

Abstract
Polymyxin is the last-line therapy against Gram-negative ‘superbugs’; however, dose-limiting nephrotoxicity can occur in up to 60% of patients after intravenous administration. Understanding the accumulation and concentration of polymyxin within renal tubular cells is essential for the development of novel strategies to ameliorate its nephrotoxicity and to develop safer, new polymyxins. We designed and synthesized a novel dual-modality iodine-labeled fluorescent probe for quantitative mapping of polymyxin in kidney proximal tubular cells. Measured by synchrotron X-ray fluorescence microscopy, polymyxin concentrations in single rat (NRK-52E) and human (HK-2) kidney tubular cells were approximately 1930- to 4760-fold higher than extracellular concentrations. Our study is the first to quantitatively measure the significant uptake of polymyxin in renal tubular cells and provides crucial information for the understanding of polymyxin-induced nephrotoxicity. Importantly, our approach represents a significant methodological advancement in determination of drug uptake for single-cell pharmacology.
The world is facing a significant global medical challenge from Gram-negative ‘superbugs’, particularly Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae, which are resistant to almost all currently available antibiotics.1 The dry antibiotic discovery pipeline means that there will be no new antibiotics available against these superbugs for many years to come.1 ‘Old’ polymyxins, namely polymyxin B and colistin, are increasingly used as the last-line therapy against these very problematic pathogens.2,3 Unfortunately, polymyxin-induced nephrotoxicity occurs in up to 60% of patients when administered intravenously and is the major dose-limiting factor for their optimal clinical use.4,5 Polymyxin-induced nephrotoxicity is associated with apoptosis and histopathological damage to kidney tubular cells.6−8 After filtration by glomeruli, polymyxins undergo extensive tubular reabsorption9,10 and accumulate in the kidney as indicated by analysis of the concentration in tissue homogenate.7 In addition, our very recent clinical pharmacokinetic study reported, for the first time, the very significant degree of exposure of renal tubular cells to polymyxin B in humans.10 Accordingly, quantifying the accumulation and concentration of polymyxin within tubular cells is very important for the development of novel strategies to ameliorate polymyxin-induced nephrotoxicity, as well as for the discovery of safer, new-generation polymyxins. Unfortunately, to date there are no experimental data on the disposition of polymyxin at the tubular cellular level. Critical barriers include the design and synthesis of a bona fide polymyxin probe, coupled with the lack of methodology to determine polymyxin concentrations in a single cell.
Quantification of intracellular drug concentrations is very challenging but crucial for understanding the mechanisms of drug toxicity.11−14 Current methodologies for quantification of drug concentrations in mammalian cells are based on radiometry, bioassays, UV absorbance, fluorescence emission, and mass spectrometry; however, none of these methods is able to accurately measure the intracellular concentration in single cells. The intracellular concentration of the highly nephrotoxic cancer therapeutic cisplatin was measured using synchrotron X-ray fluorescence microscopy (XFM); however, not all drugs contain platinum.15,16 Hence, innovative approaches to measure intracellular drug concentrations are desperately needed. In the present study, a novel dual-modality iodine-labeled fluorescent probe (FADDI-096) was designed and synthesized, and a correlative microscopy approach was employed for quantitatively mapping the intracellular accumulation of polymyxin in single kidney tubular cells.
Experimental Section
Design of FADDI-096 Using Molecular Modeling
The model of FADDI-096 in complex with LPS from Escherichia coli was constructed.17 The coordinates of the NMR solution structure of polymyxin B in interaction with E. coli LPS in solution were kindly provided by Pristovsek and Kidric,18 and the dansyl-Gly substitution and iodine were modeled onto the structure. The crystal structure of the receptor of ferrichrome-iron in E. coli FhuA (PDB code: 1QFF) was employed to derive the coordinates of LPS in E. coli.19 FADDI-096 was docked onto the LPS molecule.17 POVRAY (Version 3.6; http://www.povray.org) was employed to render molecular representations.17
Reagents for Synthesis of FADDI-096
Piperidine, diisopropylethylamine (DIPEA), trifluoroacetic acid (TFA), and 1H-benzotriazolium-1-[bis(dimethylamino)methylene]-5-chloro-hexafluorophosphate-(1-),3-oxide (HCTU) were obtained from Auspep (Melbourne, Australia). Fmoc-Dab(Boc)-OH, Fmoc-d-Phe(4-Iodo)-OH, and Fmoc-Dab(ivDde)-OH were from Chem-Impex International (U.S.A.). Fmoc-Thr(tBu)-OH was purchased from Mimotopes (Melbourne, Australia). Fmoc-OctGly-OH was obtained from Try-lead Chem (Hangzhou, China). Dimethylformamide (DMF), methanol (CH3OH), diethyl ether, dichloromethane (DCM), hydrochloric acid, and acetonitrile were from Merck (Melbourne, Australia). Fmoc-Thr(tBu)-TCP-resin was obtained from Intavis Bioanalytical Instruments (Koeln, Germany). Dansylglycine, triisopropylsilane (TIPS), and diphenylphosphorylazide (DPPA) were from Sigma-Aldrich (Castle Hill, Australia).
Synthesis of FADDI-096
FADDI-096 was synthesized using standard Fmoc solid-phase peptide synthesis techniques as described previously.20 FADDI-096 TFA salt was obtained as a white solid in a yield of 19.0 mg with purity of >95% (estimated by RP-HPLC at 214 nm). The compound was confirmed as having the correct molecular weight by ESI-MS analysis: m/z (monoisotopic) calculated: C71H116N19O16IS [M + H]+ 1649.76, [M + 2H]2+ 825.38; observed: [M + H]+ 1649.25, [M + 2H]2+ 825.50.
Antibacterial Activity of FADDI-096
Minimum inhibitory concentration (MIC) of FADDI-096 was determined using the broth microdilution method against P. aeruginosa ATCC 27853 (American Type Culture Collection, Manassas, VA).
Cell Culture and Preparation of Cells for Microscopy
NRK-52E and HK-2 cells were obtained from ATCC. NRK-52E cells were cultured in the presence of 10% fetal bovine serum (FBS) in DMEM (GlutaMAX-I). HK-2 cells were cultured in keratinocyte serum-free medium (K-SFM). Bovine pituitary extract (BPE, 0.05 mg/mL) and human recombinant epidermal growth factor (EGF, 5 ng/mL) were used as supplements. All components of the growth medium were purchased from Invitrogen (Life Technologies, VIC, Australia). Both types of cells were grown at 37 °C in a humidified atmosphere containing 5% CO2. NRK-52E cells were passaged three times per week and twice per week for HK-2 cells. Cells were harvested by trypsinization and washed with phosphate-buffered saline (PBS, pH 7.4; Invitrogen) prior to use. Two milliliters of NRK-52E (0.5 × 105 cells/mL) and HK-2 (0.25 × 105 cells/mL) cells were seeded on silicon nitride windows manufactured by Silson (Blisworth, U.K.) in 12-well plates in growth medium and conditioned for 24 and 48 h, respectively. The medium was then discarded by aspiration, and the cells were washed twice with PBS. For the accumulation study, NRK-52E cells on the silicon nitride windows were incubated with 5.0 and 50.0 μM FADDI-096 in DMEM supplemented with 0.1% FBS, whereas HK-2 cells were incubated with 10.0 μM of FADDI-096 in K-SFM supplemented with BPE (0.05 mg/mL) and EGF (5 ng/mL). After treatment, cells were fixed with a high-purity solution of 4% paraformaldehyde (PFA) prepared in phosphate-buffered saline (PBS; pH 7.4). After drying under negative pressure inside the biosafety cabinet, windows were used for fluorescence microscopy, XFM and SEM.21
Oxidative Stress Induced by FADDI-096 and Polymyxin B
Rat (NRK-52E) and human (HK-2) kidney proximal tubular cells were seeded on 8-well microslide chambers, and oxidative stress induced by FADDI-096 and polymyxin B was studied using MitoSOX red (Molecular Probes).22
Acquisition of Elemental Maps using XFM
The elemental content was determined using the Fresnel zone-plate hard X-ray microscope at the Australian Synchrotron XFM beamline.23 The silicon nitride windows were mounted on an aluminum stalk and scanned through an incident X-ray beam of 10.5 keV. This energy was chosen to induce K-shell ionization of elements with atomic numbers below 30 (Z ≤ Zn). The specimen was 15° normal to the incoming X-ray beam and scanned through the focal plane in steps of 800 nm (horizontal axis, x) and 800 nm (vertical axis, y) orthogonal to the beam axis using an integration of 1–1.5 s per scan point to ensure adequate counting statistics. A silicon-drift diode detector (Vortex, SII NanoTechnology, CA) oriented at 90° to the incident beam was used to detect the resulting X-ray fluorescence. Elemental maps were generated from the three-dimensional data sets (x, y, energy) using the MAPS software suite (v1.6.5).24 This procedure fitted the contribution of the K emission lines for the elements: P, S, P, Cl, K, Ca, Fe, Cu and Zn along with the iodine L lines, where appropriate. The absolute calibration to areal densities (μg/cm2) was achieved by fitting the fluorescence signal from thin-film standards SRM-1832 and SRM-1833 (National Institute of Standards and Technology, Gaithersburg, MD). The spectra of the standard reference materials (SRM-1832 and SRM-1833) were acquired. These spectra were employed to directly calculate the actual element concentration of these measured elements in the sample and the approximate concentration of other elements using known X-ray fluorescence yield and absorption values.25 For each treatment, regions of interest containing 10 whole cells map using XFM and the resulting quantity of iodine was used to establish the equivalent amount of FADDI-096 on a per cell basis. An average cell volume of 1000 μm3 was employed for calculation of the average intracellular concentrations of FADDI-096 and other elements.26
Accumulation of FADDI-096 Examined by Fluorescence Microscopy and Identification of Cells with SEM
For fluorescence imaging, a Deltavision Elite microscope (Applied Precision) equipped with a TruLight Illumination System was utilized. Fluorescence images (Ex: 381–399 nm; Em: 523 ± 18 nm) of cells on silicon nitride windows with or without treatments were acquired using a CoolSNAP HQ2 camera with a 20× (0.75 NA) lens and softWoRx software (Applied Precision). After acquisition of an XFM or the fluorescence image, the approximate location of the cell on the silicon nitride windows was chartered. A low magnification (20× or 40×) optical image was also taken to document the local cell morphology for the subsequent SEM imaging. The silicon nitride windows were mounted on standard aluminum stubs with double-sided carbon tape for SEM imaging. Sputter coating was performed with a sputter coater (Emitech K550X, Quorum Technologies Ltd., U.K.) to add a thin layer of gold to reduce charging. The samples were then transferred to an FEI Helios NanoLab 600 DualBeam FIB-SEM electron microscope (FEI Company, Hillsboro, OR). During SEM acquisition, low accelerating voltage 2 kV was selected, and electron signals were acquired using a secondary electron (SE) detector or a mixed mode of SE and backscattering electron (BSE) signals.
Prior to correlation, images acquired with different correlative microscopy methods were first resized to the same scale/pixel size. Markers (e.g., cluster of cells and corner of the silicon nitride window) were employed to identify the target single cells for imaging with multiple microscopy approaches. Rotation and translation of a cell, if necessary, were achieved either manually or through cross-correlation algorithm.
Results and Discussion
Design and Synthesis of the Polymyxin Probe
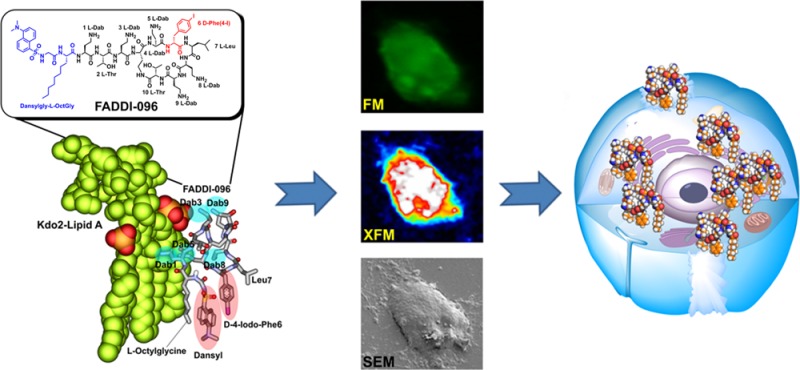
In the present study, a novel strategy was employed for the creation of a dual-modality polymyxin probe (FADDI-096) based upon our polymyxin structure–activity relationship (SAR) model.17,27 We hypothesized that labeling the core polymyxin scaffold (Figure 1) with nonradioactive iodine (XFM contrast) and a dansyl fluorophore (for fluorescence microscopy) would generate a molecular probe which made possible the quantitative mapping of the intracellular accumulation of polymyxin in single kidney tubular cells. It was a formidable challenge to design such a polymyxin probe, while not interfering with the structural elements of the polymyxin molecule required for its antimicrobial and nephrotoxic activities. Iodine was chosen as a contrast agent for XFM in the present study because of its strong anomalous diffraction.28 The most viable option for the incorporation of the iodine was via the d-phenylalanine residue at position 6 (Figure 1). This is because aryl halides are inherently more stable than alkyl halides, an important consideration for an iodine-containing probe used to map distribution in a biological system. Furthermore, as the phenyl ring is involved in the hydrophobic interaction with the fatty acyl chains of lipid A in the outer membrane of Gram-negative bacterial cells, it appeared to be the ideal position for accommodating the hydrophobic iodine atom.17,27 The incorporation was achieved using D-4-iodophenylalanine that was commercially available in its Fmoc-protected form. For the fluorescent component of the probe, a dansyl group was utilized due to its strong fluorescence and relatively small size that would not impose a significant steric effect.29 Commercially available dansyl-polymyxin B is not suitable for investigations on the mechanisms of polymyxin-induced nephrotoxicity, as it is inactive against Gram-negative pathogens due to the attachment of up to five dansyl moieties to the diaminobutyric acid (Dab) residues (Figure 1a).29 Based upon our SAR model of the interaction of polymyxin B with lipid A, modeling of FADDI-096 with lipid A from E. coli was performed (Figure 1b, c) and clearly envisaged that the most appropriate point of attachment of the dansyl group would be at the N-terminus of the polymyxin structure.17,29 Therefore, our strategy was to replace the N-terminal fatty acyl group with L-octylglycine. The eight-carbon side chain of this residue mimicked the N-terminal fatty acyl chain of polymyxin B. To avoid the need for additional orthogonal protection during the synthesis, the N-α-amino group was employed as a convenient point of attachment for the dansyl group. A glycine linker was incorporated between the dansyl group and the L-octylglycine to reduce the overall rigidity of the N-terminal construct, which appears to be more favorable for binding with lipid A (fluorescent spectra of the probe is provided as Supporting Information, Figure S1).29
Figure 1.

Structures of polymyxin B1 and FADDI-096 and molecular model of FADDI-096 with LPS: (a) polymyxin B1, (b) FADDI-096, (c) molecular model of the complex between Escherichia coli LPS with FADDI-096.
Our novel molecular probe was synthesized using optimized Fmoc solid-phase methodology that we developed in our laboratory for the routine total synthesis of new polymyxin analogues. Electrospray ionization mass spectrometry (ESI-MS) analysis in positive ion mode confirmed that the end product had a molecular weight consistent with the structure of FADDI-096 (purity >95%). FADDI-096 displayed antibacterial activity with a minimum inhibitory concentration (MIC) of 8 mg/L against a wild-type polymyxin-susceptible reference strain P. aeruginosa ATCC 27853 (polymyxin B MIC 1–2 mg/L). Furthermore, similar to polymyxin B, FADDI-096 induced oxidative stress in both rat and human kidney proximal tubular cells (Figure 2a, b), which is associated with polymyxin-induced nephrotoxicity.6,30,31 Together, these results demonstrate that the modifications introduced into the polymyxin B scaffold to produce FADDI-096 did not compromise the antibacterial activity or the ability to induce oxidative stresses that are associated with the native polymyxin B molecule.
Figure 2.
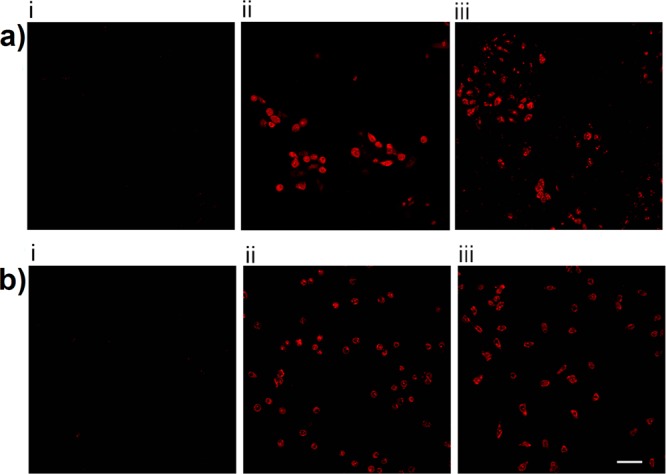
Oxidative stress in kidney tubular cells after 24-h treatment. (a) NRK-52E cells (i) without treatment and treated with (ii) 0.75 mM polymyxin B and (iii) 0.75 mM FADDI-096. (b) HK-2 cells (i) without treatment and treated with (ii) 0.5 mM polymyxin B and (iii) 0.5 mM FADDI-096. Scale bar: 50 μm.
Quantitative Mapping of Polymyxin in Kidney Tubular Cells
To quantitatively map the intracellular accumulation of FADDI-096 in single rat and human kidney tubular cells, we first conducted fluorescence microscopy followed by XFM imaging of the same samples. Furthermore, SEM was employed to correlate the individual cells with the intracellular distribution of FADDI-096 detected by fluorescence microscopy and XFM (Figure 3a–c). Our fluorescence microscopy results revealed that FADDI-096 localized throughout the cytoplasm and nuclear region (Figure 3a). We further investigated the drug localization and accumulation on a single-cell level using XFM which allowed for the monitoring of the migration of FADDI-096 into the cells (Figure 3a). The intracellular distribution of FADDI-096 observed via XFM was highly consistent with the fluorescence microscopy imaging results (Figure 3a), demonstrating the overlap of the dansyl and iodine signals from FADDI-096 itself.
Figure 3.
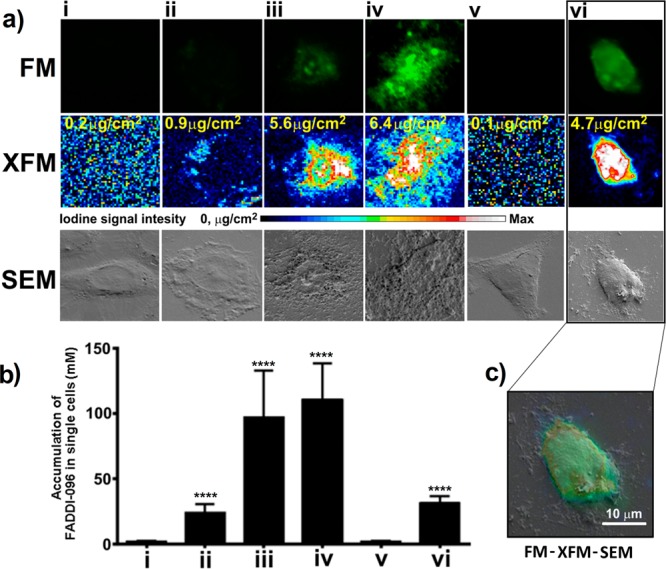
Accumulation of FADDI-096 in NRK-52E and HK-2 cells determined with fluorescence microscopy (FM), X-ray fluorescence microscopy (XFM), and scanning electron microscopy (SEM). (a) NRK-52E cells (i) without treatment, (ii) treated with 5 μM FADDI-096 for 4 h, (iii) treated with 50 μM FADDI-096 for 1 h, (iv) treated with 50 μM FADDI-096 for 4 h; and HK-2 cells (v) without treatment, (vi) treated with 10 μM FADDI-096 for 4 h. The row labeled with “XFM” shows the iodine distribution within the same NRK-52E and HK-2 cells as described in panel a. Iodine signal intensities are shown using a linear scale from zero to the maximum value; the yellow numbers at the top left of the relevant panels note the maximum pixel value (μg/cm2) in each sample. (b) Accumulation of FADDI-096, in single NRK-52E and HK-2 cells as described in panel a, measured via iodine content using XFM (mean ± SD; n = 10). ****p < 0.0001 compared with control samples. (c) Correlation of signals from different microscopes including FM (green), XFM (blue to red), and SEM (gray). Scale bar: 10 μm.
With the intracellular concentrations determined by the high sensitivity of synchrotron XFM, it is evident that the uptake of FADDI-096 by rat kidney tubular NRK-52E cells was dependent on the concentration and duration of the treatment (Figure 3b). Uptake at 4 h was significantly higher in the rat kidney tubular cells treated with 50 μM of FADDI-096 (overall intracellular concentration 110 ± 28.2 mM), compared to the cells treated with 5 μM of FADDI-096 (23.8 ± 6.63 mM). The approximately 4.62-fold increase in overall intracellular concentration resulting from a 10-fold increase in the concentration of the bathing solution is consistent with a saturable (carrier-mediated) component of the cellular uptake process of polymyxin.32 The overall concentration of FADDI-096 in human kidney tubular HK-2 cells was 31.0 ± 5.69 mM after treatment at a concentration of 10 μM for 4 h. Interestingly, the XFM results also revealed a significant increase in the intracellular calcium concentration (Figure 4), which is a potential stimulus to trigger apoptosis.33 Cellular distributions of other elements were also evaluated for all samples (detailed results are provided in the Supporting Information, Figure S2). The exceptional intracellular accumulation of the polymyxin probe in rat and human kidney tubular cells supports the recent finding of the time- and concentration-dependence of apoptosis caused by polymyxin.34 For example, the overall concentration of FADDI-096 in HK-2 cells (31.0 ± 5.69 mM) was approximately 3100-fold higher than the concentration in the bathing solution (10 μM) (Figure 3b). Collectively, our XFM data on events at the cellular level are consistent with the very substantial tubular reabsorption of polymyxin observed at the whole-body level in rats and humans.9,10 While those previous observations provide valuable information on mass transfer events in vivo, our results for the first time offer insight on the spatial and temporal accumulation of polymyxin in single tubular cells.
Figure 4.

Correlation between intracellular calcium concentration and accumulation of FADDI-096 in single rat kidney tubular NRK-52E cells. Cells were treated without or with 5 μM FADDI-096 for 4 h, 50 μM FADDI-096 for 1 h, and 50 μM FADDI-096 for 4 h (r = 0.62; p ≤ 0.0001 from Pearson correlation).
Conclusion
In the battle against life-threatening infections caused by Gram-negative ‘superbugs’, we cannot rely on the discovery of new antibiotics. We must optimize the clinical use of the last-line polymyxin and ameliorate the dose-limiting nephrotoxicity through a better understanding of the mechanism. The quantitative aptitude of XFM presented here expands the horizon of measuring drug concentrations in cells. Importantly, our study is the first to conceptualize the chemistry of a unique dual-modality probe for the combined use of correlative microscopy to determine the uptake of drugs in individual cells. This approach represents a significant methodological advancement for single-cell pharmacology.
Acknowledgments
J.L., R.L.N. and T.V. are supported by an Australian National Health and Medical Research Council (NHMRC) project grant (ID 1026109). J.L., T.V., R.L.N., P.E.T., and K.D.R. are funded by a grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI098771). J.L. is an Australian NHMRC Senior Research Fellow, and T.V. is an Australian NHMRC Industry CDA Fellow. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. Synchrotron-XFM research was undertaken on the XFM beamline at the Australian Synchrotron, Victoria, Australia. This work was conducted in part at the Melbourne Centre for Nanofabrication (MCN), an initiative partly funded by the Commonwealth of Australia and the Victorian Government.
Supporting Information Available
Additional information as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org/.
Author Contributions
# Joint senior authors.
The authors declare no competing financial interests.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Boucher H. W.; Talbot G. H.; Benjamin D. K. Jr.; Bradley J.; Guidos R. J.; Jones R. N.; Murray B. E.; Bonomo R. A.; Gilbert D. Clin. Infect. Dis. 2013, 56, 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Nation R. L.; Turnidge J. D.; Milne R. W.; Coulthard K.; Rayner C. R.; Paterson D. L. Lancet Infect. Dis. 2006, 6, 589–601. [DOI] [PubMed] [Google Scholar]
- Bergen P. J.; Landersdorfer C. B.; Zhang J.; Zhao M.; Lee H. J.; Nation R. L.; Li J. Diagn. Microbiol. Infect. Dis. 2012, 74, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garonzik S. M.; Li J.; Thamlikitkul V.; Paterson D. L.; Shoham S.; Jacob J.; Silveira F. P.; Forrest A.; Nation R. L. Antimicrob. Agents Chemother. 2011, 55, 3284–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin C. J.; Ellman T. M.; Phadke V.; Haynes L. J.; Calfee D. P.; Yin M. T. J. Infect. 2012, 65, 80–87. [DOI] [PubMed] [Google Scholar]
- Yousef J. M.; Chen G.; Hill P. A.; Nation R. L.; Li J. J. Antimicrob. Chemother. 2012, 67, 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef J. M.; Chen G.; Hill P. A.; Nation R. L.; Li J. Antimicrob. Agents Chemother. 2011, 55, 4044–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell J. D.; Neff R.; Ake J.; Howard R.; Olson S.; Paolino K.; Vishnepolsky M.; Weintrob A.; Wortmann G. Clin. Infect. Dis. 2009, 48, 1724–1728. [DOI] [PubMed] [Google Scholar]
- Li J.; Milne R. W.; Nation R. L.; Turnidge J. D.; Smeaton T. C.; Coulthard K. Antimicrob. Agents Chemother. 2003, 47, 1766–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri A. M.; Landersdorfer C. B.; Jacob J.; Boniatti M. M.; Dalarosa M. G.; Falci D. R.; Behle T. F.; Bordinhao R. C.; Wang J.; Forrest A.; Nation R. L.; Li J.; Zavascki A. P. Clin. Infect. Dis. 2013, 57, 524–531. [DOI] [PubMed] [Google Scholar]
- Dollery C. T. Clin. Pharmacol. Ther. 2013, 93, 263–266. [DOI] [PubMed] [Google Scholar]
- Malucelli E.; Iotti S.; Gianoncelli A.; Fratini M.; Merolle L.; Notargiacomo A.; Marraccini C.; Sargenti A.; Cappadone C.; Farruggia G.; Bukreeva I.; Lombardo M.; Trombini C.; Maier J. A.; Lagomarsino S. Anal. Chem. 2014, 86, 5108–5115. [DOI] [PubMed] [Google Scholar]
- Moore K. L.; Lombi E.; Zhao F. J.; Grovenor C. R. M. Anal. Bioanal. Chem. 2012, 402, 3263–3273. [DOI] [PubMed] [Google Scholar]
- Ortega R.; Deves G.; Carmona A. J. R. Soc. Interface 2009, 6, S649–S658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilinski P.; Lai B.; Cai Z.; Yun W.; Legnini D.; Talarico T.; Cholewa M.; Webster L. K.; Deacon G. B.; Rainone S.; Phillips D. R.; Stampfl A. P. Cancer Res. 2003, 63, 1776–1779. [PubMed] [Google Scholar]
- Chen K. G.; Valencia J. C.; Lai B.; Zhang G.; Paterson J. K.; Rouzaud F.; Berens W.; Wincovitch S. M.; Garfield S. H.; Leapman R. D.; Hearing V. J.; Gottesman M. M. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9903–9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov T.; Thompson P. E.; Nation R. L.; Li J. J. Med. Chem. 2010, 53, 1898–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pristovsek P.; Kidric J. Curr. Top. Med. Chem. 2004, 4, 1185–1201. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Welte W.; Hofmann E.; Lindner B.; Holst O.; Coulton J. W.; Diederichs K. Struct. Fold. Des. 2000, 8, 585–592. [DOI] [PubMed] [Google Scholar]
- Deris Z. Z.; Swarbrick J. D.; Roberts K. D.; Azad M. A.; Akter J.; Horne A. S.; Nation R. L.; Rogers K. L.; Thompson P. E.; Velkov T.; Li J. Bioconjugate Chem. 2014, 25, 750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S. A.; Myers D. E.; de Jonge M. D.; Vogt S.; Ryan C. G.; Sexton B. A.; Hoobin P.; Paterson D.; Howard D. L.; Mayo S. C.; Altissimo M.; Moorhead G. F.; Wilkins S. W. Anal. Bioanal. Chem. 2011, 401, 853–864. [DOI] [PubMed] [Google Scholar]
- Shah A.; Xia L.; Goldberg H.; Lee K. W.; Quaggin S. E.; Fantus I. G. J. Biol. Chem. 2013, 288, 6835–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson D.; de Jonge M. D.; Howard D. L.; Lewis W.; McKinlay J.; Starritt A.; Kusel M.; Ryan C. G.; Kirkham R.; Moorhead G.; Siddons D. P. 10th Int. Conf. X-Ray Microsc. 2011, 1365, 219–222. [Google Scholar]
- Vogt S. J. Phys. IV 2003, 104, 635–638. [Google Scholar]
- Grieken R. V.; Markowicz A.. Handbook of X-ray spectrometry, 2nd ed.; Marcel Dekker: New York, 2002. [Google Scholar]
- Frame K. K.; Hu W. S. Biotechnol. Bioeng. 1990, 36, 191–197. [DOI] [PubMed] [Google Scholar]
- Velkov T.; Roberts K. D.; Nation R. L.; Thompson P. E.; Li J. Future Microbiol. 2013, 8, 711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauter Z. Nat. Biotechnol. 2004, 22, 1239–1240. [DOI] [PubMed] [Google Scholar]
- Soon R. L.; Velkov T.; Chiu F.; Thompson P. E.; Kancharla R.; Roberts K.; Larson I.; Nation R. L.; Li J. Anal. Biochem. 2011, 409, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozyilmaz E.; Ebinc F. A.; Derici U.; Gulbahar O.; Goktas G.; Elmas C.; Oguzulgen I. K.; Sindel S. Intensive Care Med. 2011, 37, 141–146. [DOI] [PubMed] [Google Scholar]
- Fonseca C. D.; Watanabe M.; Vattimo M. D. F. Antimicrob. Agents Chemother. 2012, 56, 5082–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Yamaguchi H.; Ogura J.; Kobayashi M.; Yamada T.; Iseki K. Antimicrob. Agents Chemother. 2013, 57, 6319–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P.; Chan S. L. Nat. Cell Biol. 2003, 5, 1041–1043. [DOI] [PubMed] [Google Scholar]
- Azad M. A.; Finnin B. A.; Poudyal A.; Davis K.; Li J.; Hill P. A.; Nation R. L.; Velkov T. Antimicrob. Agents Chemother. 2013, 57, 4329–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.