

Abstract
Multiplex isobaric tags (e.g., tandem mass tags (TMT) and isobaric tags for relative and absolute quantification (iTRAQ)) are a valuable tool for high-throughput mass spectrometry based quantitative proteomics. We have developed our own multiplex isobaric tags, DiLeu, that feature quantitative performance on par with commercial offerings but can be readily synthesized in-house as a cost-effective alternative. In this work, we achieve a 3-fold increase in the multiplexing capacity of the DiLeu reagent without increasing structural complexity by exploiting mass defects that arise from selective incorporation of 13C, 15N, and 2H stable isotopes in the reporter group. The inclusion of eight new reporter isotopologues that differ in mass from the existing four reporters by intervals of 6 mDa yields a 12-plex isobaric set that preserves the synthetic simplicity and quantitative performance of the original implementation. We show that the new reporter variants can be baseline-resolved in high-resolution higher-energy C-trap dissociation (HCD) spectra, and we demonstrate accurate 12-plex quantitation of a DiLeu-labeled Saccharomyces cerevisiae lysate digest via high-resolution nano liquid chromatography–tandem mass spectrometry (nanoLC–MS2) analysis on an Orbitrap Elite mass spectrometer.
Quantitative mass spectrometry (MS)-based strategies featuring stable isotope labeling have become popular in recent years for comparative studies of different biological states. In these approaches, heavy isotopes are differentially incorporated into a set of samples metabolically or chemically to enable relative quantitation of the pooled samples upon MS analysis. Mass difference labeling techniques such as metabolic stable isotope labeling by amino acids in cell culture (SILAC),1,2 amino acid-coded tagging,3,4 mass differential tags for relative and absolute quantification (mTRAQ),5 and reductive dimethylation6−10 introduce mass shifts for heavy-labeled peptides to allow multiplex comparisons to be made in parallel at the MS1 level by comparing peak intensities of heavy- and light-labeled peptides. An inherent drawback to these methods is that increasing the number of quantitative channels also increases mass spectral complexity, resulting in reduced signal-to-noise ratios (S/N) and reduced instrument duty-cycle efficiency during data-dependent acquisition which negatively impact proteomic coverage and quantitation.11
Isobaric labeling techniques such as tandem mass tags (TMT)12,13 and isobaric tags for relative and absolute quantification (iTRAQ),14 on the other hand, achieve multiplex quantitation without increasing spectral complexity. As a result, isobaric labeling strategies offer greater multiplexing capacity and analytical throughput compared to mass difference labeling. In these methods, each label in the multiplexed set is identical in mass, differing only in the unique arrangement of isotopes distributed between the reporter and balance groups of the chemical structure. Thus, like peptides labeled differentially between biological samples possess the same mass and are detected as single precursors during the MS1 scan but yield distinct reporter ions in the low m/z region upon MS2 fragmentation. The intensities of these reporter ions in MS2 spectra reflect the labeled peptide’s abundance in each sample and can be compared to allow relative quantitation. Isobaric labeling reagents are essentially limited in their multiplexing capacity by the number of isotopic isoforms permitted by the reporter and balance group structures. The first generation 4-plex iTRAQ reagent was modified with a larger balancing group in the 8-plex iTRAQ reagent to support additional isotopes and afford additional quantitative channels.15 However, a study by Pichler et al. compared 4-plex iTRAQ, 6-plex TMT, and 8-plex iTRAQ using a Thermo Scientific LTQ Orbitrap with Proteome Discoverer software and revealed that the most peptides were identified when using the smaller 4-plex iTRAQ reagents, whereas the fewest were identified when using the large 8-plex iTRAQ reagents, indicating that bulkier labels compromise peptide identification.16 A later study by Pottiez et al. called these results into question through a comparison of 4-plex iTRAQ and 8-plex iTRAQ using an AB Sciex 4800 MALDI-TOF/TOF with ProteinPilot software. They determined that 8-plex iTRAQ provided more accurate quantitation over 4-plex iTRAQ without sacrificing protein identification rates.17 While the two studies used different instruments and data processing software, the conflicting observations indicate that the impact of label size on quantitative accuracy and identification rates is uncertain and requires further investigation.
Other efforts to increase the multiplexing capacity of quantitative strategies have combined isobaric labeling and mass difference labeling. One hyperplexing strategy joins triplex metabolic mass difference labeling with 6-plex TMT labeling to achieve 18-plex quantitation,18 and with the addition of medium and heavy sets of 6-plex TMT, 54-plex quantitation was demonstrated.19 Another approach, called combined precursor isotopic labeling and isobaric tagging (cPILOT), uses N-terminal-specific dimethylation at low pH followed by 6-plex TMT labeling of lysine residues at high pH to achieve 12-plex quantitation.20
Recently, the multiplexing capacity of TMT reagents was increased by exploiting subtle relative mass differences between 12C/13C and 14N/15N isotopes rather than by incorporating additional isotopes.21,22 The differences in mass between elements and their isotopes, called mass defects, arise from differences in nuclear binding energy and vary from element to element.23 By substituting an 15N in place of an 14N instead of a 13C in place of a 12C in the reporter group, the resulting reporter is lighter by 6.32 mDa. This small mass difference can be baseline-resolved at high resolution, using an Orbitrap mass analyzer, for example, requiring a minimum MSn resolving power of 30k (at 400 m/z). With the addition of four TMT isotopologue variants, TMT reagents are currently offered as a neutron encoded 10-plex set for use with high-resolution MSn platforms.24 Mass defects have also been used in MS1-level multiplexed quantitative proteomics approaches such as NeuCode SILAC,25−28 NeuCode amine-reactive labels,29 and mass defect-based pseudoisobaric dimethyl labeling (pIDL).30
While isobaric labeling approaches have come into favor, their routine use for MS-based quantitative proteomics has been stifled by their high cost. A primary contributor to the high cost is that the complex, multistep syntheses involved in producing commercial TMT and iTRAQ reagents lead to moderate to low yields. As the multiplexing capacity of commercial reagents increases, so does the cost of admission for these strategies. Currently, a TMTsixplex reagent set (Thermo Scientific) sufficient for a single experiment (100 μg of protein digest per channel) costs over $500, while a single-experiment TMT10plex reagent set costs over $900. A cost-efficient solution that overcomes this barrier to entry is beneficial to increasing the practicality and widespread application of isobaric labeling strategies.
Previously, we described the design, synthesis, and application of a novel 4-plex set of N,N-dimethyl leucine (DiLeu) isobaric labeling reagents featuring reporter ions at m/z 115, 116, 117, and 118.31 Our DiLeu reagents show comparable protein sequence coverage and quantitative accuracy to commercial isobaric tags with the benefit of significant cost savings over such offerings in that they are readily synthesized at high yield (∼80%) using commercially available isotopic reagents. Additional benefits of the DiLeu reagent include the modest mass of the tag, the greater intensity of generated reporter ions compared to iTRAQ and TMT, and enhanced collision-induced MSn fragmentation of labeled peptides at reduced collision energies, all of which can lead to increased confidence in quantitative accuracy and peptide sequence identification.
In this work, we expand upon the original DiLeu concept by describing the design, synthesis, and application of a 12-plex set of DiLeu isobaric labeling reagents made possible by subtle mass differences imparted by mass defects between 12C/13C, 14N/15N, and 1H/2H. In this way, a 3-fold increase in multiplexing capacity has been achieved while preserving the synthetic simplicity of the 4-plex set of reagents. The additional neutron encoded reporter isotopologues differ in mass by intervals of ∼6 mDa and can be resolved using high-resolution, accurate mass instrumentation. To demonstrate the strong performance of these reagents, we employ the Thermo Scientific Orbitrap Elite to accurately quantify mixtures of 12-plex DiLeu-labeled Saccharomyces cerevisiae tryptic peptides via high-resolution liquid chromatography–tandem mass spectrometry (LC–MS2).
Methods
Chemicals
All isotopic reagents used for the synthesis of labels were purchased from Isotec (Miamisburg, OH). Mass spec grade trypsin/Lys C mix and dithiothreitol (DTT) were purchased from Promega (Madison, WI). Urea, ammonium bicarbonate, ACS grade methanol (MeOH), ACS grade dichloromethane (DCM), ACS grade acetonitrile (ACN), Optima UPLC grade ACN, Optima UPLC grade water, and Optima LC/MS grade formic acid were purchased from Fisher Scientific (Pittsburgh, PA). Sodium cyanoborohydride (NaBH3CN), l-leucine, formaldehyde (CH2O), hydrogen chloride gas (HCl), iodoacetamide (IAA), tris hydrochloride, trifluoroacetic acid (TFA), triethylammonium bicarbonate (TEAB), N,N-dimethylformamide (DMF), 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate (DMTMM), N-methylmorpholine (NMM), heptafluorobutyric acid (HFBA), dimethyl sulfoxide (DMSO), and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). Hydroxylamine solution was purchased from Alfa Aesar (Ward Hill, MA).
Synthesis of DiLeu Reporter
Detailed syntheses can be found in the Supporting Information (Scheme S-1). l-Leucine or isotopic l-leucine (l-leucine-1-13C,15N, l-leucine-3-13C,15N, or l-leucine-1,2-13C) and sodium cyanoborohydride (NaBH3CN) or sodium cyanoborodeuteride (NaBD3CN) (2.5× molar excess to leucine) were suspended in H2O or D2O, and the mixture was cooled in an ice–water bath. Formaldehyde (CH2O, 37% w/w) or isotopic formaldehyde (CD2O or 13CH2O, 20% w/w) (2.5× molar excess to leucine) was added dropwise, and the mixture was stirred in an ice–water bath for 30 min. The target product was purified by flash column chromatography (MeOH/DCM) and dried in vacuo.
18O Exchange
Each isotopologue of reporter 115 and 116 requires 18O exchange prior to reductive dimethylation. l-Leucine or isotopic l-leucine was dissolved in 1 N HCl H218O solution (pH 1) and stirred on a hot plate at 65 °C for 4 h. Following evaporation of HCl from the solution in vacuo, trace amounts of acid were removed with StratoSpheres PL-HCO3 MP resin (Agilent Technologies) to obtain 18O l-leucine in free base form.
Activation of DiLeu
DiLeu reporter in anhydrous DMF was combined with DMTMM and NMM at 0.9× molar ratios to DiLeu reporter and vortexed at room temperature for 30 min. The mixture was centrifuged at 14 000g for 1 min, and the supernatant was used immediately for peptide labeling.
Yeast Lysate Enzymatic Digestion
S. cerevisiae lysate was provided by Promega (Madison, WI). Proteins were digested with trypsin/Lys C mix (Promega) according the manufacturers protocol and desalted using a SepPak C18 SPE cartridge (Waters, Milford, MA). Digested peptides were divided into 12 equal aliquots (in triplicate), dried in vacuo, and dissolved in 60:40 ACN/0.5 M TEAB pH 8.5 prior to labeling.
Protein Digest Labeling
12-Plex DiLeu labeling was performed in triplicate by addition of labeling solution at a 20:1 label to peptide digest ratio by weight and vortexing at room temperature for 2 h. The labeling reaction was quenched by addition of hydroxylamine to a concentration of 0.25%, and the labeled peptide samples were dried in vacuo. Labeled peptide samples were then dissolved in 30:70 ACN/H2O, combined in 1:1:1:1:1:1:1:1:1:1:1:1 or 16:8:4:2:1:10:10:1:2:4:8:16 ratios, and dried in vacuo. For the multiplexing comparison, labeled peptide samples were combined in 10:1 ratios as 4-plex, 8-plex, and 12-plex mixtures. The combined samples were then acidified with HFBA to a concentration of 0.5%, cleaned with Omix SCX pipet tips (Agilent Technologies, Santa Clara, CA) or SCX SpinTips (Protea Biosciences, Morgantown, WV), and desalted with Omix C18 pipet tips (Agilent Technologies, Santa Clara, CA).
LC–MS2
Samples were analyzed using a Waters nanoAcquity UPLC system (Milford, MA) coupled to a Thermo Scientific Orbitrap Elite mass spectrometer (San Jose, CA). Labeled tryptic peptide samples were dried in vacuo and dissolved in 3% ACN, 0.1% formic acid in water. Peptides were loaded onto a 75 μm inner diameter microcapillary column fabricated with an integrated emitter tip and packed with 15 cm of Bridged Ethylene Hybrid C18 particles (1.7 μm, 130 Å, Waters). Mobile phase A was composed of water, 5% DMSO, and 0.1% formic acid. Mobile phase B was composed of ACN, 5% DMSO, and 0.1% formic acid. Separation was performed using a gradient elution of 5% to 35% mobile phase B over 120 min at a flow rate of 300 nL/min. Survey scans of peptide precursors from 380 to 1600 m/z were performed at a resolving power of 120k (at 400 m/z) with an AGC target of 5 × 105 and maximum injection time of 150 ms. The top 10 precursors were then selected for higher-energy C-trap dissociation tandem mass spectrometry (HCD MS2) analysis with an isolation width of 2.0 Da, a normalized collision energy (NCE) of 27, a resolving power of 60k, an AGC target of 3 × 104, a maximum injection time of 250 ms, and a lower mass limit of 110 m/z. Precursors were subject to dynamic exclusion for 15 s with a 10 ppm tolerance.
Data Analysis
Mass spectra were processed using Proteome Discoverer (version 1.4.0.288, Thermo Scientific). Raw files were searched in Proteome Discover against UniProt S. cerevisiae complete database (September, 2013) using Sequest HT algorithm with trypsin selected as the enzyme and two missed cleavages allowed. Searches were performed with a precursor mass tolerance of 25 ppm and a fragment mass tolerance of 0.03 Da. Static modifications consisted of DiLeu labels on peptide N-termini (+145.12801 Da) and carbamidomethylation of cysteine residues (+57.02146 Da). Dynamic modifications consisted of DiLeu labels on lysine residues, oxidation of methionine residues (+15.99492 Da), deamidation of asparagine and glutamine residues (+0.98402 Da), and methylation of C termini and aspartic acid, glutamic acid, histidine, lysine, arginine, serine, and threonine residues (+14.01565 Da). Peptide spectral matches (PSMs) were validated based on q-values to 1% FDR (false discovery rate) using percolator. Quantitation was performed in Proteome Discoverer with a reporter ion integration tolerance of 20 ppm for the most confident centroid. Only the PSMs that contained all 12 reporter ions were considered, and protein quantitative ratios were determined using a minimum of one quantified peptide. Reporter ion ratio values for protein groups were exported to Excel workbook format. Isotopic interference correction factors (Supporting Information) were calculated using PTC Mathcad 14 (Needham, MA) and applied in Microsoft Excel (Redmond, CA).
Results and Discussion
The structure of the DiLeu isobaric labeling reagent follows that of other isobaric reagents in that it is composed of a reporter group, a balance group, and an amine-reactive group: an N,N-dimethylated leucine makes up the reporter and the balance groups, and a triazine ester amine-reactive moiety enables selective modification of peptide N-termini and lysine side chains (Figure 1A). The inspiration for using dimethylated leucine as a reporter group began with our previous observation that MS2 fragmentation of dimethylated peptides containing N-terminal leucine yielded the most intense immonium a1 ions compared to other N-terminal amino acids. N-terminal dimethylation also provides the added benefit of enhancing peptide fragmentation and aiding in de novo sequencing,32,33 and the compact size of dimethylated leucine results in a modest nominal mass addition to the peptide of 145 Da per label (Supporting Information Figure S-1). Consequently, the optimal collision energy required to produce both abundant reporter ions and a wealth of peptide backbone fragment ions during collision-induced MSn fragmentation of labeled peptides is lower than that which is required for unlabeled peptides.34 This is in contrast to TMT-labeled peptides, which require higher collision energy to yield adequate reporter ion and peptide backbone fragment signals compared to unlabeled peptides.35 While the DiLeu reagents incorporate deuteriums into the reporter group, DiLeu-labeled peptides do not suffer from significant shifts in LC retention time between the labels.31 By grouping the deuteriums around the polar dimethylated amine functional group, the probability of their interaction with reversed-phase stationary phase is low—as opposed to the strongly favored interaction of the hydrophobic leucine side chain with the stationary phase—and the deuterium effect is minimized, in agreement with previous research.36 The triazine ester was chosen as the amine-reactive group because it activates quickly (within 1 h), does not require purification prior to labeling, and labels amines with high efficiency.
Figure 1.
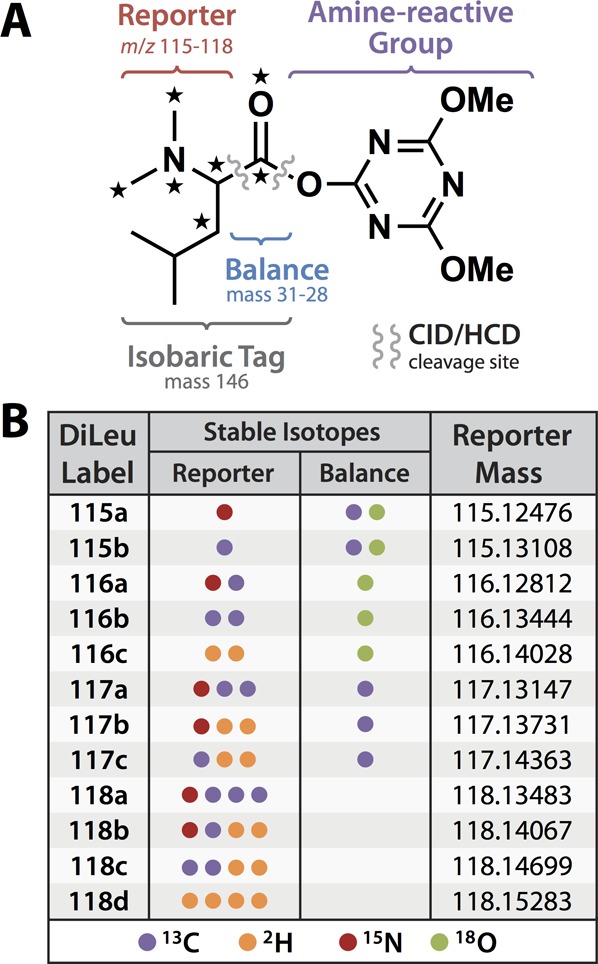
The 12-plex DiLeu general structure. (A) The DiLeu isobaric labeling reagent consists of a reporter group, balance group, and amine-reactive triazine ester group. Stars indicate positions of isotopic substitution. (B) Stable isotopes (13C, 2H, and 15N) incorporated into the reporter group are mass-balanced by stable isotopes (13C, 18O) in the carbonyl balance group. Unique combinations of isotopes incorporated into the reporter group yield two 115 variants, three 116 variants, three 117 variants, and four 118 variants whose isotopologues differ in mass by approximately 6 mDa.
DiLeu reagents are synthesized in high yield (∼80%) in only a few steps using established and relatively simple chemistry. Reductive formaldehyde dimethylation of leucine followed by activation with DMTMM yields half of the isobaric set, while the other half requires an initial 18O exchange of leucine prior to dimethylation. All isotopic reagents and chemicals are commercially available, making the synthesis of DiLeu accessible to any lab and scalable to the needs of any experiment. Dimethyl leucines can be stored for several years prior to activation, but labeling should be carried out soon after activation for optimal labeling efficiency. A typical complex tryptic digest sample can be labeled completely within 1–2 h, making same day activation and labeling convenient.
The first generation of DiLeu reagents was originally developed as a 4-plex set featuring reporter ions spaced one Da apart at m/z 115, 116, 117, and 118. Isotopic substitutions of 1H/2H and 14N/15N in the reporter group are offset by 12C/13C and 16O/18O in the carbonyl balance group to create isobaric structures. Because the carbonyl balance group offers only two isotopic substitution positions with four possible isotopic variants (12C=16O, 12C=18O, 13C=16O, and 13C=18O), the number of isobaric structures giving rise to reporters spaced by a single Da is limited to four.
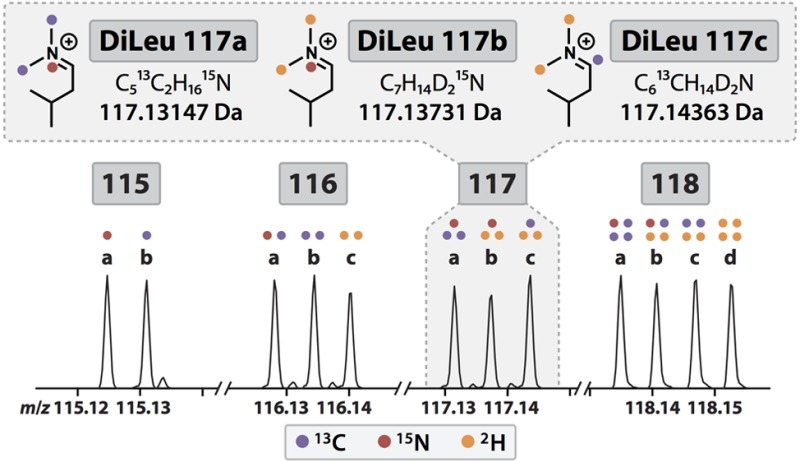
It was demonstrated recently with the TMT reagents that additional isotopologues could be created by incorporation of 15N instead of 13C in the reporter groups. In doing so, the two reporter ions differ by 6.32 mDa, which can be baseline-resolved using high-resolution MSn acquisition.21,22 Given the similarities between TMT and DiLeu reagents, it stood to reason that additional DiLeu reporters could be developed in a similar manner using alternative combinations of 12C/13C, 14N/15N, and 1H/2H. Through calculated substitution of these isotopes in the DiLeu reporter structure, we designed eight new reporter isotopologues with unique “pseudoisobaric” masses differing from the original four by intervals of 5.84 mDa or 6.32 mDa to bring the total number of reporters to 12 (Figure 2). The resulting 12-plex set of isobaric DiLeu reagents is composed of two 115 variants, three 116 variants, three 117 variants, and four 118 variants (Figure 1B). In designing the new reporters, no synthetic steps were added, and no custom isotopic reagents were needed.
Figure 2.
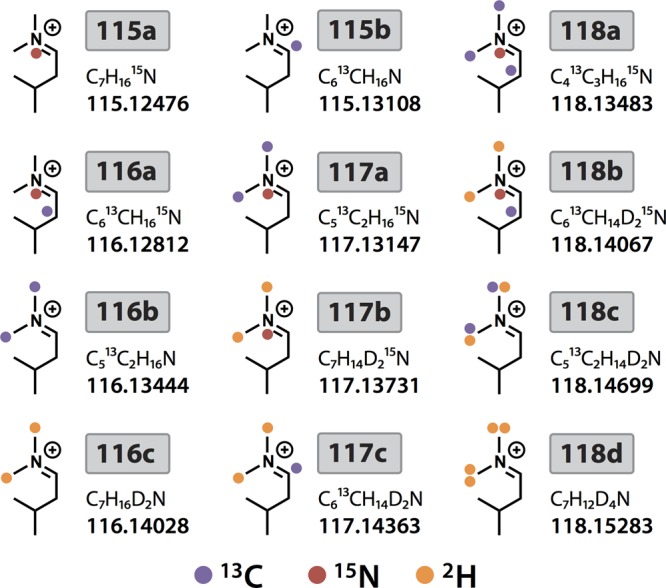
The 12-plex DiLeu reporter ion structures showing stable isotope positions.
In order to determine the resolving power at which the 12 reporter ions could be baseline-resolved, we combined each of the 12 reporters at equal concentrations and infused the mixture into an Orbitrap Elite mass spectrometer using HCD MS2 acquisition in the Orbitrap at resolving powers ranging from 15k to 240k (at m/z 400) (Figure 3). At a resolving power of 15k, the separation between neighboring reporter variants is insufficient for accurate intensity measurements for quantitation. At a resolving power of 30k, reporters are baseline-resolved into unique peaks that are suitable for accurate quantitation. At a resolving power of 60k and greater, the −1 isotopic peaks from channels 116c, 117b, 117c, 118b, and 118c are also baseline-resolved between the surrounding reporters. Because these resolved isotopic peaks no longer interfere with the surrounding primary reporter ion peaks at 60k resolving power, the most accurate quantitation can be achieved at this resolution following isotopic interference correction.
Figure 3.

The 12-plex DiLeu reporter ion peaks. The 12-plex reporters were mixed in a 1:1 ratio and infused directly into the Orbitrap Elite mass spectrometer, subjected to HCD MS2 fragmentation, and acquired at resolving powers 15–240k.
As is common with all stable isotope labeling reagents, the purities of the isotopes incorporated into the DiLeu reporter groups are not 100%. This is because the isotopic starting reagents used in the reporter syntheses contain stable isotopes in 98–99% purities. As a result, each primary DiLeu reporter ion peak is accompanied by low-intensity isotopic impurity peaks that are greater or lesser in mass by one neutron. For each type of stable isotope (13C, 15N, 2H) incorporated into the reporter group, a discrete −1 isotopic peak is observed. For example, the 118b reporter (m/z 118.14067), which contains 13C, 15N, and 2H, has three −1 isotope peaks at m/z 117.13494 (2H → 1H), 117.13786 (13C → 12C), and 117.14363 (15N → 14N). The fractional intensities of each channel’s primary reporter ion peak and isotopic peaks were determined at resolving powers of 30k and 60k via independent LC–MS2 analysis of BSA tryptic digest labeled separately with each of the 12-plex DiLeu reagents. The fractional intensities of each of the 12-plex DiLeu primary reporter ion peaks and isotopic peaks are shown as percentages of the combined intensity in Table S-1 (Supporting Information), and the interferences of isotopic peaks to neighboring primary reporter ion peaks are shown in Table S-2 (Supporting Information). At a resolving power of 30k, the exact interferences of the unresolved −1 isotopic peaks from channels 117b, 117c, 118b, and 118c to the surrounding reporter ion peaks are difficult to ascertain (Figure S-2, Supporting Information). Still, applying isotopic interference correction to data acquired at 30k resolving power is recommended with the understanding that quantitative accuracy will be lower for channels 116a–c and 117a–c than if the data had been acquired at 60k resolving power due to additional interference by unresolved isotopic peaks.
If an experiment does not require the full multiplexing capacity, a reduction in multiplexing allows acquisition at lower resolving power. Omitting the 117 channels eliminates the ambiguous isotopic interferences at 30k resolving power and allows 9-plex quantitation at a faster acquisition speed. Using only the 115 and 118 channels enables 6-plex quantitation at 30k resolving power and obviates the need for any isotopic interference correction. Selecting channels 115a, 116a, 116c, 117a, 117c, 118a, and 118c results in 12.16 mDa spacing between reporter isotopologues that can be baseline-resolved at 15k resolving power, permitting 7-plex quantitation at even shorter Orbitrap transient times or with a moderate resolving power quadrupole time-of-flight (QTOF) instrument.
Next, we aimed to demonstrate the quantitative precision, accuracy, and dynamic range of the 12-plex DiLeu reagents for bottom-up protein quantitation by labeling a complex mixture of S. cerevisiae lysate tryptic peptides and analyzing by high-resolution LC–MS2. Yeast lysate was digested with trypsin/Lys C, desalted, split into equal aliquots, and labeled in triplicate with each of the 12 DiLeu reagents. The 12-plex DiLeu-labeled yeast peptide samples were then prepared by combining at 1:1:1:1:1:1:1:1:1:1:1:1 and 16:8:4:2:1:10:10:1:2:4:8:16 ratios (115a–118d). Samples were acquired on the Orbitrap Elite using a data-dependent top 10 method with HCD MS2 acquisition at a resolving power of 60k. While 30k resolving power is sufficient for baseline separation of the reporters, 60k resolving power was chosen for this experiment because it further resolves several interfering isotopic peaks from the surrounding reporters and allows for more accurate isotopic interference correction. Data from the triplicate LC–MS2 runs were combined in Proteome Discover to calculate reporter ion ratios for 663 and 712 identified protein groups from the 1:1 and 16:1 samples, respectively. After isotopic interference corrections were applied in Excel (Figure S-1 in the Supporting Information), the 12-plex DiLeu ratios for all quantified proteins were plotted against each other (Figure 4). Across all channels, the median ratios measure within 10% of the expected values with average coefficients of variation (CVs) of 7.9% for the 1:1 ratio sample and 11.5% for the 16:1 ratio sample. Reproducibility and variance of the protein quantitative ratios between 16:1 replicates were compared and showed excellent correlation with each other (Figure 5). To also characterize the quantitative performance across the measured peptide dynamic range within a sample, reporter ion ratios of PSMs were plotted as a function of precursor ion signal intensity for a 12-plex DiLeu-labeled yeast lysate digest sample labeled in 10:1 ratios between neighboring channels (Figure S-3 in the Supporting Information). Variability of reporter ion ratios was fairly consistent across the 5 orders of magnitude of precursor intensity. These results show that the overall accuracy and precision remains excellent for highly multiplexed, complex proteomics experiments across a usable dynamic range. Furthermore, the increase in multiplexing also does not negatively impact peptide backbone fragmentation. An example HCD MS2 spectrum of a yeast lysate peptide yielding high coverage of b- and y-ions is shown (Figure 6).
Figure 4.
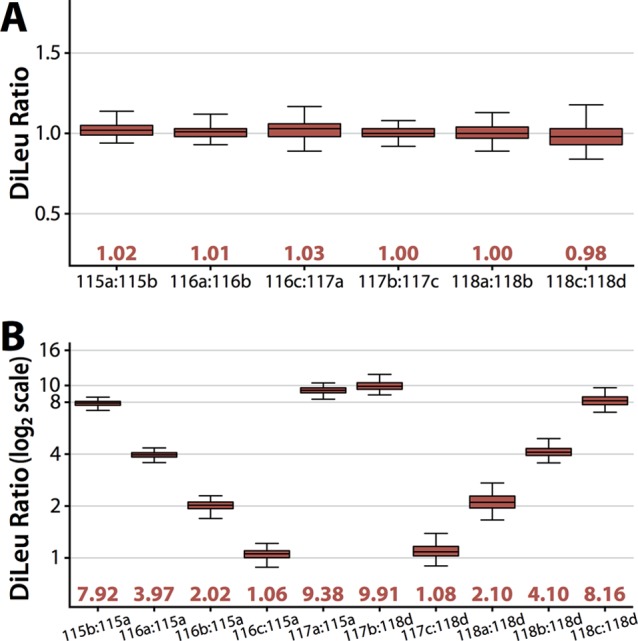
Quantitative performance. The 12-plex DiLeu-labeled yeast digest samples were combined in 1:1 ratios across all channels and in 16:8:4:2:1:10:10:1:2:4:8:16 ratios (115a–118d) and analyzed by LC–MS2 at 60k resolving power. Measured quantitative ratios of identified proteins (box and whiskers) are shown for (A) the 1:1 mixture in relation to neighboring channels and (B) the 16:1 mixture in relation to 16× channels (115a and 118d). Box plots demarcate the median (line), the 25th and 75th percentile (box), and the 5th and 95th percentile (whiskers).
Figure 5.
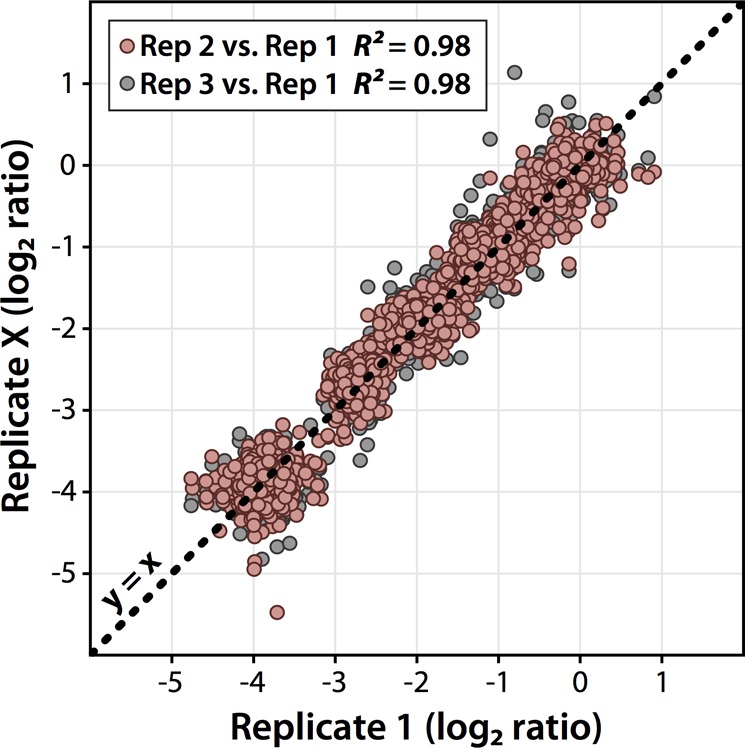
Replicate variance and reproducibility. Measured quantitative ratios of identified proteins shared between three technical replicates of the 16:1 sample were plotted against each other. Log2 ratios between replicates closely track the y = x function and show excellent correlation across the 16-fold dynamic range.
Figure 6.
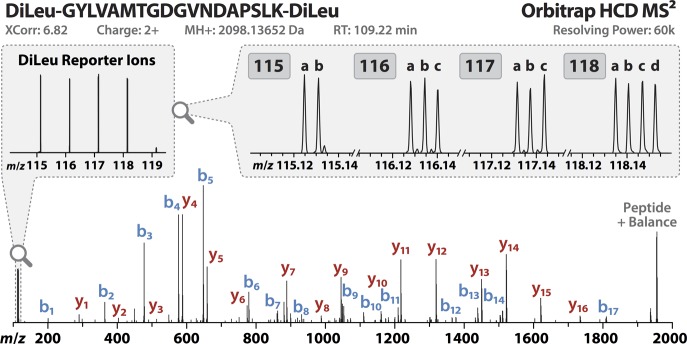
An MS2 spectrum of a 12-plex DiLeu-labeled yeast tryptic peptide acquired in the Orbitrap at 60k resolving power following HCD fragmentation (NCE 29). DiLeu reporter ion signals (1:1 ratio) are fully resolved in the low-mass range, and a wealth of b- and y-ions are observed for confident peptide sequence identification.
We also explored the effect of multiplexing on peptide and protein identification and quantitative precision. Three DiLeu-labeled yeast peptide samples were prepared: (1) a 4-plex mixture of 115b, 116a, 117c, and 118d combined at a 10:1:10:1 ratio; (2) an 8-plex mixture of 115a, 115b, 116b, 116c, 117a, 117b, 118c, and 118d combined at a 10:1:10:1:10:1:10:1 ratio; (3) a 12-plex mixture of 115a through 118d combined at a 10:1:10:1:10:1:10:1:10:1:10:1 ratio. The 4-plex sample contained channels separated by 1 Da while the 8-plex contains four pairs of channels separated by ∼6 mDa, providing a good indication of the impact of increasing the multiplexing with closely spaced reporter isotopologues. Peptide concentration and injection volume were equal across the three samples. Overall on-column sample load was greater than that used for the 1:1 and 16:1 experiments. Samples were acquired in triplicate on the Orbitrap Elite using a data-dependent top 10 method with HCD MS2 acquisition at a resolving power of 60k (at 400 m/z). We chose to keep the resolving power and acquisition speed constant for all three experiments in order to evaluate only the effect of increasing multiplexing with closely spaced reporters on identification rates and quantitative precision. As such, the 4-plex experiment is limited by the slower MS2 acquisition speed at 60k resolving power. In practice, a typical 4-plex experiment with 1 Da-spaced reporters would be acquired at the lowest MS2 resolution to achieve the fastest acquisition speed, yielding significantly more MS2 spectra and greater numbers of identified PSMs, peptides, and proteins than we observe in this comparison. Across triplicate runs, the 4-, 8-, and 12-plex experiments resulted in 1116, 1008, and 985 identified protein groups, respectively, and 5451, 4874, and 4437 identified peptides, respectively. This represents a 12% reduction in protein identification rate and a 19% reduction in peptide identification for the 12-plex experiment. The reporter ratios of quantified proteins from neighboring 10:1 channels were then plotted against each other (Figure 7). The 8-plex and 12-plex distributions are broader than the 4-plex distributions, and median values deviate by varying degrees from the expected value. Average CVs for the protein ratios of the 4-, 8-, and 12-plex were 9.9%, 16.2%, and 14.2%, respectively; average CVs for PSM reporter ion ratios were 18.7%, 31.6%, and 28.0%, respectively. While the reductions in protein and peptide identification rate and in quantitative accuracy and precision are not insignificant, we feel that these concessions are acceptable given the increase in analytical throughput.
Figure 7.
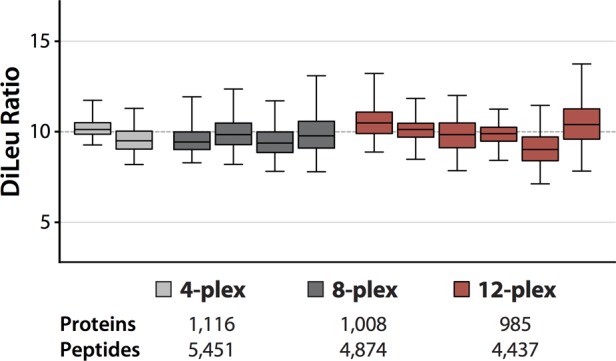
Multiplexing comparison. DiLeu-labeled yeast lysate digest samples were prepared as 4-, 8-, and 12-plex mixtures in 10:1 ratios between neighboring channels and analyzed by LC–MS2 at 60k resolving power. Measured quantitative ratios of identified proteins (box and whiskers) are shown. Box plots demarcate the median (line), the 25th and 75th percentile (box), and the 5th and 95th percentile (whiskers).
A similar decrease in quantitation precision resulting from an increase in the number of quantitative channels was observed for TMT by McAlister et al.22 They reasoned that, since bond energetics are constant regardless of the number of channels, the overall population of reporter ions produced during fragmentation is finite. Thus, increasing the number of channels reduces the population of reporter ions per channel, and the distribution of reporter ion ratios broadens. They determined that increasing the injection time in proportion to the decrease in ion population could compensate for this effect; when ion populations were equal between lower and higher multiplexed experiments, the distributions of reporter ratios were in close agreement. We anticipate that a similar tuning of injection times could have the same impact upon DiLeu quantitative precision when comparing differently multiplexed samples.
It has been shown that isobaric labeling strategies suffer from reporter ion ratio distortion for complex protein digest samples.37−39 The cause of this effect is coisolation of interfering near-isobaric ions along with the target precursor ion during MS2 fragmentation. One approach solves this problem by performing an MS3 isolation and fragmentation event on the highest intensity fragment or fragments from the MS2 scan and using MS3 reporter ion ratios for quantitation.40,41 While MS2 analysis was sufficient to benchmark the quantitative performance of 12-plex DiLeu using known samples, the MS3 quantitation strategy can be employed to overcome ratio distortion when quantifying unknown complex protein digest mixtures.
A recently discovered caveat to 10-plex TMT quantitation of complex samples is that the ∼6 mDa spaced reporter ions become prone to coalescence into a single peak at high abundance in Orbitrap mass analyzers, and this coalescence has an adverse effect on reporter ion quantitation.42 On the Q-Exactive, it was determined that decreasing the MS2 AGC target value from 1 × 106 to 2 × 105 eliminated the problem entirely without impairing protein identification or quantitation. Because the tendency to coalesce decreases as field strength increases,43 the Orbitrap Elite was found to be far less susceptible even at high AGC target values.42,44 At the MS2 AGC target setting of 3 × 104 used in our experiments, we did not observe coalescence of the 12-plex DiLeu reporter ions.
Another limitation of current multiplexed isobaric labeling strategies is the high cost of entry. Broad quantitative analyses of many biological states over several time points supported by an adequate number of biological or technical replicates can quickly become financially impractical. Likewise, experiments that require labeling large amounts of sample material necessitate expensive bulk orders of labeling reagent. In some cases, the laboratory may need to make sacrifices by adjusting research goals, reducing scale, or preparing fewer replicates in order to stay within budget. In developing the DiLeu reagents and extending them to the high-resolution enabled 12-plex set, cost-effectiveness has been of paramount concern. On a per-experiment basis, we calculate that for a labeling of 100 μg of protein digest per channel, a 12-plex DiLeu labeling costs under $23. An 8-plex DiLeu labeling costs under $12 by omitting the four most expensive labels, and a 4-plex DiLeu labeling costs under $5 by using the original 1 Da spaced labels. The reagents needed to synthesize the original 4-plex DiLeu set (115a, 116c, 117b, 118d) can be purchased at the present time for under $1500 and provide enough material to synthesize at least 200 mg of each channel, which is sufficient for 200 labeling experiments of 100 μg of protein digest. Reagents sufficient for synthesis of the 12-plex set, which adds two isotopic leucines and another isotopic version of formaldehyde, can be purchased for just over 3 times the cost of the 4-plex. A lab with basic knowledge of simple organic chemistry techniques can synthesize DiLeu reagents in house to significantly reduce the financial burden of large-scale quantitative experiments that would otherwise be unfeasible given the high cost of commercial isobaric labeling kits.
Conclusions
We have increased the multiplexing capacity of our original DiLeu isobaric labeling reagents from 4-plex to 12-plex through calculated incorporation of 12C/13C, 14N/15N, and 1H/2H stable isotopes in the reporter groups, without any other alterations to the original DiLeu structure. The additional reporter isotopologues were synthesized in house using commercially available, noncustom stable isotopic reagents. By retaining the original structure, several benefits remain. First, synthesis of each of the 12-plex DiLeu reporters is accomplished at high yield in only two or three steps using established and simple chemical reactions. Second, the dimethylated leucine label adds a modest amount of mass to labeled peptides and does not produce abnormal cleavages or interfering fragmentation artifacts in MS2 spectra that can negatively impact peptide sequence identification. Rather, the dimethylated leucine label enhances electrospray ionization by increasing hydrophobicity and promotes native fragmentation pathways by increasing proton affinity at N-termini and lysine side chains, which can improve peptide sequence identification.45 Third, the isotope-encoded dimethylated leucine reporters are as stable as their leucine counterparts, allowing storage for several years prior to activation. Finally, the high labeling efficiency of the DiLeu reagent allows complete peptide N-terminus and lysine side chain labeling of complex protein digest samples within 1–2 h.
The small mass difference of ∼6 mDa separating the 115, 116, 117, and 118 variants of the 12-plex DiLeu reporters can be baseline-resolved for accurate quantitation at an MSn resolving power of 30k (at 400 m/z), which is achievable on Orbitrap, FTICR, and some QTOF instruments. Acquiring at a resolving power of 60k baseline resolves isotopic peaks and allows more accurate isotopic interference correction at full multiplexing capacity. Reduced multiplexing configurations allow highly accurate 9-plex and 7-plex quantitation at resolving powers of 30k and 15k, respectively. We employed the Orbitrap Elite for high-resolution LC–MS2 analysis of 12-plex DiLeu-labeled yeast lysate digests combined at known concentrations and observed close agreement to the expected protein ratios at high precision with good reproducibility across a 16:1 dynamic range. Increasing the number of quantitative channels to 12 maintains quantitative performance while yielding only a modest decrease in protein and peptide identification rates.
The 12-plex DiLeu reagent set represents the highest multiplexing capacity currently available in an isobaric labeling experiment, enabling for the first time triplicate analysis of four samples in a single experiment without increasing mass spectral complexity. In the future, it is possible to further expand the multiplexing capacity of the DiLeu reagent with the inclusion of ∼3 mDa spaced reporter isotopologues to enable 21-plex quantitation at a resolving power of 60k (at m/z 400). The mainstream use of Orbitrap instrumentation has made high-resolution, accurate mass analysis an accessible new standard for most researchers. The Orbitrap Fusion and Q Exactive HF mass spectrometers, featuring ultrahigh-field Orbitrap mass analyzers that nearly double acquisition speeds compared to their predecessors, make comprehensive analysis and high-resolution enabled quantitation of complex samples even more practical. We conclude that the quantitative performance, affordability, and expansive multiplexing capability of the 12-plex DiLeu reagents establish them as a powerful tool for large-scale, high-throughput quantitative proteomics studies and make them an attractive alternative to current commercial options.
Acknowledgments
The authors thank Dr. Sergei Saveliev from Promega for providing the yeast lysate reference samples. This research was supported in part by NIH R01 DK071801, NIH EUREKA Grant (NIH R01NS071513), an Innovation and Economic Development Research Program research grant, and Wisconsin Alumni Research Foundation technology transfer grant. The Orbitrap Elite instrument was purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ong S.-E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Molina H.; Yang Y.; Ruch T.; Kim J.; Mortensen P.; Otto T.; Nalli A.; Tang Q.; Lane M. D.; Chaerkady R.; Pandey A. J. Proteome Res. 2009, 8, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.; Gu S.; Bradbury E. M.; Chen X. Anal. Chem. 2003, 75, 1316–1324. [DOI] [PubMed] [Google Scholar]
- Chen X.; Sun L.; Yu Y.; Xue Y.; Yang P. Expert Rev. Proteomics 2007, 4, 25–37. [DOI] [PubMed] [Google Scholar]
- DeSouza L. V.; Taylor A. M.; Li W.; Minkoff M. S.; Romaschin A. D.; Colgan T. J.; Siu K. W. M. J. Proteome Res. 2008, 7, 3525–3534. [DOI] [PubMed] [Google Scholar]
- Hsu J.; Huang S.; Chow N.; Chen S. Anal. Chem. 2003, 75, 6843–6852. [DOI] [PubMed] [Google Scholar]
- Ji C.; Guo N.; Li L. J. Proteome Res. 2005, 4, 2099–2108. [DOI] [PubMed] [Google Scholar]
- Boersema P. J.; Raijmakers R.; Lemeer S.; Mohammed S.; Heck A. J. R. Nat. Protoc. 2009, 4, 484–494. [DOI] [PubMed] [Google Scholar]
- Chen R.; Hui L.; Cape S. S.; Wang J.; Li L. ACS Chem. Neurosci. 2010, 1, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X.; Herbst A.; Ma D.; Aiken J.; Li L. J. Proteome Res. 2011, 10, 2687–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P.; Udeshi N. D.; Clauser K. R.; Mani D. R.; Patel J.; Ong S.; Jaffe J. D.; Carr S. A. Mol. Cell. Proteomics 2012, 11, M111–014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.; Schäfer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Neumann T.; Johnstone R.; Mohammed A. K. A.; Hamon C. Anal. Chem. 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Dayon L.; Hainard A.; Licker V.; Turck N.; Kuhn K.; Hochstrasser D. F.; Burkhard P. R.; Sanchez J. Anal. Chem. 2008, 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- Ross P. L.; Huang Y. N.; Marchese J. N.; Williamson B.; Parker K.; Hattan S.; Khainovski N.; Pillai S.; Dey S.; Daniels S.; Purkayastha S.; Juhasz P.; Martin S.; Bartlet-Jones M.; He F.; Jacobson A.; Pappin D. J. Mol. Cell. Proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- Choe L.; D’Ascenzo M.; Relkin N. R.; Pappin D.; Ross P.; Williamson B.; Guertin S.; Pribil P.; Lee K. H. Proteomics 2007, 7, 3651–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler P.; Köcher T.; Holzmann J.; Mazanek M.; Taus T.; Ammerer G.; Mechtler K. Anal. Chem. 2010, 82, 6549–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottiez G.; Wiederin J.; Fox H. S.; Ciborowski P. J. Proteome Res. 2012, 11, 3774–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N.; Gygi S. P. Sci. Signaling 2012, 5, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everley R. A.; Kunz R. C.; McAllister F. E.; Gygi S. P. Anal. Chem. 2013, 85, 5340–5346. [DOI] [PubMed] [Google Scholar]
- Evans A. R.; Robinson R. A. S. Proteomics 2013, 13, 3267–3272. [DOI] [PubMed] [Google Scholar]
- Werner T.; Becher I.; Sweetman G.; Doce C.; Savitski M. M.; Bantscheff M. Anal. Chem. 2012, 84, 7188–7194. [DOI] [PubMed] [Google Scholar]
- McAlister G. C.; Huttlin E. L.; Haas W.; Ting L.; Jedrychowski M. P.; Rogers J. C.; Kuhn K.; Pike I.; Grothe R. A.; Blethrow J. D.; Gygi S. P. Anal. Chem. 2012, 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleno L. J. Mass Spectrom. 2012, 47, 226–236. [DOI] [PubMed] [Google Scholar]
- Viner R.; Bomgarden R.; Blank M.; Rogers J.. Increasing the Multiplexing of Protein Quantitation from 6- to 10-Plex with Reporter Ion Isotopologues; Thermo Fisher Scientific: San Jose, CA, 2013; pp 1–7. [Google Scholar]
- Hebert A. S.; Merrill A. E.; Bailey D. J.; Still A. J.; Westphall M. S.; Strieter E. R.; Pagliarini D. J.; Coon J. J. Nat. Methods 2013, 10, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C. M.; Merrill A. E.; Bailey D. J.; Hebert A. S.; Westphall M. S.; Coon J. J. Anal. Chem. 2013, 85, 5129–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads T. W.; Rose C. M.; Bailey D. J.; Riley N. M.; Molden R. C.; Nestler A. J.; Merrill A. E.; Smith L. M.; Hebert A. S.; Westphall M. S.; Pagliarini D. J.; Garcia B. A.; Coon J. J. Anal. Chem. 2014, 86, 2314–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill A. E.; Hebert A. S.; MacGilvray M. E.; Rose C. M.; Bailey D. J.; Bradley J. C.; Wood W. W.; ElMasri M.; Westphall M. S.; Gasch A. P.; Coon J. J. Mol. Cell. Proteomics 2014, 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert A. S.; Merrill A. E.; Stefely J. A.; Bailey D. J.; Wenger C. D.; Westphall M. S.; Pagliarini D. J.; Coon J. J. Mol. Cell. Proteomics 2013, 12, 3360–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Shan Y.; Wu Q.; Zhang S.; Zhang L.; Zhang Y. Anal. Chem. 2013, 85, 10658–10663. [DOI] [PubMed] [Google Scholar]
- Xiang F.; Ye H.; Chen R.; Fu Q.; Li L. Anal. Chem. 2010, 82, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.; Huang S.-Y.; Shiea J.; Huang W.; Chen S. J. Proteome Res. 2005, 4, 101–108. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Li L. Anal. Chem. 2005, 77, 7783–7795. [DOI] [PubMed] [Google Scholar]
- Greer T.; Lietz C. B.; Xiang F.; Li L.. J. Am. Soc. Mass Spectrom. 2014, in press; DOI: 10.1007/s13361-014-1012-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiva C.; Sabidó E. J. Proteomics 2014, 96, 263–270. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Sioma C. S.; Thompson R. A.; Xiong L.; Regnier F. E. Anal. Chem. 2002, 74, 3662–3669. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Boesche M.; Eberhard D.; Matthieson T.; Sweetman G.; Kuster B. Mol. Cell. Proteomics 2008, 7, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Rehman I.; Wright P. C. J. Proteome Res. 2009, 8, 5347–5355. [DOI] [PubMed] [Google Scholar]
- Karp N. A.; Huber W.; Sadowski P. G.; Charles P. D.; Hester S. V.; Lilley K. S. Mol. Cell. Proteomics 2010, 9, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Nusinow D. P.; Jedrychowski M. P.; Wühr M.; Huttlin E. L.; Erickson B. K.; Rad R.; Haas W.; Gygi S. P. Anal. Chem. 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T.; Sweetman G.; Savitski M. F.; Mathieson T.; Bantscheff M.; Savitski M. M. Anal. Chem. 2014, 86, 3594–3601. [DOI] [PubMed] [Google Scholar]
- Scigelova M.; Hornshaw M.; Giannakopulos A.; Makarov A. Mol. Cell. Proteomics 2011, 10, M111–009431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A.; Smith G. T.; Sweredoski M. J.; Hess S. J. Proteome Res. 2013, 12, 3071–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L.; Xiang F.; Zhang Y.; Li L. Peptides 2012, 36, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.