

Abstract
Protein ubiquitin modifications present a vexing analytical challenge, because of the dynamic changes in the site of modification on the substrate, the number of ubiquitin moieties attached, and the diversity of linkage patterns in which they are attached. Presented here is a method to confidently assign size and linkage type of polyubiquitin modifications. The method combines intact mass measurement to determine the number of ubiquitin moieties in the chain with backbone fragmentation by 193-nm ultraviolet photodissociation (UVPD) to determine the linkage pattern. UVPD fragmentation of proteins leads to reproducible backbone cleavage at almost every inter-residue position, and in polyubiquitin chains, the N-terminally derived fragments from each constituent monomer are identical, up to the site of conjugation. The N-terminal ubiquitin fragment ions are superimposed to create a diagnostic pattern that allows easy recognition of the dominant chain linkages. The method is demonstrated by achieving almost-complete fragmentation of monoubiquitin and then, subsequently, fragmentation of dimeric, tetrameric, and longer Lys48- and Lys63-linked ubiquitin chains. The utility of the method for the analysis of mixed linkage chains is confirmed for mixtures of Lys48 and Lys63 tetramers with known relative concentrations and for an in vitro-formulated ubiquitin chain attached to a substrate protein.
Ubiquitin is a small, 76-residue protein conserved among eukaryotes,1 and its conjugation to proteins to form polyubiquitin chains of various lengths and geometries is implicated in almost every aspect of cell biology.2 Ubiquitin is covalently attached to substrates in a three-step ligation process in which the modifying ubiquitin moiety is activated via high-energy thioesters, and then joined to the substrate via an isopeptide bond between the C-terminal carboxylic acid of ubiquitin and, in most cases,3 the epsilon-amino group of an accessible lysine residue in the substrate. This process is catalyzed by a cascade of enzymes, the E1, E2, E3, and sometimes E4 ligases. In addition to the substrate protein, ubiquitin becomes ligated to itself through different primary amino groups in ubiquitin, and this polyubiquitination process codes the fate of the substrate proteins (e.g., degradation, membrane trafficking, and various signaling processes). There are seven lysine residues in ubiquitin in addition to its N-terminal primary amine for a total of eight possible sites that can be ubiquitinated,4 and Xu and co-workers observed that all possible sites of conjugation are employed in vivo in yeast.5
The ability to unravel the functional roles of different polyubiquitination patterns on proteins in vivo is impeded by the large size of the modification and the lack of methods that can deduce linkage composition of ubiquitin chains with or without an attached substrate. SDS PAGE and Western blotting with linkage specific antibodies is employed for in vitro studies, but the possibility of mixed linkage chains confounds immunoblotting experiments, and the mass resolution available from SDS PAGE is not high enough for complex samples that could contain multiple substrates.
Mass spectrometry (MS) is also used to determine lengths and linkage patterns.6−11 The amino acid sequence of ubiquitin ends in Arg-Gly-Gly, and this motif is recognized by trypsin. Hence, tryptic digestion of a ubiquitin-modified protein results in peptides that maintain a characteristic diglycine motif at the site of modification. The small size and specificity of the diglycine tag (∼114 Da)6 allows identification of the modified lysine residue using tandem mass spectrometry (MS/MS), thus providing a means to identify the presence and location of ubiquitin attachment to proteins. A similar approach using GluC digestion results in longer tags and is applied less frequently.12 Past studies have shed new light on the specific lysines involved in polyubiquitination;13,14 nevertheless two key aspects of the process have proven more difficult to decipher. First, the lengths of the polyubiquitin chains on a given substrate remain undefined; second, the linkage patterns of the polyubiquitin chains are usually uncharacterized.
Presented here is a method that exploits the unprecedented level of protein characterization attainable using top-down 193-nm ultraviolet photodissociation (UVPD) mass spectrometry15,16 to confidently identify length and linkage types within polyubiquitin chains. This method capitalizes on the fact that polyubiquitin chains have multiple N-termini, which, upon fragmentation in a mass spectrometer, create an excess of N-terminal ubiquitin fragment ion current. Different ubiquitin linkages produce characteristic ion current patterns that can be used to estimate linkage type and stoichiometry.
Materials and Methods
Ubiquitin Purification
Ubiquitin was purified as previously described.17 Briefly, ubiquitin was expressed from a pET-3a plasmid in E. coli Rosetta(DE3)pLysS (Rosetta) cells. Cultures were grown in 2× YT (microbial growth medium) under ampicillin (amp) and chloramphenicol (cl) selection at 37 °C to an OD600 of 0.6 and induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 4 h. Cells were collected by centrifugation at 6000 g for 10 min, resuspended in 50 mM Tris-HCl pH 7.6, and frozen at 80 °C. Cells were lysed by the addition of 0.02% NP-40 (cell lysis buffer) and 0.4 mg/mL lysozyme to the frozen cell pellet in the presence of Protease Inhibitor Cocktail Set V, EDTA-Free (Calbiochem No. 539137). Cells were thawed in a room-temperature water bath. To digest the DNA, 10 mM MgCl2 and 20 μg mL–1 DNase I were added to the lysate and rocked at room temperature for 10–20 min. Lysate was collected by centrifugation at 8000 g for 20 min at 4 °C. To precipitate the majority of proteins other than ubiquitin, 70% perchloric acid was added slowly to a vigorously stirring supernatant on ice to a final concentration of 0.5% (v/v), and stirring was continued for an additional 10 min. The treated lysate was centrifuged at 8000g for 20 min at 4 °C and the resulting supernatant dialyzed against 50 mM ammonium acetate pH 4.5 in 3.5 kDa molecular weight cutoff dialysis tubing (Thermo Scientific No. 68035). The dialyzed material was filtered through a 0.45-μm filter, loaded onto a 6-mL Resource S (GE No. 17-1180-01) column, washed with 2 column volumes, and eluted with a-20 column volume gradient of 0–500 mM NaCl in 50 mM ammonium acetate pH 4.5, taking 2 mL fractions. The protein was then concentrated and buffer exchanged in a 3 kDa molecular weight cutoff Amicon Ultra (Millipore No. UFC900324) into 20 mM Tris-HCl pH 7.6 to a final concentration of 50–100 mg mL–1.
Ubiquitin Chain Synthesis and Purification
Lys48 ubiquitin chains were synthesized by treating 20 mg mL–1 ubiquitin with 0.2 volume PBDM8 buffer (250 mM Tris-HCl pH8, 25 mM MgCl2, 50 mM creatine phosphate, 3 U/mL inorganic pyrophosphatase, 3 U/mL creatine phosphokinase), 2.5 mM ATP, 0.5 mM DTT, 20 μM E2–25K, and 0.1 μM His6-Ube1 at 37 °C overnight. The reaction was quenched with 5 mM DTT and 1 mM EDTA for 20 min at room temperature. To remove the enzymes, the reaction was run over a 0.5 mL Q Sepharose FF column (GE No. 17-0510-10) equilibrated with 50 mM Tris-HCl pH 7.6, 1 mM EDTA, and 5 mM DTT, and washed with four column volumes of the same buffer. The flow-through and wash were collected and acidified with 0.1 volume 2 N acetic acid to a pH of ∼4, loaded onto a 6-mL Resource S (GE No. 17-1180-01) column equilibrated with 50 mM ammonium acetate (pH 4.5), washed with 2 column volumes of 50 mM ammonium acetate, and eluted with a gradient of 1 column volume of 0–250 mM NaCl, 28 column volumes of 250–700 mM NaCl, and 1 column volume of 700–1000 mM NaCl in 50 mM ammonium acetate (pH 4.5), taking 2-mL fractions. The fractions containing Ub4(Lys48) were determined by SDS-Page analysis and concentrated in a 3 kDa molecular weight cutoff Amicon Ultra (Millipore No. UFC900324), and Ub4(Lys48) was separated on a HiLoad 16/600 Superdex 75 pg (GE No. 28-9893-33) size exclusion column equilibrated with 20 mM Tris-HCl pH 7.6, 1 mM EDTA, 2 mM DTT, 150 mM NaCl, and the fractions containing Ub4(Lys48) were pooled and concentrated.
Lys63 ubiquitin chains were created using the same protocol as for Lys48 ubiquitin chains, except that 2 μM Ubc13 and 2 μM Mms2 were used instead of E2-25K, and PBDM7.6 (pH 7.6) was used instead of PBDM8 (pH 8.0).
Enzyme Purification
His6-Ube1 (Mus musculus) was purified as previously described.18 E2-25K (Homo sapiens), Ubc13 (Saccharomyces cerevisiae), Mms2 (Saccharomyces cerevisiae), Rsp5 (Saccharomyces cerevisiae), and UbcH7 (Homo sapiens) were purified as GST-fusion proteins and the GST tag was removed by PreScission Protease. GST-E2-25K was cloned from Addgene plasmid No. 18892,17 GST-Ubc13 was cloned from Addgene plasmid No. 18894,19 and GST-Mms2 was cloned from Addgene plasmid No. 18893,19 each into a pGEX-6p-1. (The GST-Rsp5 plasmid was a generous gift from Linda Hicke.)
GST-E2-25K and GST-UbcH7 were expressed in Rosetta cells in 2× YT grown at 37 °C to an OD600 of 0.6 and induced with 0.1 mM IPTG for 4 h. GST-Ubc13 and GST-Mms2 were grown and purified as previously described.20 Briefly, GST-Ubc13 and GST-Mms2 were expressed in Rosetta cells in LB grown at 37 °C to an OD600 of 0.6 and induced with 0.4 mM IPTG overnight at 25 °C. GST-Rsp5 was expressed in Rosetta cells in 2× YT + amp + cm grown at 37 °C to an OD600 of 0.2, the cells were then grown at 18 °C to an OD600 of 0.4 and induced with 1 mM IPTG overnight at 18 °C. All cells were collected by centrifugation at 6000 g for 10 min, resuspended in PBS, and frozen at −80 °C.
For GST purification, Protease Inhibitor Cocktail Set V, EDTA-Free (Calbiochem No. 539137), 1% TritonX-100, 0.4 mg mL–1 lysozyme, 10 mM MgCl2, and 20 μg mL–1 DNase I were added to the frozen cell pellets and thawed in a room-temperature water bath. DNA was digested by rocking at room temperature for 10–20 min. Cells were lysed by an EmulsiFlex-C3 (Avestin). To aid in GST fusion protein solubility, the lysate was neutated for 30 min at 4 °C. The lysate was centrifuged twice at 14 000 g for 20 min, filtered through a 0.45-μm filter, and incubated with 2 mL of Glutathione Sepharose 4B beads (GE No. 17-0756-01). The mixture was poured into an empty PD-10 column (GE No. 17-0435-01), washed 3 times with 10 column volumes of PBS, and once with 10 column volumes of PreScission cleavage buffer (50 mM Tris-HCl pH 7.0, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT). To elute the protein, PreScission protease in a PreScission cleavage buffer was flowed into the column and incubated with the beads overnight at 4 °C. The cleaved protein was removed with 3 column volumes of PreScission cleavage buffer and concentrated (if necessary). Glycerol was added (5%–10% v/v), and the eluted protein was aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C.
Ubiquitination of Sic60-DHFR-His6 Substrate
Sic60-DHFR-His6 was expressed in Rosetta cells in 2× YT + amp + cm at 37 °C to an OD600 of 0.6 and induced with 0.4 mM IPTG for 4 h. Cells were collected by centrifugation at 6000g for 10 min, resuspended in NPI-10 (50 mM sodium phosphate, 300 mM NaCl, and 10 mM imidazole pH 8.0), and frozen at −80 °C. For purification, Protease Inhibitor Cocktail Set V, EDTA-Free (Calbiochem No. 539137), 1 mM DTT, 10 mM MgCl2, and 20 μg mL–1 DNase I were added to the frozen cells, and the pellets thawed in a room temperature water bath. To digest the DNA, cells were rocked at room temperature for 10–20 min. The cells were lysed by an EmulsiFlex-C3 (Avestin), centrifuged twice at 14 000 g for 20 min, and filtered through a 0.45-μm filter. The clarified lysate was applied to a 5-mL HisTrap FF Crude column (GE No. 17-5286-01), washed with 2 column volumes of NPI-10, washed with 10 column volumes of 4.2% NPI-250 (NPI-10 with 250 mM imidazole) in NPI-10, and then eluted with 10 column volumes of NPI-250 into 2-mL fractions. The fractions containing the protein were combined and concentrated to 1 mL in an Amicon Ultra with a molecular weight cutoff of 10 kDa. The protein was separated on a HiLoad 16/600 Superdex 75 pg (GE No. 28-9893-33) size exclusion column at 0.5 mL min–1. The fractions containing the Sic60-DHFR-His6 were combined, concentrated, and desalted into 20 mM Tris-HCl pH 7.6 in 1 mM DTT and 1 mM EDTA.
The ubiquitination procedure was modified from the method of Kraut et al.21 The protein was ubiquitinated for 1 h at 25 °C in 25 mM Tris-HCl pH 7.5, 50 mM NaCl, 4 mM MgCl2, 0.25 μM His6-Ube1, 2.5 μM UbcH7, 3.25 μM Ubiquitin, 4 mM ATP, 1 mM DTT, and 1.5 μM Rsp5. The reaction was diluted into 5 mL of 20 mM Tris-HCl pH 7.6, 5 mM DTT, and 1 mM EDTA and loaded onto a MonoQ column (GE No. 17-5166-01) equilibrated with the same buffer. The protein was then eluted with a linear gradient of 0–500 mM NaCl in the same buffer over 30 column volumes, taking 1-mL fractions. The fractions containing the Ub3(Lys63)-Sic60-DHFR-His6 were pooled, concentrated to 500 μL in an Amicon Ultra with a molecular weight cutoff of 10 kDa, and separated on a HiLoad 16/600 Superdex 75 pg (GE No. 28-9893-33) size exclusion column equilibrated with 20 mM Tris-HCl pH 7.6, 300 mM NaCl, 1.5 mM DTT, and 1 mM EDTA at 0.3 mL min–1 into 2-mL fractions.
Top-Down Ultraviolet Photodissociation (UVPD) Mass Spectrometry
Protein concentrations were based on the theoretical molar absorptivity of yeast ubiquitin. All ubiquitin chains and the ubiquitinated Sic60-DHFR fusion were buffer-exchanged three times into LC-MS grade water and diluted to a final concentration of 20 μM in a 50/49/1 (v/v/v) mixture of acetonitrile/water/formic acid. Dimeric recombinant human Lys48 and Lys63 chains were purchased from Boston Biochem (Boston, MA) and resuspended in the infusion solution directly, without further purification. All protein solutions were infused at a rate of 3 μL min–1 directly into an Orbitrap Elite mass spectrometer (Thermo Fisher, San Jose, CA) that was modified to accommodate an ArF 193-nm excimer laser (Coherent Existar XS), as previously described.22 All spectra were acquired at a resolving power of 240 000× and were an average of 500 total scans. Spectra were deconvoluted using the Xtract algorithm (Thermo Fisher) with a signal-to-noise ratio of 5:1. Spectra were interpreted manually and, using a version of ProSightPC 3.0, modified to accommodate the ion types associated with UVPD.15 Ions were matched against all predicted a-, b-, and c-type ions. Detection of solely N-terminally derived ions was based on the assumption that each constituent monomer in the chain is equally likely to fragment upon irradiation. Ion abundances of N-terminal ions for the stoichiometric comparison of Lys48- and Lys63-linked chains in known ratios were derived from the ions matched by ProSightPC 3.0.
Results and Discussion
This analysis is facilitated by the conjugation of each ubiquitin’s C-terminus to the substrate or to another ubiquitin molecule. In the case of a polyubiquitin chain, this means that fragmentation along any ubiquitin monomer in the chain results in conventional N-terminal ubiquitin fragments, as well as complementary C-terminal product ions that are appended to the remainder of the protein. For larger polyubiquinated proteins, the greater sizes of the C-terminal ions result in broader isotopic profiles, lower average ion current, and significant m/z overlap, diminishing their detection and simplifying interpretation of the resulting MS/MS spectra. In all cases, measurement of the length of the ubiquitin chains stems from the MS1 spectrum (i.e., the mass measurement of the intact ubiquitinated protein). The intact mass unambiguously defines the length of the polyubiquitin moiety, and this is regardless of linkage type. Indeed, all possible linkage combinations of the same length will have the same intact mass. While the different linkage types will undoubtedly have different three-dimensional (3D) structures, this has no effect on intact mass, since all polyubiquitin chains of equal length have identical empirical formulas. For example, Lys63 ubiquitin chains have a more-linear configuration, whereas Lys48 chains have a more “closed” configuration, so these two different chain types span a range of potential topologies. However, the proteins are analyzed under denaturing ESI conditions, not native ESI conditions, and topologies of proteins under denaturing conditions have not been found to significantly influence the resulting MS/MS fragmentation patterns.
Upon UVPD, the resulting MS/MS spectra are thus composed of predominantly N-terminal ions that are representative of the linkages. The N-termini of ubiquitin moieties in different polyubiquitin chains are identical up to the first Lys residue whose epsilon amino group is modified by forming the pseudo-peptide (isopeptide) bond with the C-terminus of the next ubiquitin moiety in the chain. Cleavage of the polypeptide backbone in these N-terminal regions during fragmentation creates identical ions from each ubiquitin in the chain that collectively increase the ion current for the fragments shared by each monomer. Once the modified Lys is reached, the mass of the fragments increases (by at least the mass of an entire ubiquitin moiety), removing its contribution to the N-terminal fragments shared by each monomer. In addition, different branching patterns in the polyubiquitin chain result in distinct characteristic ion current patterns that can, in principle, be assigned unambiguously (see Figure 1).
Figure 1.
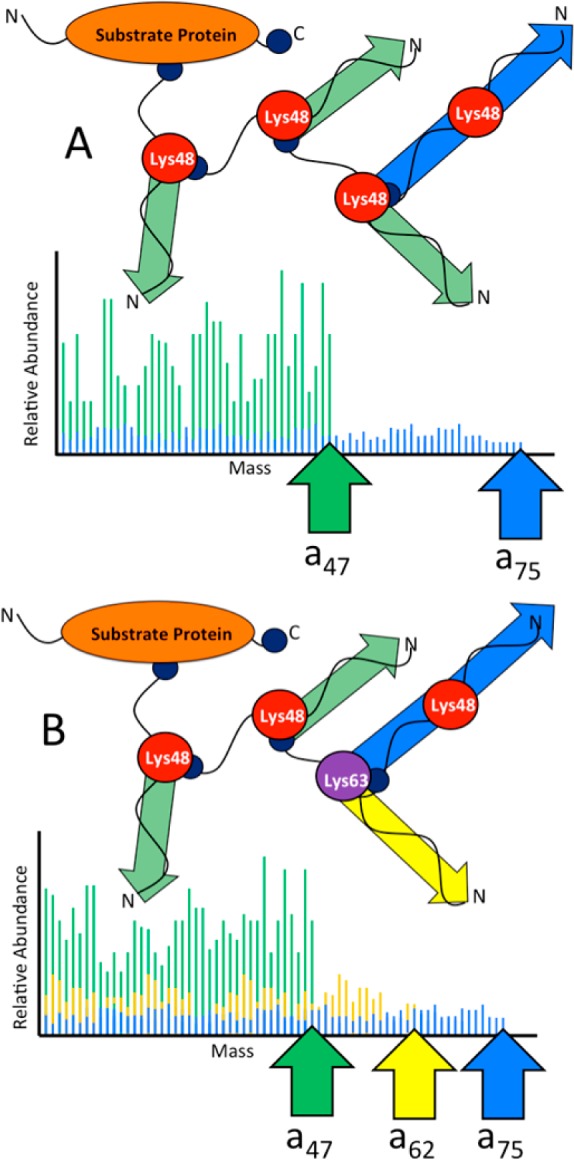
Schematic representations of a (A) homogeneously Lys48-linked and (B) a mixed linkage tetraubiquitin chain composed of three Lys48 linkages and one Lys63 linkage. N-termini are labeled, and C-termini are depicted with dark blue circles. Shown on the lower portion of panels (A) and (B) are the corresponding theoretical UVPD mass spectra depicting the N-terminally derived ions from the ubiquitin chains. The green-shaded peaks in the spectra highlight regions of the protein that result in isobaric N-terminal fragment ions from each monomeric Ub unit. In panel (B), the N-terminal ions that are specific to the Lys63 linkage portion are shown in yellow, and the ions derived from the most distal ubiquitin in the chain, which extend all the way to the C-terminus (the a75 ion), are shown in blue.
In practice, this strategy is dependent on the creation of ions representing every (or almost every) inter-residue position from the N-terminus of ubiquitin to the position preceding the Lys residue at the branch point to ensure that linkage types can be confidently differentiated from each other. In contrast to other methods, UVPD results in a reproducible and almost-complete series of N-terminal ions for most intact proteins.23,24 Indeed, previous work from this laboratory15 demonstrated that UVPD of monoubiquitin resulted in cleavage at every inter-residue position (see Figure S1 in the Supporting Information), providing 100% product ion sequence coverage. This level of characterization is crucial for defining the linkage type in ubiquitin chains. Fragmentation between every residue allows one to resolve the site of conjugation to a single-candidate Lys residue. Of the seven available Lys residues on ubiquitin, the results from the present study indicate (via observation of an almost-complete sequence of a-type ions) that top-down UVPD is capable of characterizing the ubiquitination of five of them. The characterization of two other linkage sites (Lys6 and Lys11) is not expected to be feasible due to the m/z overlap of the diagnostic ions arising from these linkages with the multiply charged artifacts left over following deconvolution. This study serves as a proof of concept and focus on the two most common linkage types: Lys48 and Lys63.
To test whether the hypothetical spectra depicted in Figure 1 can, in fact, be produced by UVPD of polyubiquitin chains, Lys63 and Lys48 dimers, tetramers, and longer multimers were analyzed using UVPD. Examples of the resulting UVPD mass spectra are shown in Figure 2, and charge-deconvoluted, monoisotopic UVPD mass spectra are shown in Figure 3 for Lys48- and Lys63-linked tetraubiquitin to illustrate the striking array of a-type ions. As observed in Figure 3, the contiguous series of a-type ions is easily assigned, with the series terminating at a47 for the Lys48-linked tetraubiquitin (Figure 3A), relative to the longer series of a-type ions for the Lys63-linked tetramer (Figure 3B). Complementary C-terminal x-type ions are not identified, which represents an outcome attributed to their much larger sizes and higher charge states than the a-type ions. These features (large sizes and high charge states of the C-terminal ions) result in each fragment ion being distributed among a greater array of isotopes (i.e., low S/N) and with substantial m/z overlap (i.e., difficult to resolve), ultimately obscuring their assignment. Potential secondary dissociation of these large ions (and formation of unassignable internal ions) may also contribute to their absence.
Figure 2.

Charge-deconvoluted UVPD product ion spectra of (A) Lys48-linked diubiquitin (10+) and (B) Lys63-linked diubiquitin (10+). The schematic depictions to the right show diubiquitin linked through either Lys48 or Lys63, and the green arrows indicate the regions that would yield N-terminal ion produced by cleavage between Lys 48 and Lys63. The portion of the UVPD mass spectrum shaded in green represents ions formed by backbone cleavage between the N-terminus and Lys48, and the portion of the UVPD mass spectrum shaded in gold represents ions produced by cleavage occurring between Lys48 and Lys63. The significant decrease in abundance of N-terminal fragments ions beyond a47 in panel (A), relative to panel (B), indicates that the larger an fragment ions evolve from only one of the two ubiquitin chains in the diubiquitin in panel (A). Also shown are UVPD spectra of Lys48 linked to Ubi2 (10+) (panel (C)), Ubi4 (20+) (panel (D)), Ubi5 (30+) (panel (E)), and a mixture of Ubi7 and Ubi8 (multiple charge states) (panel (F)). The regions shaded in green represent the array of all fragment ions up to the a47 ion. For each spectrum, the purified ubiquitin chains were infused and activated via a single 5-ns laser pulse.
Figure 3.

Representative charge deconvoluted, monoisotopic UVPD mass spectra of (A) Lys48- and (B) Lys63-linked tetraubiquitin (24+), obtained using 1 laser pulse (at 1 mJ per pulse). Ladder-type sequencing up to the isopeptide bond provides evidence for linkage type determination.
The Lys63 and Lys48 dimers resulted in N-terminal ion series clearly indicative of their respective linkage types (Figures 2A and 2B). The green shaded regions in Figures 2A and 2B represent all fragment ions from the N-terminus through the a47 ion (the longest possible N-terminal ion before Lys 48); the yellow shaded region represents fragment ions from a48 to a62 for two diubiquitin species. In the case of Lys48-linked ubiquitin (Figure 2A), the total fragment ion current in the diagnostic region decreased dramatically after the a47 ion, as expected for species with isopeptide bonds at the 48th residue. For the Lys63-linked dimer (Figure 2B), the ion current decreased after the a62 ion. The pattern becomes even more pronounced for longer homogeneously linked ubiquitin chains (see Figures 2C–F), and the change in ion current from regions in the protein that are specifically indicative of the sites of conjugation can be used in a semiquantitative manner (see discussion below).
The correlation between the N-terminal product ion current and the number of constituent monomers contributing to that ion current can be exploited to characterize the linkages quantitatively after empirical calibration. To test the feasibility of top-down UVPD-MS for determining linkage stoichiometry, homogeneously linked Lys48 and Lys63 ubiquitin tetramers were purified and mixed in known molar ratios. While these are not actual mixed-linkage polyubiquitins, the fragmentation patterns obtained for samples containing both tetramers would reflect the stoichiometry of fragment ions that are uniquely representative of each linkage type. The method requires that the two oligomers have similar ionization efficiencies, and analysis of the homogeneously linked tetraubiquitin chains showed that this is indeed the case (see Figure S2 in the Supporting Information). The two tetrameric chains were then mixed in different molar ratios and analyzed via UVPD-MS (see Figure 4). The ion current of the N-terminal fragments is easily detected and the total product ion current decreased for the ions specific to the Lys63 linkage type (ions a48–a62), relative to the much greater ion current attributed to Lys48 (and common Lys63) fragment ions as the ratio of Lys48 to Lys63 linkages increased in the sample. The UVPD mass spectra in Figure 4A show the gradual change in abundance of the ions that are uniquely derived from the Lys63 tetramer (a48–a62), relative to the higher abundance fragments that are derived from both tetramers (a1–a47). Even for the equimolar Lys48:Lys63 samples, there is an intrinsic excess of ions, prior to Lys48, because these ions are produced from both tetramers, while the N-terminal ions representative of residues 48–62 are unique to the Lys63 tetramer. This phenomenon is depicted in the theoretical spectrum shown in Figure 1B (where yellow lines indicate the contribution from Lys63 linkage-containing monomer fragments to those attributed to the Lys48-derived fragments). To accommodate this overlap, the average abundance of all N-terminally derived ions present between Lys48 and Lys63 must be subtracted from the average abundance of N-terminally derived ions up until Lys48, as shown below:
where ((∑i1–47)/47) is the average intensity of all ions from a1 to a47, and ((∑i48–62)/15) is the average intensity of all ions from a48 to a62 (and the denominator 15 or 47 represents the number of residues corresponding to the different linkage positions). This treatment accounts for the contribution of fragment ions derived from the Lys63 tetramer to the ion current shared by both species and is readily adapted for other polyubiquitin linkages. Plotting the derived linkage ratios against their expected ratios results in a linear relationship that provides an estimate of the ratio of one linkage type to the other (Figure 4B). (Refer to Tables S1–S5 in the Supporting Information for the tabulations of mass assignments of fragment ions and abundances used for the calculations.)
Figure 4.
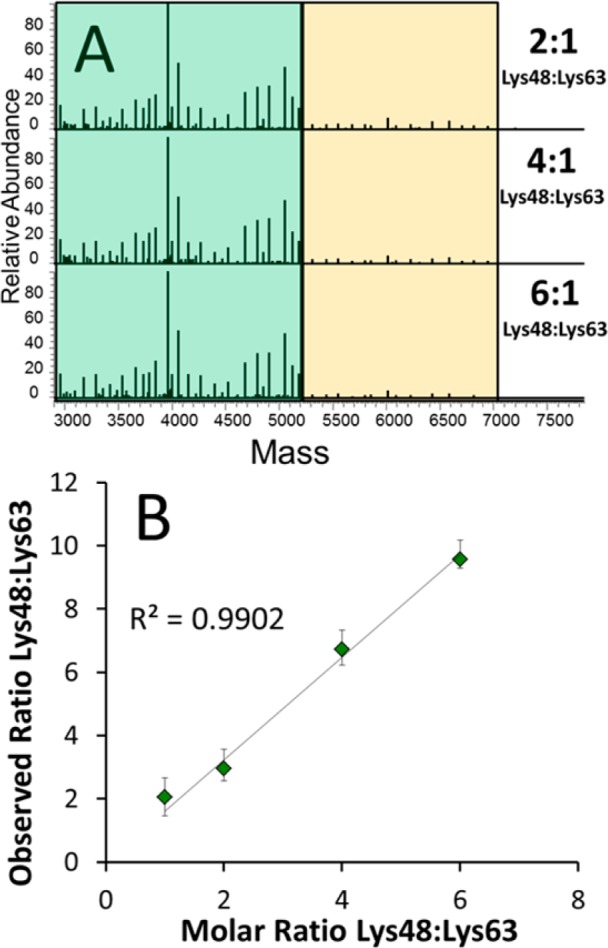
(A) Representative charge-deconvoluted UVPD mass spectra for solutions containing different molar ratios of Lys48-linked tetraubiquitin to Lys63-linked tetraubiquitin. Tetraubiquitin chains were mixed in the molar ratios 1:1, 2:1, 4:1, and 6:1 (Lys 48-linked: Lys 63-linked chains) and infused into the mass spectrometer. The z = 25 charge state was isolated (containing both Lys48 and Lys63 tetraubiquitin populations) and subjected to UVPD (one pulse, 1.5 mJ). Ions attributed to the Lys48 linkage are shaded in green and ions attributed to the Lys63 linkage are shaded in yellow. (B) Graphical depiction of the correlation between expected and observed ratios of Lys48 to Lys63, based on summation of linkage-specific product ions.
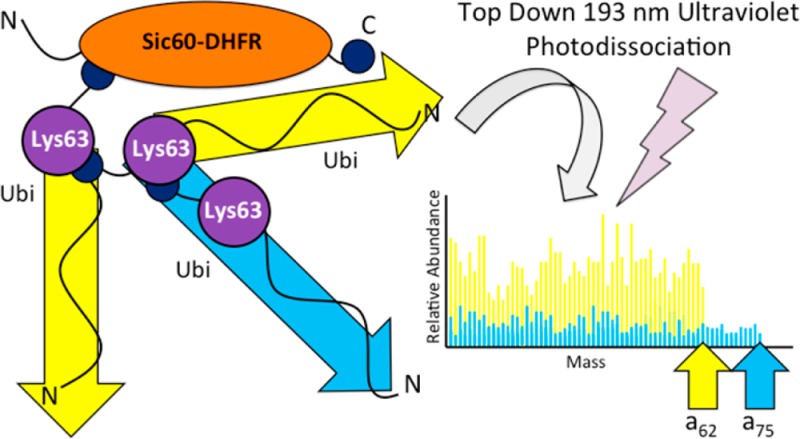
A final test for the method used an in vitro-formulated substrate, a fusion construct of amino acids 1 to 60 of yeast Sic1 protein, modified by the addition of a PY motif to accept ubiquitination by Rsp5,25 followed by E. coli dihydrofolate reductase and then a hexahistidine tag. The total mass of the fusion protein prior to ubiquitination is ∼24.9 kDa. Ubiquitin moieties were then ligated to amino acid 38 of the substrate via the HECT E3 ligase Rsp5.26 Intact mass spectra revealed that several species were produced, including diubiquitinated (for a total mass of ∼43 kDa) and triubiquitinated species (for a total mass of ∼52 kDa), and these were observed both in their N-terminally acetylated and nonacetylated forms, as is common for proteins expressed in E. coli (see Figure 5). Top-down UVPD-MS of ubiquitin chains attached to nonubiquitin substrates provides the opportunity for discriminating the substrate from the polyubiquitin chain based on ion type alone, because ubiquitin-derived fragment ions will match only predicted N- terminal ions (a-, b-, and c-type ions) in the sequence, but fragment ions derived from the substrate can be matched to predicted sequence ions from either terminus (a-, b-, c-, x-, y-, and z-type ions). Since ubiquitin is ligated to the protein via its C-terminus, there will be no C-terminally derived ions produced from a ubiquitin moiety ligated to the substrate. This allows the set of observed fragment ions from a given spectrum to be searched against specific predicted ion types from candidate protein sequences that will be uniquely attributed to either the substrate or the modifying ubiquitin moiety. Ideally, a two-step search method will be exploited in which only C-terminally derived ions are detected to identify the substrate, and only N-terminally derived ions from ubiquitin are used to deduce linkage type. First, UVPD mass spectra derived from monoubiquitin and the unmodified Sic60-DHFR construct were compared to confirm that there was no significant fragment ion overlap that would prevent assignment of diagnostic fragment ions and that could not be overcome by the high mass accuracy used in this study (see Figure 6). Figure 6 shows a representative deconvoluted section from 5000 Da to 6000 Da, and the spectra are summarized in Tables S8 and S9 in the Supporting Information. The top-down MS/MS spectra for intact proteins are congested and require high-mass-accuracy capabilities to assign the fragment ions; however, this can be readily done using an Orbitrap mass spectrometer. Two isotopic envelopes are expanded in Figure S3 in the Supporting Information, showing the region of the a50 ion for monoubiquitin (10+ precursor) and the Y47/y47 region for Sic60-DHFR (23+ precursor). The mass difference between these two is readily differentiated in the high-resolution/high-accuracy mass spectrum. Overall, there appears to be no ambiguity in assignment of ubiquitin-related fragment ions and fragment ions from the substrate protein. Next, the bioinformatic approach outlined above was applied to the Sic60-DHFR-Ubn construct, following infusion of the pure protein and UVPD-MS analysis, and revealed high ion current from N-terminally derived ions matched to the ubiquitin primary sequence up to the a62 ion (see Table S6 in the Supporting Information). Within the same spectrum, C-terminally derived ions were matched to the Sic60-DHFR construct well into the interior of the protein’s primary sequence (up until the Y134 ion (see Table S7 in the Supporting Information)). Comparison of the average ion current derived from ions that preceded the isopeptide bond at Lys63 to those that came after it (from a63 up to the a75 ion) revealed a 3.4:1 excess of the Lys63-specific region over the ions representative of the most distal ubiquitin (from a63 up to the a75 ion). This ratio, taken together with the chain length deduced from the intact mass measurement (i.e., corresponding to the net mass of three attached ubiquitins; Figure 5), shows that the modifying ubiquitin moieties were linked via Lys63. The same approach was used to compare the ion current of the regions specific to the Lys48 linkage to that of those ions specific for Lys63. After applying the proposed spectral subtraction, the Lys48:Lys63 ratio was empirically derived to be 1.5:1 (see Table S6 in the Supporting Information), which is consistent with the slight fragmentation bias for detection of the Lys48-specific fragments observed in the diubiquitin and tetraubiquitin studies (Figures 2B and 4). Using these data in conjunction with the intact mass of the precursor and high observed ion current up to and including the a62 ion, there was ample evidence to confirm that this substrate was homogeneously linked via Lys63 linkages and contains three ubiquitins.
Figure 5.

(A) Mass spectrum obtained for a mixture of ubiquitinated Sic60-DHFR fusion proteins. An asterisk highlights the z = 42 charge state, which was isolated for UVPD MS/MS. (B) Deconvoluted mass spectrum from panel (A). The mass of the expected translated sequence was observed (middle peak in each spectrum), as well as an acetylated version. The double dagger symbol (‡) denotes an alternative form of the fusion protein in which the first two N-terminal residues were removed. Shown in the right section are the corresponding species that have three, rather than two, ubiquitins attached.
Figure 6.

Shown are expanded regions of charge-deconvoluted UVPD mass spectra of (A) monoubiquitin (based on UVPD of the 10+ precursor) and (B) unmodified Sic60-DHFR (based on UVPD of the 23+ precursor) demonstrating little ion overlap. Panels (C) and (D) show comparisons of the expanded isotopic envelopes of the a47 ion from monoubiquitin and the combination of the Y47 and y47 ions from Sic60-DHFR (expanded peaks are shaded in gray in panels (A) and (B)). The majority of matched ions were matched at <2 ppm and none were matched at >10 ppm. For reference, the mass difference shown between two adjacent peaks in panels (C) and (D) is >50 ppm, thus confirming the ability to readily differentiate ubiquitin-derived and DHFR-derived ions.
The present method was demonstrated on relatively concentrated (20 μM) ubiquitin samples to demonstrate feasibility; however, in practice, the sensitivity is identical to that of any top-down experiment. Top-down UVPD-MS has been previously demonstrated in an online chromatographic workflow.23 In the earlier study, as little as 3.8 pmol of ribosomal proteins (on the order of 0.1 ug total protein) were loaded on column and analyzed. Top-down methods have not yet routinely achieved the sensitivity and detection limits of bottom-up methods. However, the superior capabilities of top-down MS/MS methods for characterization of proteoforms of proteins make it well-suited for the type of application reported in the present study.
Conclusions
The method outlined above demonstrates the power of top-down UVPD-MS for determining the size and linkage type of polyubiquitin chains. Although the order of connectivity remains undefined, the potential for relative stoichiometry determination based on ion current measurements within a mixture of multiple chain types provides an unprecedented level of characterization from a single experiment. This arises from the systematic change in ion current associated with N-terminally derived ions that precede or follow the site of isopeptide conjugation. This method capitalizes on both the critically important intact mass measurement of top-down MS to describe chain length and the almost-complete backbone fragmentation provided by N-terminally derived ions after UVPD to detect the ubiquitin linkage pattern, especially for homogeneously linked chains. To further advance this strategy, additional effort will be directed at making it quantitative, as well as implementing it in a chromatographic workflow to dissect mixtures of ubiquinated substrates. Moreover, heterogeneous sets of conjugates of the same size are currently intractable, and protein-level online separation methods do not offer sufficient resolution to resolve them at this point.
Acknowledgments
Financial support was provided by the Welch Foundation (No. F-1155, to J.S.B., and No. F-1817, to A.M.), the National Science Foundation (NSF) (No. CHE-1402753, to J.S.B.), the National Institutes of Health (NIH) (No. 1K12GM102745, to J.S.B., with fellowship to J.R.C.; and No. U54GM105816, to A.M.).
Supporting Information Available
Supporting Information includes the 193-nm UVPD mass spectrum of ubiquitin (12+); mass spectra showing the charge state distributions of Lys48-linked tetraubiquitin and Lys63-linked tetraubiquitin; and tables that shows the theoretical and identified ions found in the UVPD mass spectrum of Sic60-DHFR, ubiquitinated Sic60-DHFR, the possible product ions from the Sic60-DHFR protein, the ion assignments and abundances obtained for the mixtures of Lys48:Lys63 tetraubiquitin, and the relationship between the ratios of Lys48:Lys63 tetraubiquitin mixtures and known molar ratios. This material is available free of charge via the Internet at http://pubs.acs.org/.
Author Contributions
‡ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sutter M.; Damberger F. F.; Imkamp F.; Allain F. H.-T.; Weber-Ban E. J. Am. Chem. Soc. 2010, 132, 5610–5612. [DOI] [PubMed] [Google Scholar]
- Pickart C. M. Cell 2004, 116, 181–190. [DOI] [PubMed] [Google Scholar]
- McDowell G. S.; Philpott A. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [DOI] [PubMed] [Google Scholar]
- Nakasone M. A.; Livnat-Levanon N.; Glickman M. H.; Cohen R. E.; Fushman D. Structure 2013, 21, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P.; Duong D. M.; Seyfried N. T.; Cheng D.; Xie Y.; Robert J.; Rush J.; Hochstrasser M.; Finley D.; Peng J. Cell 2009, 137, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J.; Schwartz D.; Elias J. E.; Thoreen C. C.; Cheng D.; Marsischky G.; Roelofs J.; Finley D.; Gygi S. P. Nat. Biotechnol. 2003, 21, 921–926. [DOI] [PubMed] [Google Scholar]
- Xu P.; Peng J. Anal. Chem. 2008, 80, 3438–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan J.; Roach L.; Sokratous K.; Tooth D.; Long J.; Garner T. P.; Searle M. S.; Oldham N. J.; Layfield R. J. Proteome Res. 2012, 11, 1969–1980. [DOI] [PubMed] [Google Scholar]
- Jung J.; Pierson N.; Marquardt A.; Scheffner M.; Przybylski M.; Clemmer D. J. Am. Soc. Mass Spectrom. 2011, 22, 1463–1471. [DOI] [PubMed] [Google Scholar]
- Cannon J.; Nakasone M.; Fushman D.; Fenselau C. Anal. Chem. 2012, 84, 10121–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkevich E. M.; Sanchez N. A.; Ge Y.; Strieter E. R. Biochemistry 2014, 53, 4979–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban Warren M. R.; Parker C. E.; Mocanu V.; Klapper D.; Borchers C. H. Rapid Commun. Mass Spectrom. 2005, 19, 429–437. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick D. S.; Hathaway N. A.; Hanna J.; Elsasser S.; Rush J.; Finley D.; King R. W.; Gygi S. P. Nat. Cell Biol. 2006, 8, 700–710. [DOI] [PubMed] [Google Scholar]
- Mirzaei H.; Rogers R. S.; Grimes B.; Eng J.; Aderem A.; Aebersold R. Mol. Biosyst. 2010, 6, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Li W.; Holden D. D.; Zhang Y.; Griep-Raming J.; Fellers R. T.; Early B. P.; Thomas P. M.; Kelleher N. L.; Brodbelt J. S. J. Am. Chem. Soc. 2013, 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbelt J. S. Chem. Soc. Rev. 2014, 43, 2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart C. M.; Raasi S. In Ubiquitin and Protein Degradation; Deshaies R. J., Ed.; Methods in Enzymology, Vol. 399; Academic Press: Amsterdam, 2005; pp 21–36. [DOI] [PubMed] [Google Scholar]
- Carvalho A.; Pinto M.; Grou C.; Vitorino R.; Domingues P.; Yamao F.; Sá-Miranda C.; Azevedo J. Mol. Biotechnol. 2012, 51, 254–261. [DOI] [PubMed] [Google Scholar]
- Raasi S.; Pickart C. In Ubiquitin–Proteasome Protocols; Patterson C.; Cyr D., Eds.; Methods in Molecular Biology, Vol. 301; Humana Press: Totowa, NJ, 2005; pp 47–55. [DOI] [PubMed] [Google Scholar]
- Ptak C.; Varelas X.; Moraes T.; McKenna S.; Ellison M. J. In Ubiquitin and Protein Degradation; Deshaies R. J., Ed.; Methods in Enzymology, Vol. 399; Academic Press: Amsterdam, 2005; pp 43–54. [DOI] [PubMed] [Google Scholar]
- Kraut D. A.; Israeli E.; Schrader E. K.; Patil A.; Nakai K.; Nanavati D.; Inobe T.; Matouschek A. ACS Chem. Biol. 2012, 7, 1444–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasicek L. A.; Ledvina A. R.; Shaw J.; Griep-Raming J.; Westphall M. S.; Coon J. J.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2011, 22, 1105–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. R.; Cammarata M. B.; Robotham S. A.; Cotham V. C.; Shaw J. B.; Fellers R. T.; Early B. P.; Thomas P. M.; Kelleher N. L.; Brodbelt J. S. Anal. Chem. 2014, 86, 2185–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. R.; Kluwe C.; Ellington A.; Brodbelt J. S. Proteomics 2014, 14, 1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki Y.; Isono E.; Toh-E A. In Ubiquitin and Protein Degradation; Deshaies R. J., Ed.; Methods in Enzymology, Vol. 399; Academic Press: Amsterdam, 2005; pp 215–227. [DOI] [PubMed] [Google Scholar]
- Park S.; Ntai I.; Thomas P.; Konishcheva E.; Kelleher N. L.; Statsuk A. V. Biochemistry (Moscow) 2012, 51, 8327–8329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.