet PJ, Papa SM, Metman LV, Mouradian MM, Chase TN. Modulation of levodopa-induced motor response complications by NMDA antagonists in Parkinson's disease. Neurosci Biobehav Rev. 1997;21:447–453. doi: 10.1016/s0149-7634(96)00038-3. [DOI] [PubMed] [Google Scholar]
- Breit S, Bouali-Benazzouz R, Benabid AL, Benazzouz A. Unilateral lesion of the nigrostriatal pathway induces an increase of neuronal activity of the pedunculopontine nucleus, which is reversed by the lesion of the subthalamic nucleus in the rat. Eur J Neurosci. 2001;14:1833–1842. doi: 10.1046/j.0953-816x.2001.01800.x. [DOI] [PubMed] [Google Scholar]
- Breit S, Lessmann L, Benazzouz A, Schulz JB. Unilateral lesion of the pedunculopontine nucleus induces hyperactivity in the subthalamic nucleus and substantia nigra in the rat. Eur J Neurosci. 2005;22:2283–2294. doi: 10.1111/j.1460-9568.2005.04402.x. [DOI] [PubMed] [Google Scholar]
- Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson's disease. Mov Disord. 2003;18:357–363. doi: 10.1002/mds.10358. [DOI] [PubMed] [Google Scholar]
- Brown P, Williams D. Basal ganglia local field potential activity: character and functional significance in the human. Clin Neurophysiol. 2005;116:2510–2519. doi: 10.1016/j.clinph.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Brownell AL, Canales K, Chen YI, Jenkins BG, Owen C, Livni E, Yu M, Cicchetti F, Sanchez-Pernaute R, Isacson O. Mapping of brain function after MPTP-induced neurotoxicity in a primate Parkinson's disease model. NeuroImage. 2003;20:1064–1075. doi: 10.1016/S1053-8119(03)00348-3. [DOI] [PubMed] [Google Scholar]
- Burbaud P, Gross C, Benazzouz A, Coussemacq M, Bioulac B. Reduction of apomorphine-induced rotational behaviour by subthalamic lesion in 6-OHDA lesioned rats is associated with a normalization of firing rate and discharge pattern of pars reticulata neurons. Exp Brain Res. 1995;105:48–58. doi: 10.1007/BF00242181. [DOI] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Synchronization of low-frequency rhythms in corticothalamic networks. Neuroscience. 1997;76:11–24. doi: 10.1016/s0306-4522(96)00393-4. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M. Spatiotemporal analysis of local field potentials and unit discharges in cat cerebral cortex during natural wake and sleep states. J Neurosci. 1999;19:4595–4608. doi: 10.1523/JNEUROSCI.19-11-04595.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue JP, Sanes JN, Hatsopoulos NG, Gaal G. Neural discharge and local field potential oscillations in primate motor cortex during voluntary movements. J Neurophysiol. 1998;79:159–173. doi: 10.1152/jn.1998.79.1.159. [DOI] [PubMed] [Google Scholar]
- Florio T, Scarnati E, Confalone G, Minchella D, Galati S, Stanzione P, Stefani A, Mazzone P. High-frequency stimulation of the subthalamic nucleus modulates the activity of pedunculopontine neurons through direct activation of excitatory fibres as well as through indirect activation of inhibitory pallidal fibres in the rat. Eur J Neurosci. 2007;25:1174–1186. doi: 10.1111/j.1460-9568.2007.05360.x. [DOI] [PubMed] [Google Scholar]
- Fontanini A, Spano P, Bower JM. Ketamine-xylazine-induced slow (< 1.5 Hz) oscillations in the rat piriform (olfactory) cortex are functionally correlated with respiration. J Neurosci. 2003;23:7993–8001. doi: 10.1523/JNEUROSCI.23-22-07993.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford B, Holmes CJ, Mainville L, Jones BE. GABAergic neurons in the rat pontomesencephalic tegmentum: codistribution with cholinergic and other tegmental neurons projecting to the posterior lateral hypothalamus. J Comp Neurol. 1995;363:177–196. doi: 10.1002/cne.903630203. [DOI] [PubMed] [Google Scholar]
- Garcia-Rill E, Houser CR, Skinner RD, Smith W, Woodward DJ. Locomotion-inducing sites in the vicinity of the pedunculopontine nucleus. Brain Res Bull. 1987;18:731–738. doi: 10.1016/0361-9230(87)90208-5. [DOI] [PubMed] [Google Scholar]
- Garcia-Rill E, Skinner RD, Fitzgerald JA. Chemical activation of the mesencephalic locomotor region. Brain Res. 1985;330:43–54. doi: 10.1016/0006-8993(85)90006-x. [DOI] [PubMed] [Google Scholar]
- Gnanalingham KK, Milkowski NA, Smith LA, Hunter AJ, Jenner P, Marsden CD. Short and long-term changes in cerebral [14C]-2-deoxyglucose uptake in the MPTP-treated marmoset: relationship to locomotor activity. J Neural Transm Gen Sect. 1995;101:65–82. doi: 10.1007/BF01271546. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Boraud T, Maraton S, Haber SN, Vaadia E, Bergman H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson's disease. J Neurosci. 2002;22:4639–4653. doi: 10.1523/JNEUROSCI.22-11-04639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Rokni U, Boraud T, Vaadia E, Bergman H. Spike synchronization in the cortex/basal-ganglia networks of Parkinsonian primates reflects global dynamics of the local field potentials. J Neurosci. 2004;24:6003–6010. doi: 10.1523/JNEUROSCI.4848-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gallego M, Fernandez-Villalba E, Fernandez-Barreiro A, Herrero MT. Changes in the neuronal activity in the pedunculopontine nucleus in chronic MPTP-treated primates: an in situ hybridization study of cytochrome oxidase subunit I, choline acetyl transferase and substance P mRNA expression. J Neural Transm. 2007;114:319–326. doi: 10.1007/s00702-006-0547-x. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, O'Brien CF. N-methyl-D-aspartate antagonists in the treatment of Parkinson's disease. Arch Neurol. 1991;48:977–981. doi: 10.1001/archneur.1991.00530210109030. [DOI] [PubMed] [Google Scholar]
- Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg. 2002;94:313–318. doi: 10.1097/00000539-200202000-00015. [DOI] [PubMed] [Google Scholar]
- Hassani OK, Mouroux M, Feger J. Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience. 1996;72:105–115. doi: 10.1016/0306-4522(95)00535-8. [DOI] [PubMed] [Google Scholar]
- Hazrati LN, Parent A. Projection from the deep cerebellar nuclei to the pedunculopontine nucleus in the squirrel monkey. Brain Res. 1992;585:267–271. doi: 10.1016/0006-8993(92)91216-2. [DOI] [PubMed] [Google Scholar]
- Heise CE, Mitrofanis J. Fos immunoreactivity in some locomotor neural centres of 6OHDA-lesioned rats. Anat Embryol (Berl) 2006;211:659–671. doi: 10.1007/s00429-006-0130-0. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Graybiel AM, Duyckaerts C, Javoy-Agid F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A. 1987;84:5976–5980. doi: 10.1073/pnas.84.16.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollerman JR, Grace AA. Subthalamic nucleus cell firing in the 6-OHDA-treated rat: basal activity and response to haloperidol. Brain Res. 1992;590:291–299. doi: 10.1016/0006-8993(92)91108-q. [DOI] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, Arbuthnott GW. Spine density on neostriatal neurones changes with 6-hydroxydopamine lesions and with age. Brain Res. 1989;503:334–338. doi: 10.1016/0006-8993(89)91686-7. [DOI] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, Taggart P, Arbuthnott GW. Plasticity of synapses in the rat neostriatum after unilateral lesion of the nigrostriatal dopaminergic pathway. J Neurosci. 1998;18:4732–4743. doi: 10.1523/JNEUROSCI.18-12-04732.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, van Maldegem B, Weenink A, Arbuthnott GW. Morphological changes in the rat neostriatum after unilateral 6-hydroxydopamine injections into the nigrostriatal pathway. Exp Brain Res. 1993;93:17–27. doi: 10.1007/BF00227776. [DOI] [PubMed] [Google Scholar]
- Inglis WL, Winn P. The pedunculopontine tegmental nucleus: where the striatum meets the reticular formation. Prog Neurobiol. 1995;47:1–29. doi: 10.1016/0301-0082(95)00013-l. [DOI] [PubMed] [Google Scholar]
- Jenkinson N, Nandi D, Miall RC, Stein JF, Aziz TZ. Pedunculopontine nucleus stimulation improves akinesia in a Parkinsonian monkey. NeuroReport. 2004;15:2621–2624. doi: 10.1097/00001756-200412030-00012. [DOI] [PubMed] [Google Scholar]
- Jeon MF, Ha Y, Cho YH, Lee BH, Park YG, Chang JW. Effect of ipsilateral subthalamic nucleus lesioning in a rat parkinsonian model: study of behavior correlated with neuronal activity in the pedunculopontine nucleus. J Neurosurg. 2003;99:762–767. doi: 10.3171/jns.2003.99.4.0762. [DOI] [PubMed] [Google Scholar]
- Kang Y, Kitai ST. Electrophysiological properties of pedunculopontine neurons and their postsynaptic responses following stimulation of substantia nigra reticulata. Brain Res. 1990;535:79–95. doi: 10.1016/0006-8993(90)91826-3. [DOI] [PubMed] [Google Scholar]
- Kojima J, Yamaji Y, Matsumura M, Nambu A, Inase M, Tokuno H, Takada M, Imai H. Excitotoxic lesions of the pedunculopontine tegmental nucleus produce contralateral hemiparkinsonism in the monkey. Neurosci Lett. 1997;226:111–114. doi: 10.1016/s0304-3940(97)00254-1. [DOI] [PubMed] [Google Scholar]
- Lavoie B, Parent A. Pedunculopontine nucleus in the squirrel monkey: cholinergic and glutamatergic projections to the substantia nigra. J Comp Neurol. 1994a;344:232–241. doi: 10.1002/cne.903440205. [DOI] [PubMed] [Google Scholar]
- Lavoie B, Parent A. Pedunculopontine nucleus in the squirrel monkey: distribution of cholinergic and monoaminergic neurons in the mesopontine tegmentum with evidence for the presence of glutamate in cholinergic neurons. J Comp Neurol. 1994b;344:190–209. doi: 10.1002/cne.903440203. [DOI] [PubMed] [Google Scholar]
- Lavoie B, Parent A. Pedunculopontine nucleus in the squirrel monkey: projections to the basal ganglia as revealed by anterograde tract-tracing methods. J Comp Neurol. 1994c;344:210–231. doi: 10.1002/cne.903440204. [DOI] [PubMed] [Google Scholar]
- Leblois A, Boraud T, Meissner W, Bergman H, Hansel D. Competition between feedback loops underlies normal and pathological dynamics in the basal ganglia. J Neurosci. 2006;26:3567–3583. doi: 10.1523/JNEUROSCI.5050-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Rinne JO, Marsden CD. The pedunculopontine nucleus: its role in the genesis of movement disorders. Yonsei Med J. 2000;41:167–184. doi: 10.3349/ymj.2000.41.2.167. [DOI] [PubMed] [Google Scholar]
- Luginger E, Wenning GK, Bosch S, Poewe W. Beneficial effects of amantadine on L-dopa-induced dyskinesias in Parkinson's disease. Mov Disord. 2000;15:873–878. doi: 10.1002/1531-8257(200009)15:5<873::aid-mds1017>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- MacLeod NK, Ryman A, Arbuthnott GW. Electrophysiological properties of nigrothalamic neurons after 6-hydroxydopamine lesions in the rat. Neuroscience. 1990;38:447–456. doi: 10.1016/0306-4522(90)90041-2. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Bolam JP, Bevan MD. Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus-globus pallidus network. Neuroscience. 2001;106:313–330. doi: 10.1016/s0306-4522(01)00281-0. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Bolam JP, Bevan MD. Relationship of activity in the subthalamic nucleus-globus pallidus network to cortical electroencephalogram. J Neurosci. 2000;20:820–833. doi: 10.1523/JNEUROSCI.20-02-00820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masdeu JC, Alampur U, Cavaliere R, Tavoulareas G. Astasia and gait failure with damage of the pontomesencephalic locomotor region. Ann Neurol. 1994;35:619–621. doi: 10.1002/ana.410350517. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Kojima J. The role of the pedunculopontine tegmental nucleus in experimental parkinsonism in primates. Stereotact Funct Neurosurg. 2001;77:108–115. doi: 10.1159/000064614. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Nambu A, Yamaji Y, Watanabe K, Imai H, Inase M, Tokuno H, Takada M. Organization of somatic motor inputs from the frontal lobe to the pedunculopontine tegmental nucleus in the macaque monkey. Neuroscience. 2000;98:97–110. doi: 10.1016/s0306-4522(00)00099-3. [DOI] [PubMed] [Google Scholar]
- Mazzone P, Lozano A, Stanzione P, Galati S, Scarnati E, Peppe A, Stefani A. Implantation of human pedunculopontine nucleus: a safe and clinically relevant target in Parkinson's disease. NeuroReport. 2005;16:1877–1881. doi: 10.1097/01.wnr.0000187629.38010.12. [DOI] [PubMed] [Google Scholar]
- McAllister AK. Cellular and molecular mechanisms of dendrite growth. Cereb Cortex. 2000;10:963–973. doi: 10.1093/cercor/10.10.963. [DOI] [PubMed] [Google Scholar]
- Mena-Segovia J, Ross HM, Magill PJ, Bolam JP. The pedunculopontine nucleus: towards a functional integration with the basal ganglia. In: Bolam JP, Ingham CA, Magill PJ, editors. The Basal Ganglia VIII. Springer Science+Business Media, Inc; New York: 2005. pp. 523–532. [Google Scholar]
- Mitchell IJ, Clarke CE, Boyce S, Robertson RG, Peggs D, Sambrook MA, Crossman AR. Neural mechanisms underlying parkinsonian symptoms based upon regional uptake of 2-deoxyglucose in monkeys exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience. 1989;32:213–226. doi: 10.1016/0306-4522(89)90120-6. [DOI] [PubMed] [Google Scholar]
- Moran A, Bar-Gad I, Bergman H, Israel Z. Real-time refinement of subthalamic nucleus targeting using Bayesian decision-making on the root mean square measure. Mov Disord. 2006;21:1425–1431. doi: 10.1002/mds.20995. [DOI] [PubMed] [Google Scholar]
- Munro-Davies LE, Winter J, Aziz TZ, Stein JF. The role of the pedunculopontine region in basal-ganglia mechanisms of akinesia. Exp Brain Res. 1999;129:511–517. doi: 10.1007/s002210050921. [DOI] [PubMed] [Google Scholar]
- Murer MG, Riquelme LA, Tseng KY, Pazo JH. Substantia nigra pars reticulata single unit activity in normal and 60HDA-lesioned rats: effects of intrastriatal apomorphine and subthalamic lesions. Synapse. 1997;27:278–293. doi: 10.1002/(SICI)1098-2396(199712)27:4<278::AID-SYN2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Murer MG, Tseng KY, Kasanetz F, Belluscio M, Riquelme LA. Brain oscillations, medium spiny neurons, and dopamine. Cell Mol Neurobiol. 2002;22:611–632. doi: 10.1023/a:1021840504342. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Fetz EE. Synchronization of neurons during local field potential oscillations in sensorimotor cortex of awake monkeys. J Neurophysiol. 1996a;76:3968–3982. doi: 10.1152/jn.1996.76.6.3968. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Fetz EE. Oscillatory activity in sensorimotor cortex of awake monkeys: synchronization of local field potentials and relation to behavior. J Neurophysiol. 1996b;76:3949–3967. doi: 10.1152/jn.1996.76.6.3949. [DOI] [PubMed] [Google Scholar]
- Musizza B, Stefanovska A, McClintock PV, Palus M, Petrovcic J, Ribaric S, Bajrovic FF. Interactions between cardiac, respiratory and EEG-delta oscillations in rats during anaesthesia. J Physiol. 2007;580:315–326. doi: 10.1113/jphysiol.2006.126748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthusamy KA, Aravamuthan BR, Kringelbach ML, Jenkinson N, Voets NL, Johansen-Berg H, Stein JF, Aziz TZ. Connectivity of the human pedunculopontine nucleus region and diffusion tensor imaging in surgical targeting. J Neurosurg. 2007;107:814–820. doi: 10.3171/JNS-07/10/0814. [DOI] [PubMed] [Google Scholar]
- Nandi D, Aziz TZ, Giladi N, Winter J, Stein JF. Reversal of akinesia in experimental parkinsonism by GABA antagonist microinjections in the pedunculopontine nucleus. Brain. 2002;125:2418–2430. doi: 10.1093/brain/awf259. [DOI] [PubMed] [Google Scholar]
- Ni ZG, Bouali-Benazzouz R, Gao DM, Benabid AL, Benazzouz A. Time-course of changes in firing rates and firing patterns of subthalamic nucleus neuronal activity after 6-OHDA-induced dopamine depletion in rats. Brain Res. 2001;899:142–147. doi: 10.1016/s0006-8993(01)02219-3. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Rodriguez M, Macias R, Alvarez L, Guridi J, Vitek J, Delong MR. Pathophysiologic basis of surgery for Parkinson's disease. Neurology. 2000;55:S7–12. [PubMed] [Google Scholar]
- Olsson M, Nikkhah G, Bentlage C, Bjorklund A. Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J Neurosci. 1995;15:3863–3875. doi: 10.1523/JNEUROSCI.15-05-03863.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahapill PA, Lozano AM. The pedunculopontine nucleus and Parkinson's disease. Brain. 2000;123:1767–1783. doi: 10.1093/brain/123.9.1767. [DOI] [PubMed] [Google Scholar]
- Palombo E, Porrino LJ, Bankiewicz KS, Crane AM, Kopin IJ, Sokoloff L. Administration of MPTP acutely increases glucose utilization in the substantia nigra of primates. Brain Res. 1988;453:227–234. doi: 10.1016/0006-8993(88)90162-x. [DOI] [PubMed] [Google Scholar]
- Papa SM, Boldry RC, Engber TM, Kask AM, Chase TN. Reversal of levodopa-induced motor fluctuations in experimental parkinsonism by NMDA receptor blockade. Brain Res. 1995;701:13–18. doi: 10.1016/0006-8993(95)00924-3. [DOI] [PubMed] [Google Scholar]
- Papa SM, Chase TN. Levodopa-induced dyskinesias improved by a glutamate antagonist in Parkinsonian monkeys. Ann Neurol. 1996;39:574–578. doi: 10.1002/ana.410390505. [DOI] [PubMed] [Google Scholar]
- Parr-Brownlie LC, Poloskey SL, Flanagan KK, Eisenhofer G, Bergstrom DA, Walters JR. Dopamine lesion-induced changes in subthalamic nucleus activity are not associated with alterations in firing rate or pattern in layer V neurons of the anterior cingulate cortex in anesthetized rats. Eur J Neurosci. 2007;26:1925–1939. doi: 10.1111/j.1460-9568.2007.05814.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1986. [Google Scholar]
- Perier C, Agid Y, Hirsch EC, Feger J. Ipsilateral and contralateral subthalamic activity after unilateral dopaminergic lesion. NeuroReport. 2000;11:3275–3278. doi: 10.1097/00001756-200009280-00045. [DOI] [PubMed] [Google Scholar]
- Plaha P, Gill SS. Bilateral deep brain stimulation of the pedunculopontine nucleus for Parkinson's disease. NeuroReport. 2005;16:1883–1887. doi: 10.1097/01.wnr.0000187637.20771.a0. [DOI] [PubMed] [Google Scholar]
- Rasch MJ, Gretton A, Murayama Y, Maass W, Logothetis NK. Inferring spike trains from local field potentials. J Neurophysiol. 2008;99:1461–1476. doi: 10.1152/jn.00919.2007. [DOI] [PubMed] [Google Scholar]
- Reese NB, Garcia-Rill E, Skinner RD. The pedunculopontine nucleus-auditory input, arousal and pathophysiology. Prog Neurobiol. 1995;47:105–133. doi: 10.1016/0301-0082(95)00023-o. [DOI] [PubMed] [Google Scholar]
- Rohlfs A, Nikkhah G, Rosenthal C, Rundfeldt C, Brandis A, Samii M, Loscher W. Hemispheric asymmetries in spontaneous firing characteristics of substantia nigra pars reticulata neurons following a unilateral 6-hydroxydopamine lesion of the rat nigrostriatal pathway. Brain Res. 1997;761:352–356. doi: 10.1016/s0006-8993(97)00475-7. [DOI] [PubMed] [Google Scholar]
- Rolland AS, Herrero MT, Garcia-Martinez V, Ruberg M, Hirsch EC, Francois C. Metabolic activity of cerebellar and basal ganglia-thalamic neurons is reduced in parkinsonism. Brain. 2007;130:265–275. doi: 10.1093/brain/awl337. [DOI] [PubMed] [Google Scholar]
- Rosenberg JR, Amjad AM, Breeze P, Brillinger DR, Halliday DM. The Fourier approach to the identification of functional coupling between neuronal spike trains. Prog Biophys Mol Biol. 1989;53:1–31. doi: 10.1016/0079-6107(89)90004-7. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- Rye DB, Saper CB, Lee HJ, Wainer BH. Pedunculopontine tegmental nucleus of the rat: cytoarchitecture, cytochemistry, and some extrapyramidal connections of the mesopontine tegmentum. J Comp Neurol. 1987;259:483–528. doi: 10.1002/cne.902590403. [DOI] [PubMed] [Google Scholar]
- Salinas E, Sejnowski TJ. Correlated neuronal activity and the flow of neural information. Nat Rev Neurosci. 2001;2:539–550. doi: 10.1038/35086012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson P, Mavoungou R, Albe-Fessard D. Changes in substantia nigra pars reticulata activity following lesions of the substantia nigra pars compacta. Neurosci Lett. 1986;67:25–30. doi: 10.1016/0304-3940(86)90202-8. [DOI] [PubMed] [Google Scholar]
- Sceniak MP, Maciver MB. Cellular actions of urethane on rat visual cortical neurons in vitro. J Neurophysiol. 2006;95:3865–3874. doi: 10.1152/jn.01196.2005. [DOI] [PubMed] [Google Scholar]
- Schwartzman RJ, Alexander GM, Ferraro TN, Grothusen JR, Stahl SM. Cerebral metabolism of parkinsonian primates 21 days after MPTP. Exp Neurol. 1988;102:307–313. doi: 10.1016/0014-4886(88)90224-5. [DOI] [PubMed] [Google Scholar]
- Sharott A, Magill PJ, Bolam JP, Brown P. Directional analysis of coherent oscillatory field potentials in the cerebral cortex and basal ganglia of the rat. J Physiol. 2005;562:951–963. doi: 10.1113/jphysiol.2004.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shink E, Bevan MD, Bolam JP, Smith Y. The subthalamic nucleus and the external pallidum: two tightly interconnected structures that control the output of the basal ganglia in the monkey. Neuroscience. 1996;73:335–357. doi: 10.1016/0306-4522(96)00022-x. [DOI] [PubMed] [Google Scholar]
- Shink E, Sidibe M, Smith Y. Efferent connections of the internal globus pallidus in the squirrel monkey: II. Topography and synaptic organization of pallidal efferents to the pedunculopontine nucleus. J Comp Neurol. 1997;382:348–363. [PubMed] [Google Scholar]
- Solis O, Limon DI, Flores-Hernandez J, Flores G. Alterations in dendritic morphology of the prefrontal cortical and striatum neurons in the unilateral 6-OHDA-rat model of Parkinson's disease. Synapse. 2007;61:450–458. doi: 10.1002/syn.20381. [DOI] [PubMed] [Google Scholar]
- Spann BM, Grofova I. Origin of ascending and spinal pathways from the nucleus tegmenti pedunculopontinus in the rat. J Comp Neurol. 1989;283:13–27. doi: 10.1002/cne.902830103. [DOI] [PubMed] [Google Scholar]
- Spann BM, Grofova I. Nigropedunculopontine projection in the rat: an anterograde tracing study with phaseolus vulgaris-leucoagglutinin (PHA-L) J Comp Neurol. 1991;311:375–388. doi: 10.1002/cne.903110308. [DOI] [PubMed] [Google Scholar]
- Spann BM, Grofova I. Cholinergic and non-cholinergic neurons in the rat pedunculopontine tegmental nucleus. Anat Embryol (Berl) 1992;186:215–227. doi: 10.1007/BF00174143. [DOI] [PubMed] [Google Scholar]
- Stefani A, Lozano AM, Peppe A, Stanzione P, Galati S, Tropepi D, Pierantozzi M, Brusa L, Scarnati E, Mazzone P. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson's disease. Brain. 2007;130:1596–1607. doi: 10.1093/brain/awl346. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F. A novel slow (< 1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CH, Boraud T, Bezard E, Bioulac B, Gross C, Benazzouz A. Electrophysiological and metabolic evidence that high-frequency stimulation of the subthalamic nucleus bridles neuronal activity in the subthalamic nucleus and the substantia nigra reticulata. FASEB J. 2003;17:1820–1830. doi: 10.1096/fj.03-0163com. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Pazo JH, Murer MG, Riquelme LA. Subthalamic nucleus lesions reduce low frequency oscillatory firing of substantia nigra pars reticulata neurons in a rat model of Parkinson's disease. Brain Res. 2001a;904:93–103. doi: 10.1016/s0006-8993(01)02489-1. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Riquelme LA, Murer MG. Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J Neurosci. 2001b;21:6430–6439. doi: 10.1523/JNEUROSCI.21-16-06430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Riquelme LA, Belforte JE, Pazo JH, Murer MG. Substantia nigra pars reticulata units in 6-hydroxydopamine-lesioned rats: responses to striatal D2 dopamine receptor stimulation and subthalamic lesions. Eur J Neurosci. 2000;12:247–256. doi: 10.1046/j.1460-9568.2000.00910.x. [DOI] [PubMed] [Google Scholar]
- Verhagen ML, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson's disease. Neurology. 1998;50:1323–1326. doi: 10.1212/wnl.50.5.1323. [DOI] [PubMed] [Google Scholar]
- Vila M, Perier C, Feger J, Yelnik J, Faucheux B, Ruberg M, Raisman-Vozari R, Agid Y, Hirsch EC. Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur J Neurosci. 2000;12:337–344. doi: 10.1046/j.1460-9568.2000.00901.x. [DOI] [PubMed] [Google Scholar]
- Walters JR, Hu D, Itoga CA, Parr-Brownlie LC, Bergstrom DA. Phase relationships support a role for coordinated activity in the indirect pathway in organizing slow oscillations in basal ganglia output after loss of dopamine. Neuroscience. 2007;144:762–776. doi: 10.1016/j.neuroscience.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters JR, Hu D, Itoga CA, Parr-Brownlie LC, Bergstrom DA. Do local field potentials reflect synchronized spiking activity of neuronal populations in the basal ganglia? Studies in a rodent model of Parkinson's disease. In: Bolam JP, Ingham CA, Magill PJ, editors. The Basal Ganglia VIII. Springer Science+Business Media, Inc; New York: 2005. pp. 523–532. [Google Scholar]
- Wichmann T, Delong MR. Deep brain stimulation for neurologic and neuropsychiatric disorders. Neuron. 2006;52:197–204. doi: 10.1016/j.neuron.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Wolff K, Winstock AR. Ketamine: from medicine to misuse. CNS Drugs. 2006;20:199–218. doi: 10.2165/00023210-200620030-00003. [DOI] [PubMed] [Google Scholar]
- Zaidel A, Moran A, Marjan G, Bergman H, Israel Z. Prior pallidotomy reduces and modifies neuronal activity in the subthalamic nucleus of Parkinson's disease patients. Eur J Neurosci. 2008;27:483–491. doi: 10.1111/j.1460-9568.2008.06019.x. [DOI] [PubMed] [Google Scholar]
- Zold CL, Ballion B, Riquelme LA, Gonon F, Murer MG. Nigrostriatal lesion induces D2-modulated phase-locked activity in the basal ganglia of rats. Eur J Neurosci. 2007;25:2131–2144. doi: 10.1111/j.1460-9568.2007.05475.x. [DOI] [PubMed] [Google Scholar]