

Abstract
Several retrospective epidemiological studies report that utilization of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) inhibitors called statins at mid-life can reduce the risk of developing sporadic Alzheimer’s disease (AD) by as much as 70%. Conversely, administration of these inhibitors in clinically diagnosed subjects with AD confers little or no benefits over time. Here, we investigated the association between AD and HMGCR rs3846662, a polymorphism known to be involved in regulation of HMGCR exon 13 skipping, in a founder population and in two distinct mixed North American populations of converting mild cognitively impaired (MCI) subjects [ADCS and ADNI cohorts]. Targeting more specifically women, the G allele negative (G−) AD subjects exhibit delayed age of onset of AD [P = 0.017] and significantly reduced risk of AD [O.R.: 0.521; P = 0.0028], matching the effect size reported by the APOE2 variant. Stratification for APOE4 in a large sample of MCI patients from the ADCS cohort revealed a significant protective effect of G negative carriers on AD conversion three years after MCI diagnosis [O.R.: 0.554; P = 0.041]. Conversion rate among APOE4 carriers with the HMGCR’s G negative allele was markedly reduced [from 76% to 26.97%] to levels similar to APOE4 non-carriers [27.14%], which strongly indicate protection. Conversion data from the independent ADNI cohort also showed significantly reduced MCI or AD conversion among APOE4 carriers with the protective A allele [P = 0.005]. In conclusion, HMGCR rs3846662 act as potent genetic modifier for AD risk, age of onset and conversion.
Keywords: Alzheimer’s disease, mild cognitively impaired, HMGCR, rs3846662, genetic association
INTRODUCTION
Alzheimer’s disease (AD) is an adult-onset chronic neurodegenerative disorder that occurs predominantly later in life. It is the commonest cause of dementia and represents the fourth most common cause of death in the developed world (1). The two most famous pathological features of AD are the extracellular senile plaques, primarily composed of Aβ peptides, and the intracellular neurofibrillary tangles (NFT), resulting from the truncation and/or hyperphosphorylation of the microtubule-stabilizing Tau proteins [reviewed in (2)]. In recent years, patients diagnosed with mild cognitive impairment (MCI), a transitional stage between normal ageing and AD (3, 4), received overwhelming attention from the AD scientific community. It is estimated from previous research that nearly 80% of amnesic MCI patients, the dominant MCI subtype with a primary memory component (5), will have converted to AD within the course of six years (6) at an annual conversion rate of 10–15% (7). Given the absence of curative treatment, elucidation of factors affecting conversion of MCI to AD represents one of the most challenging and urgent medical mysteries affecting our ageing population.
About 5% of all AD cases show an autosomal dominant inheritance (8), whereas a greater challenge lies in discovering the causes of the more common form of AD – dubbed sporadic AD. Indeed, the concordance rate of AD among identical twins was shown to vary from 60 to 72% (9, 10), highlighting the existence of interplay between genetic, environmental and health factors (11). Apolipoprotein E (APOE) encodes the main lipid carrier protein in the central nervous system (CNS) and is the most robustly and consistently associated gene with AD risk, with the ε4 (referred to here as APOE4) and ε2 (referred to here as APOE2) alleles substantially increasing and decreasing the risk level, respectively (12–17). The APOE4 allele is also known to increase the likelihood of cognitive impairments in clinically normal 50+ years old over time (18) and to precipitate conversion to AD among MCI patients (5). Despite being present in about 50% of AD cases, the APOE4 allele is neither necessary nor sufficient for the development of AD (12, 14, 15). The search for the identification of additional genes contributing to AD led to the identification of 695 candidate genes (19) of which a surprising number are directly involved in lipid metabolism at the level of transport, synthesis, storage and internalization of lipoproteins (20). These includes BIN1 (21), PICALM (22), ABCA7 (21) and CLU (22, 23). While the genetic heterogeneity of these large genome-wide association studies (GWAS) have an increase power to detect risk genes with smaller effect sizes, population-relevant signal will likely go undetected (24). The use of isolated populations with a few founders, such as the French Canadians of Quebec (25), reduces the genetic background noise and allows the detection of population-specific signals (24). Moreover, targeted testing of polymorphisms known to strongly associate with altered transcript levels may be a powerful way to identify genetic associations with diseases that would otherwise be difficult to detect (26). Here, we evaluate a functional polymorphism (rs3846662) in HMGCR for association with AD in an isolated population, the Quebec founder population (QFP) (25), and corroborate our findings in two other well-characterized cohorts: the ADCS (3) and the ADNI cohorts (27).
HMGCR is a strong functional AD candidate gene because it encodes the 3-hydroxy-3-methylglutaryl-CoA reductase enzyme, which serves as the rate-limiting step in cholesterol synthesis in all mammalian cells. Cholesterol requirement of most brain cells are met by two separate yet interrelated processes: synthesis by HMGCR and internalization of lipoproteins via the APOE/LDL receptor cascade (28, 29), which is compromise in AD. Alterations in lipid homeostasis are known to severely impair neuronal function and elicit neurodegenerative disorders such as Niemann-Pick type C disease, a fatal cholesterol storage disorder characterized by the presence of AD-like NFTs in the brain (30). Elevated plasma cholesterol levels are known among vascular risk factors of AD (31, 32). Although not a universal finding, treatment of hypercholesterolemia with HMGCR inhibitors (statins) in middle-aged individuals confers some level of neuroprotection against late-life development of AD (33–36). Additionally, statin treatment was shown to reduce the cerebrospinal fluid phospho-Tau content (37). This finding is consistent with the quasi-absence of cortical NFTs in autopsy-confirmed cognitively intact subjects who have used statins for several years as opposed to non-users (38). Furthermore, whole-genome scans of late-onset AD cases reported the presence of several linkage hot spots across the genome, including one in the vicinity of the HMGCR gene on chromosome 5 (19, 39). Accordingly, two recent studies found an association between HMGCR polymorphisms and AD (40, 41). One of HMGCR most important co-regulator of cholesterol synthesis, 3-hydroxy-3-methylglutaryl-CoA synthase (42), was also shown to be significantly associated with sporadic AD in stratified populations enriched in APOE4–negative subjects; pointing toward a possible cholesterol metabolism dysfunction in AD subjects born without the APOE4 allele risk factor.
Overall, studies provide evidences of an association between HMGCR and AD, but this association is not compelling because none of the twenty-one GWAS performed to date substantiated HMGCR as a susceptibility gene for AD (current as of January 31, 2014: http://www.alzgene.org/largescale.asp). Interestingly, HMGCR undergoes alternative splicing of exon 13, which encodes part de the catalytic domain of the enzyme. Two independent group reported the involvement of the intron 13 single nucleotide polymorphism (SNP) rs3846662 in regulation of HMGCR exon 13 skipping (43, 44) by altering the binding motif of a molecule that regulate HMGCR alternative splicing: heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) (45). Since the Δ13 transcript could not rescue HMGCR activity in UT2 HMGCR-deficient cells upon transfection (44), has been associated with lower levels of plasma LDL-cholesterol (LDL-C) (46) and is one of the single most informative molecular marker of LDL-C response to statins (43, 47, 48), the rs3846662 represented the ideal functional polymorphism to study in our three cohorts study. The rs3846662 is even more important to study in the context of AD and MCI given our original preliminary report of the presence of multiple genetic risk factors in the HMGCR gene and their impacts on AD pathology in a small cohort of autopsy-confirmed cases (49).
MATERIALS AND METHODS
Quebec founder population (QFP, cohort 1): HMGCR gene in controls and AD cases
Human Subjects Demographics
Patients demographic characteristics are summarized in Table 1. Definite diagnosis of AD was based on histopathological confirmation of AD according to NINCDS-ADRDA criteria (50), whereas controls had to be free of neurological or psychiatric diseases and, for autopsy-confirmed cases, of brain structural lesions (tangle and plaque indices reading < 20 /mm3 and < 10 /mm2, respectively). All subjects are from the so-called QFP (French Canadians of Quebec). This population (age-matched controls: N= 250 / autopsy-confirmed AD cases: N= 324) descended in genetic isolation from several thousand founders who emigrated from France in the 17th century (25). The demographic history of the QFP, which is characterized by population bottleneck, rapid population expansion, and little admixture, makes it a valuable resource for use in genetic studies (51). The population has been well characterized as having reduced genetic heterogeneity for Mendelian diseases (52). Age at death, age at recruitment and education are not significantly different between controls and autopsy-confirmed AD subjects. APOE genotypes distribution is similar to previously reported prevalence for Eastern Canadians (12), with a strong and significant enrichment of the APOE4 allele in autopsied AD cases (Table 1). All brain and blood tissues were obtained from the Douglas Hospital Brain Bank, Montreal, Canada. Post-mortem delays generally varied from 10 to 20 hrs and were matched for control and AD subjects.
Table 1.
Quebec Founder Population (QFP), Alzheimer’s disease cooperative study (ADCS) and Alzheimer’s disease neuroimaging initiative (ADNI) Demographics
| AD autopsy-Confirmed Cases | MCI patients | AD, MCI and elderly controls | ||||
|---|---|---|---|---|---|---|
| Control n = 250 |
AD n = 324 |
Non-converters n = 271 |
AD Converters n = 138 |
Non-converters n = 935 |
Converters n = 298 |
|
| Age at death/ recruitment | 75.5 ± 11.1 | 79.2 ± 8.3 | 71.56 ± 7.48 | 74.18 ± 6.54 | 75.02 ± 7.06 | 75.43 ± 6.56 |
| mean ± SD, y | ||||||
| Age at onset/conversion | — | 71.7 ± 8.9 | – | 75.4 ± 8.43 | - | 77.27 ± 6.8 |
| mean ± SD, y | ||||||
| Sex | 124 (50) | 210 (65) | 115 (38.7) | 69 (45) | 348 (39) | 97 (35) |
| No. (%) female | ||||||
| Schooling | 8.1 ± 4.0 | 8.9 ± 4.4 | 15.13 ± 2.86 | 14.67 ± 3.09 | 14.92 ± 4.74 | 14.90 ± 5.25 |
| mean ± SD, y | ||||||
| APOE3 allele | 0.76 | 0.56 | 0.611 | 0.536 | 0.71 | 0.59 |
| frequency | ||||||
| APOE4 allele | 0.11 | 0.38 | 0.354 | 0.428 | 0.24 | 0.39 |
| frequency | ||||||
| APOE2 allele | 0.13 | 0.06 | 0.035 | 0.036 | 0.05 | 0.03 |
| frequency | ||||||
| HMGCR A allele | 0.60 | 0.53 | 0.53 | 0.49 | 0.47 | 0.46 |
| frequency | ||||||
Abbreviations: AD: Alzheimer’s disease; y: year; SD: standard deviation.
DNA Extraction
DNA was extracted from brain tissues (AD and control cases) or blood lymphocytes (control cases) using the DNeasy tissue kit (from Qiagen) and automated DNA extraction (NA-1000; AutoGen, Holliston, MA, USA), respectively.
Sequencing of the HMGCR gene and mapping of rs3846662
Complete sequencing of the coding and non-coding regions of the HMGCR gene was performed in 30 autopsy-confirmed AD cases and 15 age-matched control subjects using the Applied Biosystem 3730xl DNA analyzer from the McGill Innovation Centre. Genotype profiling of intron 13 of HMGCR was performed with PCR followed by pyrosequencing (53). The intron 13 SNP (rs3846662) was amplified using a PCR approach, with the following primer pairs: forward biotin 5′-TTTGCCAGTTTAAAAATACATCAT-3′ and reverse 5′-TTGACCCAAAAGGTA-TCACTAATT - 3′. Genomic DNA (250–500 ng) was amplified with 0.2 pM of each primer, 1X PCR buffer (Qiagen kit), 0.4 mM dNTP, 1.25 mM MgCl2, 0.05% DMSO and 0.01U of Qiagen Taq polymerase. Amplification was carried out on a Biometra T professional Basic thermocycler (Biometra, Göttingen, Germany) with the following conditions for 35 cycles: 30 sec. at 95°C, 30 sec. at 50.9°C and 1 min. at 72°C. These 35 amplification cycles were preceded by a 3-minute hot start at 95°C and followed by a final 4-minute extension at 62°C to the last cycle. PCR products were visualized on a 1.2% agarose gel. The intron 13 SNP was subsequently determined via an established pyrosequencing protocol (53) with oligo-sequencing 5′-ACTCTTCTCATTGCCTTAC - 3′. The sequence to analyze was: C/TTATGATGTAT.
Alzheimer’s disease Cooperative study (ADCS, cohort 2): HMGCR gene in MCI subjects
Human Subjects Demographics
Patients demographic characteristics are summarized in Table 1. MCI patients recruited for the purpose of the present study took part in the three years follow up, double-blind ADCS (3) and provided written informed consent for AD-related genetic screening. Information about the study design, methods to determine MCI diagnosis as well as conversion to AD can be found in the published ADCS study (3). Age at recruitment and education was equivalent between AD converters and non-converters. Consistent with previous findings (6), APOE genotypes distribution was significantly different across groups, with a significant disproportion of APOE4 allele found in AD converters (Table 1).
DNA Extraction and mapping of rs3846662
DNA extraction from blood samples was performed using Qiagen kits as described in the published ADCS study (3). Mapping of rs3846662 in intron 13 was performed as described for the QFP cohort.
Alzheimer’s disease Neuroimaging Initiative (ADNI, cohort 3): HMGCR gene in a mix population of AD, MCI and cognitively intact elderly
Human Subjects Demographics, DNA Extraction and mapping of rs3846662
Patients demographic characteristics are summarized in Table 1. Genotyping information on elderly controls from the ADNI cohort was obtained via a genome-wide whole brain analysis including 620 901 SNPs using the Human 610-Quad Bead Chip (Illumina Inc., San Diego, CA). Genotype profiling of the intron 13 of the HMGCR gene was extracted from the open-access database and data from the 1233 individuals recruited during ADNI-I were compiled for further conversion-to-deteriorated cognitive level analyses. Information about the specific GWAS protocol used to obtain genotyping information can be found in a recent report by Shen and collaborators (27), whereas the full clinical data set used in the preparation of this article were obtained from the ADNI database (adni.loni.ucla.edu). ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 subjects but ADNI has been followed by ADNI-GO and ADNI-2. To date these three protocols have recruited over 1500 adults, ages 55 to 90, to participate in the research, consisting of cognitively normal older individuals, people with early or late MCI, and people with early AD. The follow up duration of each group is specified in the protocols for ADNI-1, ADNI-2 and ADNI-GO. Subjects originally recruited for ADNI-1 and ADNI-GO had the option to be followed in ADNI-2. For up-to-date information, see www.adni-info.org.
Statistical Analyses for the three cohorts
Binary logistic regressions were computed between HMGCR polymorphism and disease status in the QFP and ADCS cohorts. Stratification by gender and APOE genotype was performed on these cohorts using Wald statistics. Odds ratios for HMGCR and APOE polymorphisms were also calculated across cohorts.
Wilcoxon chi-square rank tests derived from Kaplan-Meier survival curves were used to contrast the effects of the different genetic variants of the HMGCR and APOE polymorphisms on the age of onset of AD in autopsy-confirmed AD cases from the QFP cohort. Finally, MCI-to-AD conversion rate during the three years ADCS study and conversion rate during the 48-months ADNI study was computed as a function of the HMGCR’s intron13 allele using Wilcoxon chi-square rank tests derived from Kaplan-Meier survival curves stratified by APOE genotype.
RESULTS
In order to identify AD specific SNPs within the HMGCR gene, a complete sequencing of the coding and non-coding regions of the HMGCR gene was performed in 30 autopsy-confirmed AD cases and 15 age-matched control subjects of the QFP. The relatively homogeneous environmental exposures and reduce genetic heterogeneity associated with this population (25, 51, 52) were likely to be advantageous for the study of AD, a disease resulting from genetic and environmental interplay. The use of autopsy-confirmed cases allowed us to further reduce the false-positive background noise normally seen in clinical diagnosis. This preliminary study failed to reveal any disease-specific genetic mutations in all 20 exons. A rare polymorphism in exon 15 was detected but did not differ in terms of group incidence between AD and age-matched control subjects. Mapping of the introns, on the other hand, turned out to be more interesting as the rs3846662 SNP in intron 13 of the HMGCR gene (A or G allele) was found to significantly associate with sporadic AD.
Table 1 summarizes the frequency distribution obtained in our QFP cohort that included 250 controls and 324 AD cases. The association between HMGCR’s intron 13 G-negative polymorphism (AA) and sporadic AD was found to be significant [O.R. = 0.694; P = .024] (Table 2). Stratification by gender revealed that this association is significant only in women [Women: O.R. = 0.521; P = 0.0028; Men: O.R. = 0.890; P = .686] as is the case for the well-known APOE2 allele benefit [Women: O.R. = 0.316, P<0.001; Men: O.R. = 0.679; P = .293] in the same group of subjects (Table 2).
Table 2.
Binary logistic regression between HMGCR rs3846662 and the AD status. The regression was performed in the autopsy-confirmed AD cases of the QFP cohort. Secondary analyses were performed in the same population but stratified by gender or APOE genotype, using Wald statistic. The odds ratios are also provided.
| Allele | Overall effect | Women | Men | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Sig. (2-tailed) | OR | N | Sig (2-tailed) | OR | N | Sig (2-tailed) | OR | |
| HMGCR-G- | 574 | 0.024* | 0.694 | 334 | 0.003* | 0.521 | 240 | 0.686 | 0.890 |
| APOE4 | 573 | 0.001** | 6.180 | 333 | 0.001** | 7.204 | 240 | 0.001** | 5.253 |
| APOE2 | 573 | 0.001** | 0.447 | 333 | 0.001** | 0.316 | 240 | 0.293 | 0.679 |
| Allele | Non-APOE4 carriers | APOE4 carriers | Non-APOE2 carriers | APOE2 carriers | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Sig. (2-tailed) | OR | N | Sig (2-tailed) | OR | N | Sig (2-tailed) | OR | N | Sig (2-tailed) | OR | |
| HMGCR-G- | 308 | 0.634 | 0.881 | 262 | 0.183 | 0.713 | 469 | 0.05* | 0.634 | 101 | 0.304 | 1.558 |
Abbreviations: OR: odds ratio; N: sample size; HMGCR-G- : G negative versus G positive genotype. Asterisks represent significant risk for AD at the 95% C.I. (*) or 99% C.I. (**) level.
Analyses of the impact of the HMGCR polymorphism on age of onset in AD is summarized in Figure 1. While we did not observe any effect of this variant on the age at death [Wilcoxon Survival test: X2 1, 293= 0.950; P = .330], a significant effect of a double dose of intron 13 A allele (G negative genotype) on age of onset was detected [Wilcoxon Survival test: X2 1, 289= 4.57; P = .024]. This HMGCR protective genotype exerted a strong impact in women [Wilcoxon Survival test: X2 1, 188= 6.09; P = .017] who exhibited a delayed age of onset of about 3.6 years. This age effect in G negative subjects was not found in men [Wilcoxon Survival test: X2 1, 100= 1.88; P = .170]. Analysis of the “A” variant dose effect on age of onset reveals a significant association (p <0.03) in women; particularly between the age of 60 and 80 (Supplementary Materials).
Figure 1. Age of onset in HMGCR rs3846662 intron 13 G negative vs G positive carriers.
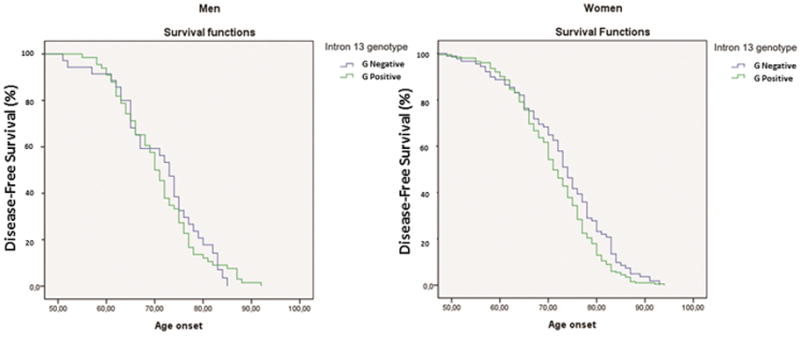
The joint table contrasts the effects of the different genetic variants of the HMGCR gene to those of APOE using a Wilcoxon chi-square rank test. N: sample size; X2= chi square; r: correlation; HMGCR G-: G negative genotype versus G positive. Asterisks represent significant association for AD at the 95% C.I. (*) or 99% C.I. (**) level.
As reported previously in several independent studies (16, 17), a very similar protective effect was observed with the APOE2 allele [Wilcoxon Survival test: X2 1, 289= 5.05; P = 0.019], women displaying again the most significant impact on age of onset [Wilcoxon Survival test: X2 1, 188= 6.43; P = 0.013] (Figure 1). In sharp contrast, the APOE4 allele in this study was strongly associated with an earlier age of onset of AD [Wilcoxon Survival test: X2 1, 289= 11.32; P < .001], particularly so in women [Wilcoxon Survival test: X2 1, 188= 7.48; P = 0.009].
In summary, studies performed in our autopsied-confirmed QFP subjects uncovered a protective association between HMGCR’s G negative genotype, AD risk and age of onset, particularly in women. The question was then to decipher whether or not the A allele could modulate the APOE4 risk in patients not affected by full-blown AD, such as in MCI patients.
In a follow-up study, a total of 409 MCI patients from the original Petersen et al., 2005 (3) MCI conversion trial (ADCS) were thus genotyped for APOE and HMGCR polymorphisms. Table 1 summarizes the frequency distribution obtained in MCI patients from the three years ADCS study that included 271 non-converters and 138 converters to AD. Conversion was determined by an expert panel from the ADCS study [refer to (3) for more details]. Associations between intron 13 A allele and AD conversion were not found to be significant [O.R. = 0.726; P =.129, table 3]. However, APOE4 genotype stratification revealed a significant protective effect in G negative carriers on AD conversion among APOE4-positive MCI patients [O.R. = 0.554; P=.041, table 3]. Interestingly, conversion rate among APOE4/HMGCR’s G negative subjects was markedly reduced [from 76% to 26.97% conversion to AD] to levels similar to APOE4 non-carriers [27.14% conversion to AD] at three years’ post-MCI diagnosis. This suggests that the HMGCR gene variant can markedly attenuate APOE4 risk, especially in the pre-dementia stages of the disease.
Table 3.
Binary logistic regression between HMGCR rs3846662 and the AD status. Regressions were computed in the MCI patients of the ADCS cohort. Secondary analyses were performed in the same cohort stratified by gender and APOE genotype using Wald Statistics. Odds ratios are also provided.
| Allele | Overall effect | Women | Men | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Sig. (2-tailed) | OR | N | Sig (2-tailed) | OR | N | Sig (2-tailed) | OR | |
| HMGCR-G- | 409 | 0.129 | 0.726 | 164 | 0.342 | 0.798 | 245 | 0.145 | 0.650 |
| APOE4 | 408 | 0.029* | 1.573 | 164 | 0.017* | 2.24 | 244 | 0.285 | 1.238 |
| APOE2 | 409 | 0.118 | 0.408 | 164 | 0.209 | 0.403 | 245 | 0.296 | 0.355 |
| Allele | Non-APOE4 carriers | APOE4 carriers | Non-APOE2 carriers | APOE2 carriers | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Sig. (2-tailed) | OR | N | Sig (2-tailed) | OR | N | Sig (2-tailed) | OR | N | Sig (2-tailed) | OR | |
| HMGCR-G- | 140 | 0.476 | 1.129 | 268 | 0.041* | 0.554 | 392 | 0.156 | 0.742 | 17 | 0.579 | 0.545 |
Abbreviations: OR: odds ratio; N: sample size; HMGCR-G- : G negative versus G positive genotype. Asterisks represent significant risk for AD at the 95% C.I. (*) level.
To extend and replicate the conversion dataset in the ADCS cohort, we decided to examine the conversion rate from normal controls/MCI/AD to MCI/AD in the ADNI cohort as a function of HMGCR’s G allele (positive vs negative) status and APOE stratification. To this end, we used genotyping data from ADNI that included 1233 individuals who were followed over a period of 48 months for conversion to MCI or AD. Table 1 summarizes the frequency distribution obtained in this mix population of cognitively intact, MCI and AD subjects that included 935 non-converters and 298 converters. Consistent with the abovementioned ADCS’ findings, associations between G negative status and MCI/AD conversion were not found to be significant [O.R. = 1.019; P =.908]. However, the G negative polymorphism exerted a very significant protective effect specific to APOE4 carriers [X21, 756= 7.751; P =.005] (Figure 2). Analysis of senile plaques and neurofibrillary tangles densities in a cohort of 118 autopsied AD cases derived from our eastern Canadian population isolate reveals that the reported apoE4-mediated increases in hippocampal and cortical plaques and tangles density in AD is actually prevented in G negative carriers as opposed to G positive subjects (supplemental material); consistent with the protective role of G negative polymorphism on age of onset and conversion rate. As initially hypothesized, these confirmatory results clearly indicate that APOE and HMGCR genes are interdependently modulating conversion to MCI or AD among an at-risk cognitively intact or MCI population.
Figure 2. Kaplan–Meier Estimates of the Rate of Progression from Normal to Mild Cognitive Impairment (MCI) and Alzheimer’s Disease (AD).
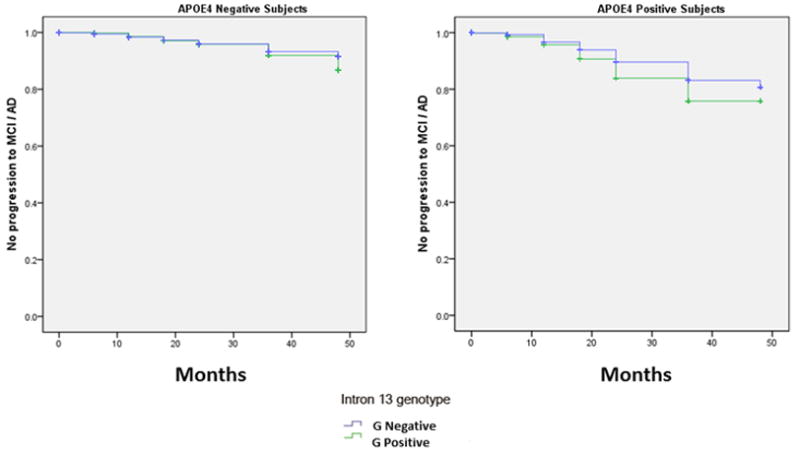
Conversion rate, stratified by APOE genotype, among subjects from the ADNI cohort as a function of the HMGCR intron13 genotype (APOE4 positive subject, P = 0.005). AA: G negative genotype; AG/GG: G positive genotype.
DISCUSSION
The association between sporadic AD and the HMGCR’s rs3846662 G negative status in this three cohorts study clearly identify the HMGCR gene as one of the most important and common protective variant ever identified for sporadic AD, second only to APOE2. It provides us with a novel lead explanation as for the discrepancies between retrospective and prospective studies dealing with the potential benefit of statins in sporadic AD. Indeed, several retrospective cross-sectional observational studies have shown that statins can reduce by up to 70% the risk of developing AD (33, 35). However, results of prospective studies have been inconsistent (54–56), and recent double blind placebo-controlled clinical trials in mild-to-moderate AD using simvastatin and atorvastatin for at least 6 months failed to show disease stabilization or improvement (57, 58), mostly due to a marked treatment response heterogeneity. This suggests that for statins to reduce risk, it must be taken during a certain critical period and for a certain length of time, preferably years prior to the expected onset of the disease. Corroborating this conclusion, the protective HMGCR’s G negative polymorphism was shown to modulate APOE4 risk in cognitively intact and MCI subjects and, to delay age of onset of AD by 3.6 years. These findings are consistent, at least in part, with a recent small case-control study which reported an interaction between the rs3761740 A allele in the promoter region of the HMGCR gene, the APOE E4 allele and an altered risk of AD (OR = 2.41; 95% CI = 0.93–6.22) (60). Analysis of the allelic distribution of this promoter variant and the rs3846662 SNP examined in the present study reveals a potent linkage disequilibrium (probability: 0.97) between the two variants in our population isolate from eastern Canada which could very well explain the complementary nature of findings reported by the two research teams (data not shown).
Whether the protective effect of statins on AD risk is mediated through reduction of vascular risk factors or through the direct modulation of CNS cholesterol homeostasis remains to be clarify. Interestingly, Wolozin and colleagues reported that statins neuroprotective properties sharply differed as a function of lipids solubility (36), an observation recently replicated in a secondary analysis of the “preventive” Ginkgo Evaluation of Memory Study (59). These studies found that the more lipid-soluble (i.e. simvastatin) HMGCR inhibitors exhibited high protective effects as opposed to the more lipophobic and less likely to cross the blood-brain-barrier statins (i.e. atorvastatin), which exhibited little to no protective effect. These observations would thus favor the hypothesis that statin mediates its neuroprotective effect through direct modulation of CNS cholesterol homeostasis. Unfortunately, none of the statin studies examined the contribution of the genetic polymorphisms of HMGCR and APOE on the extent of the protective effect.
Combined with the results of the genome-wide screening on chromosome 5 (19, 39), our findings clearly point toward a potential role of the HMGCR in the etiopathology of AD. Our study indicates that carriers of the intron 13 rs3846662 variant display a protective effect that resemble in size and gender what has been reported for APOE2 in humans. The similarity between the genetic association of APOE, the brain’s most important cholesterol transporter, and HMGCR, the rate-limiting step in cholesterol synthesis in the brain, is revealing to be quite interesting. On one hand, APOE4 is a risk factor that precipitates age of onset, markedly so in women, whereas the APOE2 variant as well as the HMGCR’s G negative polymorphism both delay age of onset of AD, more so, in women. Furthermore, APOE is perceived by lipid neurobiologists as a key extracellular lipid transport protein, while HMGCR, which is localized in the endoplasmic reticulum, is primarily an intracellular, organelle-specific protein that regulates intracellular lipid production. It is thus quite conceivable that these two proteins actually have complementary roles in the maintenance of local brain cholesterol homeostasis, particularly in presence of neurodegeneration or damage.
In conclusion, this three cohorts study provides strong evidence that HMGCR is a genetic modifier for risk, age of onset and MCI conversion to AD. Converging evidence have now confirmed the involvement of rs3846662 SNP in HMGCR exon 13 skipping in peripheral cells in vivo, the A allele being associated with an increased in exon 13 skipping (44). As reflected by its association with decreased LDL-C levels (46), the rs3846662 A allele is associated with decreased HMGCR activity. This finding is in accordance with the blunted response to statin therapy observed in G negative (or AA allele) carriers (43, 47, 48). Given that these findings were obtained mostly in the periphery, whether the rs3846662 is as important for the CNS needs to be substantiated. Studies addressing if the rs3846662 modulates mRNA splicing, protein HMGCR levels and activity in the human brain are currently underway in autopsied human brains.
Supplementary Material
Acknowledgments
This study was supported in part by the Natural Sciences and Engineering Research Council of Canada (JP), J.L. Levesque Foundation (JP) and by the Canadian Institutes of Health Research (VL/LDB/JP). The authors would also like to thank Mrs. Danielle Cécyre at the Douglas Institute/ Bell Canada Brain Bank for providing human brain tissues. Data collection and sharing for this project was supported by the ADNI National Institutes of Health (NIH) grant U01 AG024904 (PI: Michael W Weiner, MD, VA Medical Center and University of California, San Francisco, CA, USA). Funding sources for ADNI include the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering, the US Food and Drug Administration, the non-profit partners of the Alzheimer’s Association, the Alzheimer’s Drug Discovery Foundation and the Dana Foundation, and the following private sector contributors: Abbott, AstraZeneca AB, Amorfix, Bayer Schering Pharma AG, Bioclinica Inc, Biogen Idec, Bristol–Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, Innogenetics, IXICO, Janssen Alzheimer Immunotherapy, Johnson and Johnson, Eli Lilly and Co, Medpace Inc, Merck and Co, Inc, Meso Scale Diagnostic & LLC, Novartis AG, Pfizer Inc, F Hoffman–La Roche, Servier, Synarc, Inc and Takeda Pharmaceuticals. Private sector contributions to ADNI are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education. The study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego, CA, USA and ADNI data are disseminated by the Laboratory of Neuro Imaging at the University of Los Angeles, CA, USA. Additional ADNI support comes from the NIH grants P30 AG010129, K01 AG030514 and U24 AG21886. We also wish to thank all the members and funders of the ADCS who participated in the original MCI study.
Footnotes
Conflict of interest: the authors report no conflict of interest.
References
- 1.Kalaria RN, Maestre GE, Arizaga R, Friedland RP, Galasko D, Hall K, et al. Alzheimer’s disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurol. 2008 Sep;7(9):812–826. doi: 10.1016/S1474-4422(08)70169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adlard PA, Cummings BJ. Alzheimer’s disease--a sum greater than its parts? Neurobiol Aging. 2004 Jul;25(6):725–733. doi: 10.1016/j.neurobiolaging.2003.12.016. discussion 743–726. [DOI] [PubMed] [Google Scholar]
- 3.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005 Jun 9;352(23):2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 4.Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, et al. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001 Mar;58(3):397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- 5.Petersen RC, Stevens JC, Ganguli M, Tangalos EG, Cummings JL, DeKosky ST. Practice parameter: early detection of dementia: mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001 May 8;56(9):1133–1142. doi: 10.1212/wnl.56.9.1133. [DOI] [PubMed] [Google Scholar]
- 6.Petersen RC, Smith GE, Ivnik RJ, Tangalos EG, Schaid DJ, Thibodeau SN, et al. Apolipoprotein E status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. Jama. 1995 Apr 26;273(16):1274–1278. [PubMed] [Google Scholar]
- 7.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999 Mar;56(3):303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997 Apr;20( 4):154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 9.Gatz M, Fratiglioni L, Johansson B, Berg S, Mortimer JA, Reynolds CA, et al. Complete ascertainment of dementia in the Swedish Twin Registry: the HARMONY study. Neurobiol Aging. 2005 Apr;26(4):439–447. doi: 10.1016/j.neurobiolaging.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Raiha I, Kaprio J, Koskenvuo M, Rajala T, Sourander L. Alzheimer’s disease in twins. Biomed Pharmacother. 1997;51(3):101–104. doi: 10.1016/s0753-3322(97)86906-5. [DOI] [PubMed] [Google Scholar]
- 11.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006 Feb;63(2):168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 12.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993 Sep 18;342(8873):697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 13.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993 Mar 1;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993 Aug 13;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 15.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 16.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994 Jun;7(2):180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 17.Benjamin R, Leake A, McArthur FK, Ince PG, Candy JM, Edwardson JA, et al. Protective effect of apoE epsilon 2 in Alzheimer’s disease. Lancet. 1994 Aug 13;344(8920):473. doi: 10.1016/s0140-6736(94)91804-x. [DOI] [PubMed] [Google Scholar]
- 18.de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET) Proc Natl Acad Sci U S A. 2001 Sep 11;98(19):10966–10971. doi: 10.1073/pnas.191044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, et al. Results of a high-resolution genome screen of 437 Alzheimer’s disease families. Hum Mol Genet. 2003 Jan 1;12(1):23–32. doi: 10.1093/hmg/ddg007. [DOI] [PubMed] [Google Scholar]
- 20.Wollmer MA. Cholesterol-related genes in Alzheimer’s disease. Biochim Biophys Acta. 2010 Aug;1801(8):762–773. doi: 10.1016/j.bbalip.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011 May;43(5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009 Oct;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009 Oct;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 24.Bettens K, Sleegers K, Van Broeckhoven C. Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum Mol Genet. 2010 Apr 27; doi: 10.1093/hmg/ddq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laberge AM, Michaud J, Richter A, Lemyre E, Lambert M, Brais B, et al. Population history and its impact on medical genetics in Quebec. Clin Genet. 2005 Oct;68(4):287–301. doi: 10.1111/j.1399-0004.2005.00497.x. [DOI] [PubMed] [Google Scholar]
- 26.Carrasquillo MM, Belbin O, Zou F, Allen M, Ertekin-Taner N, Ansari M, et al. Concordant association of insulin degrading enzyme gene (IDE) variants with IDE mRNA, Abeta, and Alzheimer’s disease. PLoS One. 2010;5(1):e8764. doi: 10.1371/journal.pone.0008764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen L, Kim S, Risacher SL, Nho K, Swaminathan S, West JD, et al. Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in MCI and AD: A study of the ADNI cohort. Neuroimage. 2010 Nov 15;53(3):1051–1063. doi: 10.1016/j.neuroimage.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poirier J. Apolipoprotein E, cholesterol transport and synthesis in sporadic Alzheimer’s disease. Neurobiol Aging. 2005 Mar;26(3):355–361. doi: 10.1016/j.neurobiolaging.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Yu C, Youmans KL, LaDu MJ. Proposed mechanism for lipoprotein remodelling in the brain. Biochim Biophys Acta. 2010 Aug;1801(8):819–823. doi: 10.1016/j.bbalip.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohm TG, Treiber-Held S, Distl R, Glockner F, Schonheit B, Tamanai M, et al. Cholesterol and tau protein--findings in Alzheimer’s and Niemann Pick C’s disease. Pharmacopsychiatry. 2003 Sep;36(Suppl 2):S120–126. doi: 10.1055/s-2003-43060. [DOI] [PubMed] [Google Scholar]
- 31.Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet. 1997 Jan 18;349(9046):151–154. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- 32.Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, et al. The aging brain and cognition: contribution of vascular injury and abeta to mild cognitive dysfunction. JAMA Neurol. 2013 Apr;70(4):488–495. doi: 10.1001/2013.jamaneurol.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000 Nov 11;356(9242):1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 34.Rockwood K, Kirkland S, Hogan DB, MacKnight C, Merry H, Verreault R, et al. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol. 2002 Feb;59(2):223–227. doi: 10.1001/archneur.59.2.223. [DOI] [PubMed] [Google Scholar]
- 35.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000 Oct;57(10):1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 36.Wolozin B, Wang SW, Li NC, Lee A, Lee TA, Kazis LE. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007;5:20. doi: 10.1186/1741-7015-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riekse RG, Li G, Petrie EC, Leverenz JB, Vavrek D, Vuletic S, et al. Effect of statins on Alzheimer’s disease biomarkers in cerebrospinal fluid. J Alzheimers Dis. 2006 Dec;10(4):399–406. doi: 10.3233/jad-2006-10408. [DOI] [PubMed] [Google Scholar]
- 38.Li G, Larson EB, Sonnen JA, Shofer JB, Petrie EC, Schantz A, et al. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology. 2007 Aug 28;69(9):878–885. doi: 10.1212/01.wnl.0000277657.95487.1c. [DOI] [PubMed] [Google Scholar]
- 39.Kehoe P, Wavrant-De Vrieze F, Crook R, Wu WS, Holmans P, Fenton I, et al. A full genome scan for late onset Alzheimer’s disease. Hum Mol Genet. 1999 Feb;8(2):237–245. doi: 10.1093/hmg/8.2.237. [DOI] [PubMed] [Google Scholar]
- 40.Porcellini E, Calabrese E, Guerini F, Govoni M, Chiappelli M, Tumini E, et al. The hydroxy-methyl-glutaryl CoA reductase promoter polymorphism is associated with Alzheimer’s risk and cognitive deterioration. Neurosci Lett. 2007 Apr 6;416(1):66–70. doi: 10.1016/j.neulet.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Rodriguez E, Mateo I, Infante J, Llorca J, Garcia-Gorostiaga I, Vazquez-Higuera JL, et al. Interaction between HMGCR and ABCA1 cholesterol-related genes modulates Alzheimer’s disease risk. Brain Res. 2009 Jul 14;1280:166–171. doi: 10.1016/j.brainres.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 42.Wollmer MA, Sleegers K, Ingelsson M, Zekanowski C, Brouwers N, Maruszak A, et al. Association study of cholesterol-related genes in Alzheimer’s disease. Neurogenetics. 2007 Aug;8(3):179–188. doi: 10.1007/s10048-007-0087-z. [DOI] [PubMed] [Google Scholar]
- 43.Medina MW, Gao F, Ruan W, Rotter JI, Krauss RM. Alternative splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase is associated with plasma low-density lipoprotein cholesterol response to simvastatin. Circulation. 2008 Jul 22;118(4):355–362. doi: 10.1161/CIRCULATIONAHA.108.773267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burkhardt R, Kenny EE, Lowe JK, Birkeland A, Josowitz R, Noel M, et al. Common SNPs in HMGCR in micronesians and whites associated with LDL-cholesterol levels affect alternative splicing of exon13. Arterioscler Thromb Vasc Biol. 2008 Nov;28(11):2078–2084. doi: 10.1161/ATVBAHA.108.172288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu CY, Theusch E, Lo K, Mangravite LM, Naidoo D, Kutilova M, et al. HNRNPA1 regulates HMGCR alternative splicing and modulates cellular cholesterol metabolism. Hum Mol Genet. 2013 Sep 17; doi: 10.1093/hmg/ddt422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009 Jan;41(1):47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chasman DI, Posada D, Subrahmanyan L, Cook NR, Stanton VP, Jr, Ridker PM. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA. 2004 Jun 16;291(23):2821–2827. doi: 10.1001/jama.291.23.2821. [DOI] [PubMed] [Google Scholar]
- 48.Krauss RM, Mangravite LM, Smith JD, Medina MW, Wang D, Guo X, et al. Variation in the 3-hydroxyl-3-methylglutaryl coenzyme a reductase gene is associated with racial differences in low-density lipoprotein cholesterol response to simvastatin treatment. Circulation. 2008 Mar 25;117(12):1537–1544. doi: 10.1161/CIRCULATIONAHA.107.708388. [DOI] [PubMed] [Google Scholar]
- 49.Dea D, Théroux L, Legault V, Leduc V, Poirier J. HMG-COA reductase as a risk factor and modulator of Alzheimer pathology; 11th International Conference of Alzheimer’s Disease and Related Disorders; 2010; Hawai, USA: Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association; 2010. p. S191. [Google Scholar]
- 50.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985 Nov;42(11):1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 51.Gagnon A, Toupance B, Tremblay M, Beise J, Heyer E. Transmission of migration propensity increases genetic divergence between populations. Am J Phys Anthropol. 2006 Apr;129(4):630–636. doi: 10.1002/ajpa.20330. [DOI] [PubMed] [Google Scholar]
- 52.Betard C, Kessling AM, Roy M, Chamberland A, Lussier-Cacan S, Davignon J. Molecular genetic evidence for a founder effect in familial hypercholesterolemia among French Canadians. Hum Genet. 1992 Mar;88(5):529–536. doi: 10.1007/BF00219339. [DOI] [PubMed] [Google Scholar]
- 53.Royo JL, Hidalgo M, Ruiz A. Pyrosequencing protocol using a universal biotinylated primer for mutation detection and SNP genotyping. Nat Protoc. 2007;2(7):1734–1739. doi: 10.1038/nprot.2007.244. [DOI] [PubMed] [Google Scholar]
- 54.Li G, Higdon R, Kukull WA, Peskind E, Van Valen Moore K, Tsuang D, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology. 2004 Nov 9;63(9):1624–1628. doi: 10.1212/01.wnl.0000142963.90204.58. [DOI] [PubMed] [Google Scholar]
- 55.Rea TD, Breitner JC, Psaty BM, Fitzpatrick AL, Lopez OL, Newman AB, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005 Jul;62(7):1047–1051. doi: 10.1001/archneur.62.7.1047. [DOI] [PubMed] [Google Scholar]
- 56.Haag MD, Hofman A, Koudstaal PJ, Stricker BH, Breteler MM. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J Neurol Neurosurg Psychiatry. 2009 Jan;80(1):13–17. doi: 10.1136/jnnp.2008.150433. [DOI] [PubMed] [Google Scholar]
- 57.Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease. LEADe. Neurology. 2010 Mar 3;74:956–964. doi: 10.1212/WNL.0b013e3181d6476a. [DOI] [PubMed] [Google Scholar]
- 58.Arvanitakis Z, Knopman DS. Clinical trial efforts in Alzheimer disease. Why test statins? Neurology. 2010 Mar 3;74:945–946. doi: 10.1212/WNL.0b013e3181d6479a. [DOI] [PubMed] [Google Scholar]
- 59.Bettermann K, Arnold AM, Williamson J, Rapp S, Sink K, Toole JF, et al. Statins, risk of dementia, and cognitive function: secondary analysis of the ginkgo evaluation of memory study. J Stroke Cerebrovasc Dis. 2011 Aug;21(6):436–444. doi: 10.1016/j.jstrokecerebrovasdis.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keller L, Murphy C, Wang HX, Fratiglioni L, Olin M, Gafvels M, Bjorkhem I, Graff C, Meaney S. A functional polymorphism in the HMGCR promoter affects transcriptional activity but not the risk for Alzheimer disease in Swedish populations. Brain Research. 2010;1344:185–191. doi: 10.1016/j.brainres.2010.04.073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.