

Abstract
Background
O-Fucose is added to cysteine-rich domains called Thrombospondin type 1 repeats (TSRs) by Protein O-fucosyltransferase 2 (POFUT2) and is elongated with glucose by β3-glucosyltransferase (B3GLCT). Mutations in B3GLCT result in Peters Plus Syndrome (PPS), an autosomal recessive disorder characterized by eye and other developmental defects. Although 49 putative targets are known, the function of the disaccharide and its role in PPS remain unexplored.
Results
Here we show that while POFUT2 is required for secretion of all targets tested, B3GLCT only affects the secretion of a subset, consistent with the observation that B3GLCT mutant phenotypes in PPS patients are less severe than embryonic lethal phenotypes of Pofut2-null mice. O-Glycosylation occurs co-translationally, as TSRs fold. Mass spectral analysis reveals that TSRs from mature, secreted protein are stoichiometrically modified with the disaccharide, while TSRs from protein still folding in the ER are partially modified, suggesting that O-glycosylation marks folded TSRs and promotes ER exit. In vitro unfolding assays demonstrate that fucose and glucose stabilize folded TSRs in an additive manner. In vitro refolding assays under redox conditions showed that POFUT2 recognizes, glycosylates, and stabilizes the folded form of TSRs, resulting in a net acceleration of folding.
Conclusions
While known ER quality control machinery rely on identifying and tagging unfolded proteins, we find that POFUT2 and B3GLCT mediate a non-canonical ER quality control mechanism that recognizes folded TSRs and stabilizes them by glycosylation. Our findings provide a molecular basis for the defects observed in PPS and potential targets that contribute to the pathology.
Introduction
Thrombospondin type I repeats (TSR) are small protein domains characterized by the presence of six cysteines forming three conserved disulfide bonds. Seventy five percent of predicted TSRs contain the consensus sequence, CXX(S/T)CXXG, for a rare modification called O-fucosylation catalyzed by Protein O-fucosyltransferase 2 (POFUT2) [1]. POFUT2 is localized to the endoplasmic reticulum (ER), and in vitro only O-fucosylates properly folded substrates [1–3]. In cell culture, O-fucosylation is required for the secretion of two targets, ADAMTS13 and ADAMTSL1 [4, 5]. These observations led to hypothesis that POFUT2 plays a role in folding and/or quality control of proteins that contain TSRs [1, 2, 6].
TSRs with the POFUT2 consensus sequence are found in 49 proteins including secreted matrix proteins such as thrombospondin-1 and -2 (TSP1, 2), and all members of the ADAMTS and ADAMTSL families [7]. Several POFUT2 targets have known physiological functions, and mutations in several are associated with human pathologies [8]. Loss-of-function mutations in mouse Pofut2 result in early embryonic lethality that likely results from defects in one or more POFUT2 target proteins [7].
The O-fucose on TSRs can be further extended with the addition of glucose by β3-glucosyltransferase (B3GLCT) to form a Glcβ1-3Fuc disaccharide [6, 9]. The Glcβ1-3Fuc disaccharide has been confirmed on several putative targets, including ADAMTS5, ADAMTS13, ADAMTSL1, TSP1, TSP2, F-spondin and Properdin [4, 5, 8, 10–12]. Mutations in human B3GLCT cause a rare developmental disorder called Peters plus syndrome (PPS) characterized by short stature, anterior eye chamber defects, cleft palate/upper-lip amongst others [13–15]. Most disease-specific mutations in B3GLCT result in alternative splicing of the mRNA, which makes non-functional protein [15], although more recently missense and truncation mutations have also been identified [16]. Analysis of Properdin from serum of PPS patients shows a total loss of Glc from the TSRs, suggesting these patients are essentially null for B3GLCT [14].
Though the involvement of B3GLCT in PPS has been known for many years, the molecular details for how these rare post-translational modifications affect protein function are poorly understood. In this work, we use cell-based and in vitro assays to gain insight into why addition of the Glcβ1-3Fuc disaccharide to TSRs is important for their function. Our results confirm that while POFUT2 is required for secretion of all targets tested, B3GLCT affects secretion of targets to varying extents. We propose that POFUT2 and B3GLCT mediate a novel quality control mechanism for recognizing, modifying, and stabilizing a folded structure. Since PPS is less severe than the Pofut2 null phenotype, we hypothesize that defects in only a subset of POFUT2 targets contribute to the PPS pathology.
Results
POFUT2 and B3GLCT are required for the secretion of their substrates to different extents
Embryonic lethality of Pofut2 mice is seen around E7.5 and all but ten POFUT2/B3GLCT targets are expressed at this stage [7]. Prior work has demonstrated that siRNA knockdown of POFUT2 results in loss of ADAMTS13 secretion. Since ADAMTS13 is not expressed at E7.5, other targets must contribute to the Pofut2 phenotype. To examine whether the secretion defect seen in ADAMTS13 extends to other targets, we looked at the effects of POFUT2 siRNA knockdown in HEK293T cells expressing TSR1-3 from TSP1 (called TSP1(TSR1-3) henceforth), ADAMTSL1 and ADAMTSL2 (Figure 1A). Reducing POFUT2 severely impacted the secretion of all of these proteins (Figure 1B and C). In contrast, knocking down B3GLCT had a more selective effect, with very little impact on ADAMTS13 secretion but significant reduction in secretion of ADAMTSL1, ADAMTSL2, and TSP1(TSR1-3) (Figure 1). Taken together, these results suggest that POFUT2 is required for secretion of proteins with TSRs containing the consensus sequence while B3GLCT is essential for trafficking a subset of POFUT2 targets.
Figure 1. B3GLCT is required for secretion of some but not all POFUT2 targets.
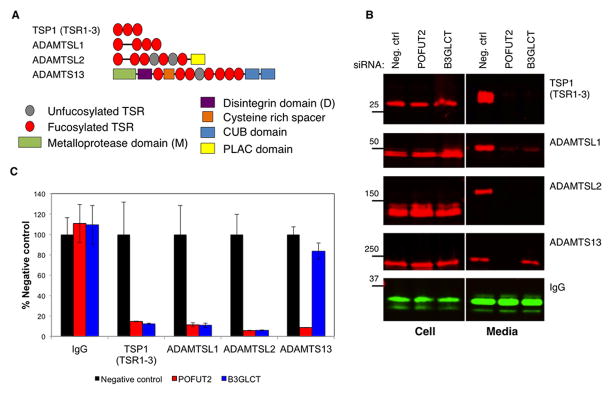
A) Domain map of POFUT2 targets tested.
B) siRNA specific for POFUT2, B3GLCT or non-specific control were co-transfected into HEK293T cells with plasmids encoding tagged POFUT2 targets (ADAMTS13, ADAMTSL1, ADAMTSL2 and TSP1(TSR1-3) or human IgG heavy chain. Media and cell lystates were evaluated by Western blot (red= anti-V5 or -myc; green= anti-IgG).
C) Quantitation of data from panel A. Data are mean +/− standard deviation (n=3).
O-Fucosylation of TSR-containing proteins occurs co-translationally in the ER
Although POFUT2 localizes to the ER, the source of the donor substrate, GDP-fucose, in the ER is unclear, at least in mammals. Hence it remains unknown where O-fucosylation occurs. If O-fucosylation by POFUT2 is part of a quality control pathway, addition of fucose to TSRs would be an ER-associated event. Both POFUT2 and B3GLCT are soluble proteins that localize to the ER [1, 6, 9]. If GDP-fucose is present in the ER, POFUT2 could O-fucosylate TSRs co-translationally, playing an active role in the folding of TSRs. Alternatively, POFUT2 could bind substrates in the ER and shuttle to the ER-Golgi intermediate compartment (ERGIC) where there is a putative GDP-fucose transporter [17], or to the Golgi where there is a well-described GDP-fucose transporter [18]. Once fucosylated, substrates could move on in the secretory pathway while POFUT2 cycles back to the ER [3].
To differentiate these possibilities, we determined the sub-cellular location of O-fucosylation using a bioorthogonal analog of fucose, 6-alkynylfucose (6AF). The alkyne group can be covalently modified with azido-biotin using “click” chemistry for easy detection [19]. We used ADAMTS13 and ADAMTSL1 as model substrates. ADAMTS13 has 10 N-glycosylation sites and 7 O-fucosylation sites [4], while ADAMTSL1 has 1 N-glycosylation site in addition to its 4 O-fucosylation sites [5]. Using Endoglycosidase H (EndoH) sensitivity as a measure of N-glycan processing, we can determine whether cell-associated ADAMTS13 or ADAMTSL1 is in the ER or has progressed further through the secretory pathway. 6AF-labeled ADAMTS13 or ADAMTSL1 purified from HEK293T cells were subjected to digestion with either EndoH or Peptide N-glycanase F (PNGaseF). Western blot analysis of the digested samples (Figure 2A) showed that the bulk of cell-associated ADAMTS13 and ADAMTSL1 was sensitive to EndoH, confirming the ER localization of these proteins. PNGaseF served as a positive control since it cleaves all types of N-glycans. Analysis of ADAMTSL1 from media showed EndoH resistant, PNGaseF sensitive, O-fucosylated protein, consistent with the acquisition of complex-type modification in the Golgi (Figure 2A, shown for ADAMTSL1; ADAMTS13 levels are low in the medium making this analysis difficult). While these data suggest that O-fucosylation of TSRs occurs in the ER, they do not rule out the possibility that the protein is O-fucosylated in the ERGIC.
Figure 2. O-Fucosylation occurs in the ER and is at least partly co-translational.

A) 6AF-labeled ADAMTS13-V5-His6 or ADAMTSL1-myc-His6 was immunoprecipitated from cell lysates or Ni-NTA enriched from media (sufficient ADAMTS13 could not be obtained from media fractions). Purified proteins were subjected to EndoH or PNGaseF digest and analyzed by western blots with indicated antibodies (red= anti-V5 or -myc; green=Streptavidin).
B) Ribosomes were isolated from HEK293T cells transfected with either empty vector or plasmid encoding 3XFlag-ADAMTS13 and labeled with 6AF. Nascent polypeptides were detected with anti-Flag (red) and 6AF was detected with Streptavidin (green). * indicates background bands that appear in the empty vector controls.
To determine whether O-fucosylation of TSRs occurs before the protein moves to the ERGIC, we examined whether O-fucosylation is a co-translational process by analyzing the fucosylation status of ribosome-associated ADAMTS13. HEK293T cells cultured in the presence of 6AF were transfected with N-terminal 3XFlag-ADAMTS13 and treated briefly with cycloheximide (CHX) to block polypeptide elongation. Ribosomes were purified using sucrose cushion ultracentrifugation, and Western blot analysis of the ribosome fraction showed distinct bands of nascent polypeptides labeled with 6AF in ADAMTS13 transfected samples, indicating the presence of O-fucose (Figure 2B). The bands labeled with 6AF were above 50 kDa, which corresponds to the size of a polypeptide translated past the first TSR on ADAMTS13. Similar results were seen when HEK293T cells expressing ADAMTS13 were treated with puromycin, a translational inhibitor that covalently attaches to the growing polypeptide chain (Figure S1) [20]. Western blot analysis of ADAMTS13 immunopurified from HEK293T cells treated with puromycin showed that truncated polypeptides labeled with 6AF also react with the anti-puromycin antibody (see overlay). These results strongly argue that O-fucosylation is co-translational and occurs in the ER, and that O-fucosylation occurs while TSRs are folding.
Glycosylation marks folded TSRs and is required for ER exit
The results in Figure 2 suggest that majority of the cell-associated TSR-containing proteins are in the ER and that O-fucosylation occurs early in protein synthesis. To determine the efficiency of O-fucosylation on individual TSRs of ADAMTS13, we performed semi-quantitative mass spectral analysis on protein purified from conditioned media and cell lysates. Consistent with previously published results [4], we found that peptides from TSR5, 7, and 8 of mature ADAMTS13 purified from the medium were efficiently modified with the major glycoform being the disaccharide, Glcβ1-3Fuc (Figures 3A and S2A, Table S1). In contrast, a mixture of unmodified and glycosylated peptides were detected on ADAMTS13 isolated from cell lysates with the majority being unmodified (Figure 3A). Interestingly, the C-terminal site (TSR8) was more poorly modified than N-terminal sites (TSR5 and 7), suggesting that glycosylation of TSR8 occurs after glycosylation of the other TSRs. These results confirm that the cell-associated protein is only partially modified.
Figure 3. Properly folded TSRs are marked with O-fucose disaccharide in cells.

A) ADAMTS13 was purified from Media or Cell Lysate fractions of HEK293T cells transfected with plasmid encoding ADAMTS13. Purified ADAMTS13 was subjected to reduction/alkylation, trypsin digestion, and analysis by nano-LC-MS/MS as described in Methods. Extracted ion chromatograms (EICs) of the ions corresponding to unmodified (black), O-fucose mono- (red) and disaccharide (blue) glycoforms of the peptides containing the O-fucosylation site from TSR5, 7 and 8 derived from ADAMTS13 are shown. Details of the ions representing each glycoform used to prepare the EICs are listed in Table S1 and Figure S2A.
B) HEK293T cells transfected with 3XFlag-ADAMTS13 were lysed in the presence of 100 mM iodoacetamide. Aliquots from the cell lysates were analyzed by Western blot under non-reducing (top panel) and reducing (bottom panel) conditions. Treating the samples with reducing agents (bottom panel) led to complete reduction of ADAMTS13, making the two populations indistinguishable. Mature form of ADAMTS13 isolated from medium is provided as a control for fully folded protein (migration position marked with *).
C) EICs (prepared as in A) of peptides from unfolded aggregates (top of gel from B) and mostly folded (migrating with mature protein at * from B) cell-associated ADAMTS13 showing predominantly unmodified peptides from TSR5, 7 and 8 in the aggregate, but majority glycosylated peptide from TSR5 and 7 in the mostly folded fraction.
D) Lec13 CHO cells were transfected with plasmids encoding the tagged POFUT2 targets shown or IgG as a negative control. Each target was immunoprecipitated from cell lysates and western blotted with anti-tag antibodies or with anti-BiP. The cells were rescued by addition of 1mM fucose exogenously. Quantitation represents relative amounts of BiP/Protein with -Fuc normalized to 1. Note that BiP was not detected in the IgG immunoprecipitate. Data are mean +/− standard deviation from 3 independent experiments.
Since POFUT2 only modifies properly folded TSRs in vitro [2], we predicted that the unmodified peptides in cells would be from incompletely folded TSRs, whereas the glycosylated peptides would be from folded TSRs. To separate populations of ADAMTS13 with unfolded TSRs containing unpaired cysteines from properly folded TSRs, we lysed cells in the presence of excess iodoacetamide. Immunopurified ADAMTS13 was then analyzed by reducing and non-reducing SDS-PAGE (Figure 3B). The mature protein from the medium migrated at its expected size under both reducing and non-reducing conditions, showing it no longer has unpaired cysteines. Under reducing conditions, cell-associated protein migrated at the predicted size regardless iodoacetamide treatment. In the absence of iodoacetamide, all of the cell-associated protein ran as an aggregate at the top of the non-reducing gel, suggesting that all cell-associated ADAMTS13 has at least one unpaired cysteine. When cells were lysed in the presence of iodoacetamide, we observed that some of the protein migrates at the size of the mature protein on the non-reducing gel. This population likely has fewer unpaired cysteines (i.e. is more completely folded) than the aggregate at the top of the gel. Mass spectral analysis of these populations revealed that the majority of the peptides derived from the aggregate are not glycosylated (Figure 3C), consistent with the TSRs being mostly unfolded in this protein population. In contrast, peptides from the band corresponding to the band co-migrating with mature protein showed almost fully glycosylated TSR5 and 7, while TSR8 was still predominantly unmodified. This suggests that the TSRs in this protein population are mostly folded and will exit the cell upon the folding and glycosylation of TSR8. These data demonstrate that glycosylation serves as a marker for the folded TSRs within the cell and that glycosylation must be complete prior to ER exit.
The presence of a “mark” implies there may be receptor that recognizes the mark which would promote ER exit, or that the lack of the mark is recognized by some mechanism that retains the unmarked TSRs in the ER. We hypothesized that there could be either a lectin-like protein that recognizes O-fucose glycans and promotes their ER exit or there could be a chaperone/oxidoreductase that actively binds to unfolded TSRs and keeps them in the ER. To address these possibilities, we used GDP-mannose 4,6 dehydratase (GMD) mutant Lec13 CHO cells that cannot synthesize GDP-fucose de novo [21]. The mutant can be rescued by adding L-fucose exogenously to the cell culture medium, which is taken up by the salvage pathway and converted to GDP-fucose. In this system, we used ADAMTS13 as bait to look for interacting proteins in the presence of a thiol-cleavable cross-linker, dithiobis [succinimidyl propionate] (DSP). We observed enrichment of a ~80 KDa protein in the absence of fucose (Figure S2B), which we identified as binding immunoglobulin protein (BiP) using mass spectrometry. We confirmed this observation by co-immunoprecipitation of several proteins (without DSP) and Western blotting. Expression of TSR-containing proteins did not affect BiP levels compared to the IgG control regardless of the presence of fucose, suggesting UPR is not being induced in this situation (Figure S2C). All proteins tested co-precipitated more BiP in the absence of fucose than in its presence (Figure 3D). These results suggest that ER retention of these proteins in the absence of O-fucosylation is mediated by enhanced interactions with ER chaperones such as BiP.
Addition of the Glcβ1-3Fuc disaccharide stabilizes folded TSRs
The data above shows that O-fucosylation occurs during folding in the ER and validates that only properly folded TSRs are fucosylated in vivo. To determine how the addition of fucose and glucose could affect the folding of the TSR-containing proteins in cells, we evaluated the effect of the sugars on the stability of a folded TSR in vitro. Previous structural studies demonstrated that O-fucosylation does not considerably alter the structure of a TSR [6]. However, the possibility that O-fucosylation may lend thermodynamic stability to the TSR has not been explored. To test this, bacterially expressed TSR3 from TSP1 (TSP1-TSR3) [2] was modified with either fucose (Fuc-TSR) or the Glcβ1-3Fuc disaccharide (GlcFuc-TSR) in vitro using purified enzymes. The TSRs were then unfolded using low concentrations of guanidine hydrochloride and DTT. Differences in hydrophobicities of unfolded and folded isoforms allow analysis of the unfolding reaction by reverse phase HPLC. Sampling at various time points showed that the Fuc-TSR is more resistant to denaturation than the unmodified TSR (Figure 4A). The addition of the glucose further increases the resistance to chemical denaturation. Quantitation of the data in Figure 4A shows that the halfway point of the unfolding reaction, indicated by the intersection of the plots of unfolded and folded populations, increases with the addition of each sugar (Figure 4B). Since unfolding rate is a reflection of the equilibrium between the unfolded and folded states (Figure 4B, schematic), these results strongly argue that addition fucose and glucose stabilize folded TSRs in an additive manner and accelerate the net folding rate of TSRs.
Figure 4. Fucose and glucose stabilize folded TSRs in an additive manner.

A) Equivalent amounts of unmodified, Fuc-TSR and GlcFuc-TSR (all TSP1-TSR3) were incubated with denaturants in an in vitro melting reaction. Aliquots were removed at the indicated times and analyzed by reverse phase HPLC. TSR levels were monitored by absorbance at 214 nm (U= unfolded isoform; F= folded isoform). Note that Fuc-TSR and GlcFuc-TSR were not completely unfolded within the 80 min time course, so were completely denatured by incubating for 80 min in 30 mM DTT.
B) U and F were quantified by determining area-under-curve (AUC) for each population of TSR in the melting reaction in panel A. Values were normalized to U at t=80min for TSR and completely denatured Fuc-TSR and GlcFuc-TSR (30 mM DTT) set to 100%. Schematic on the right shows the unfolding reactions as equilibria, with weighted arrows representing the favored reaction (orange line=disulfide bond; red triangle= fucose; blue circle= glucose). Data are mean +/− standard deviation from 3 or more independent experiments.
POFUT2 accelerates the folding of TSRs in vitro
If O-fucosylation stabilizes folded TSRs, and POFUT2 only modifies properly folded TSRs [1], O-fucosylation should shift the folding equilibrium towards the folded state, accelerating the net folding rate. We tested this using an in vitro refolding assay with bacterially expressed TSP1-TSR3, recombinant POFUT2 and GDP-fucose. TSP1-TSR3 was completely denatured and then allowed to refold in the presence of redox agents. The reaction was sampled at various time points and folding intermediates were acid-trapped. These conditions allow creation of a folding equilibrium between unfolded, intermediates, and the folded forms of the TSR (U, I, and F, respectively, in Figure 5A). Addition of POFUT2 to the redox system did not have a significant effect on the rate of folding, but when the reaction was supplemented with both POFUT2 and GDP-fucose, the rate of formation of folded TSR significantly increased (Figure 5A). Quantitation of the unfolded TSR (U) and the folded TSR (F) across the different conditions and time points showed that while the amount of unfolded protein decreased at roughly the same rate, the rate of appearance of the folded TSR was much higher in the presence of POFUT2 and GDP-fucose (Figure 5C). When the reaction mixture was analyzed by mass spectrometry, the majority of the TSP1-TSR3 population was O-fucosylated 90 min into the reaction in the presence of POFUT2 and GDP-fucose (Figure S3A, Table S2). These results demonstrate that POFUT2 accelerates the net rate of TSR folding in the presence of GDP-fucose.
Figure 5. O-Fucosylation by POFUT2 accelerates the net folding rate of TSRs.

A) Bacterially expressed (unmodified) TSP1-TSR3 was denatured and refolded in vitro in the presence of redox agents (black), redox and POFUT2 (blue) or redox, POFUT2 and GDP-fucose (red). Aliquots were removed at the indicated times and analyzed using reverse-phase HPLC (U= unfolded TSR; F=folded TSR; I= folding intermediates).
B) In-vitro refolding assay (as in A) in the presence of GDP-fucose using POFUT1 (black), a closely related O-fucosyltransferase that modifies properly folded Epidermal Growth Factor-like repeats [37], as a negative control, wt POFUT2 (red) and the catalytically inactive POFUT2-E54A (blue).
C) Quantitation of unfolded and folded products by AUC in B shows the rate of disappearance of unfolded protein (top) and rate of formation of folded protein (bottom).
Though GDP-fucose alone does not have an effect on TSR refolding (Figure S3B), it appears to be a requirement in POFUT2-mediated refolding of TSP1-TSR3. It was necessary to distinguish between two possibilities: 1) that binding GDP-fucose was a structural necessity for substrate binding and/or recognition, or 2) that fucosyltransferase activity is required for the folding of TSP1-TSR3. The POFUT2-E54A mutant has previously been shown to have no fucosyltransferase activity ([22], Figure S3C). In vitro folding assays performed using wt and E54A mutant enzyme in the presence of GDP-fucose showed that fucosyltransferase activity is necessary for altering the rate of folding (Figure 5B, C). The E54A mutant did not affect the rate of folding of the TSR. These data suggest that both the catalytic activity and GDP-fucose are required for POFUT2 to affect folding. In addition, a homolog of POFUT2 with distinct specificity, POFUT1, had no effect on TSR folding (Figure S3D, 5C) demonstrating the specificity of this reaction. Finally, addition of GDP (not GDP-fucose) and wt POFUT2 also had no effect on folding (Figure S3E). Thus, fucosylation of the TSR by POFUT2 accelerates the rate of TSR folding, suggesting that in the absence of O-fucosylation, POFUT2 target proteins fold inefficiently leading to an accumulation of misfolded protein and reduced secretion.
Discussion
Nearly thirty percent of all known proteins have disulfide bonds [23]. The appropriate pairing of the cysteines in a crowded environment like the ER poses a challenge to the folding and quality control machinery. In this complex mixture, it makes strategic sense for the cell to have dedicated quality control mechanisms for different types of cysteine-rich motifs in addition to the generic network of oxidoreductases and chaperones. POFUT2 and B3GLCT fall into this category. Each TSR has six cysteines and most POFUT2 targets have multiple tandem TSRs. A protein such as ADAMTS13 has 8 TSRs with 46 cysteines (just in the TSRs) that must pair correctly. Our data suggests that the more TSRs found in a protein, the greater the need for a specialized quality control system such as described here (Figure S4). We propose that POFUT2 and B3GLCT assist in the quality control of TSR folding to help resolve this complexity.
The main function of the POFUT2-B3GLCT machinery is to recognize and modify properly folded TSRs, stabilizing the folded structure, possibly in a processive manner (from N- to C-terminal) (Figure 6A). The most N-terminal TSR will interact with chaperones (such as BiP) and oxidoreductases (labeled PFC in Figure 6) that assist in folding when it arrives in the ER allowing it to enter a folding equilibrium much like those observed during the in vitro folding of TSP1-TSR3 in Figure 5A (indicated by the double headed arrow in Figure 6A-i). POFUT2 (previously loaded with GDP-fucose) binds a correctly folded TSR isomer (Figure 6A-ii). O-Fucosylation of the TSR stabilizes the folded structure, driving the folding equilibrium forward (indicated by the thick arrow, Figure 6A-iii), accelerating the folding of the TSR. B3GLCT then adds glucose, further stabilizing the folded TSR, shifting the equilibrium more (indicated by the second thick arrow, Figure 6A-iv). The cycle is then repeated for the next TSR (Figure 6A-v) and so on until a chain of fully folded and glycosylated TSRs emerges (Figure 6A-vi). Fully O-glycosylated protein can then exit the ER because the PFC no longer associates with the folded TSRs. Once a TSR folds, hydrophobic patches will be buried and cysteines in disulfide bonds, thus the TSR will no longer interact with the chaperones and oxidoreductases of the PFC. In the absence of POFUT2, TSR folding will stall in the folding equilibrium, causing continued interactions with the PFC. Ultimately the misfolded protein would be degraded in the cell. Although O-fucosylation on its own may be sufficient to stabilize some TSRs (e.g. those from ADAMTS13, Figure 1), other TSRs require addition of glucose by B3GLCT for efficient folding and secretion (e.g. from ADAMTSL1, ADAMTSL2, TSP1(TSR1-3), Figure 1).
Figure 6. A model for POFUT2 in the folding of TSRs.
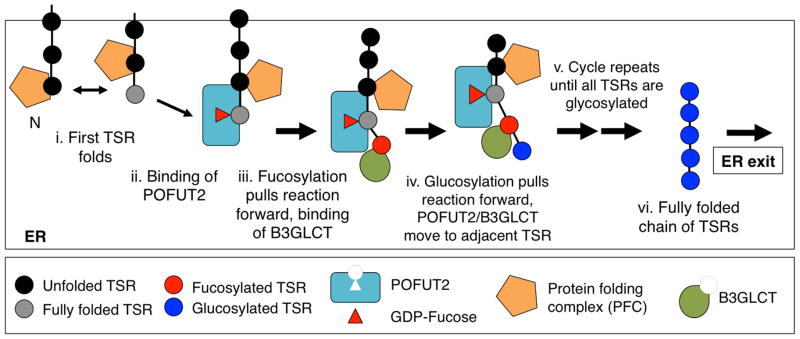
See discussion.
Recent structural studies indicate that in addition to the TSR binding site, POFUT2 has a hydrophobic patch that could interact with an adjacent TSR [22]. This would allow POFUT2 to physically separate adjacent TSRs and enable their interactions with the PFC (Figure 6A-ii).
Our results provide first insights into the role of B3GLCT in PPS. Although ADAMTS13 secretion was only minimally affected when B3GLCT was knocked down, secretion of several other proteins (ADAMTSL1, ADAMTSL2, TSP1 (TSR1-3) was profoundly reduced (Figure 1). Hess and co-workers demonstrated that serum levels of Properdin were reduced about 50% in PPS patients relative to controls [14]. These results suggest that addition of glucose affects secretion of proteins to different extents. The results also correlate nicely with the PPS phenotype. For instance, PPS patients do not show symptoms of ADAMTS13 deficiency, which results in a blood clotting disorder called Thrombotic thrombocytopenic purpura [24]. Similarly, PPS patients have no reported defects in activation of the alternative complement pathway, suggesting that serum levels of Properdin are sufficient [13]. However, patients with Geleophysic dysplasia (GD), caused by mutations in ADAMTSL2 resulting in reduced secretion [25], show several common symptoms with PPS patients such as short stature, unusual facial features and brachydactyly [13, 25]. Taken together, these independent observations suggest that PPS is a collective disorder of individual pathologies caused by secretion defects in a subset of B3GLCT target proteins.
The identification of O-fucosylated nascent proteins (Figure 2B) strongly argues in favor of GDP-fucose presence in the ER. Though an ER GDP-fucose transporter has been identified in Drosophila [18], the mammalian homolog of this protein has been demonstrated to have UDP-xylose, not GDP-fucose, transporter activity [26]. Possible models to explain GDP-fucose availability in the ER are retrograde transport of GDP-fucose from the ERGIC or Golgi, where there are known GDP-fucose transporters (Slc35C1 and C2 [17, 27]) or cycling of POFUT2 itself to these compartments where it could bind GDP-fucose. Of course, there still remains the possibility of an as yet unidentified ER-associated GDP-fucose transporter.
The fact that POFUT2 recognizes and modifies a folded structure distinguishes it from other ER quality control systems. The UDP-glucose: glycoprotein glucosyltransferase recognizes hydrophobic patches on unfolded proteins, modifying them so that they can participate in another round of folding with calnexin or calreticulin [28, 29]. In contrast, we propose that POFUT2 is able to selectively modify a properly folded TSR. O-Fucosylation of the TSR stabilizes the folded motif, preventing it from forming incorrect disulfide bonds with the adjacent newly synthesized TSR. The net result drives the folding equilibrium forward. Addition of glucose by B3GLCT further stabilizes the folded TSR, which appears to be required for proper folding of some proteins but not others. The crystal structure of TSR3 from TSP1 shows the O-fucosylated amino acid to be located in a loop preceding the first of two beta sheets [30]. Previous studies have suggested that the core N-linked N-acetylglucosamine stabilizes local loop structures in proteins, and stability is enhanced by addition of the mannoses [31]. The addition of fucose and glucose may have a similar effect on the loop they modify in TSRs.
Materials and Methods
Plasmids used in this study
The preparation of full-length pcDNA4.0 ADAMTS13-V5-His6 construct was described previously [4]. Truncation mutants were generated from this plasmid and ligated into pcDNA4.0 V5-His6 (Invitrogen). The N-terminal 3X-Flag tagged ADAMTS13 construct was generated by ligating a 3XFlag fragment containing HindIII/BamHI sites (amplified from p3XFlag-CMV (Sigma)) into pSecTag2 HygroC to generate an N-terminal 3XFlag-containing vector. Full length ADAMTS13 with XhoI overhangs was then ligated into this vector to generate the N-3XFlag-ADAMTS13 construct. Plasmids encoding Human IgG, TSP1(TSR1-3)-myc-His6, ADAMTSL1-myc-His6 and ADAMTSL2-myc-His6 constructs used are as described previously [5, 19, 32, 33]. Human POFUT2 constructs (wt and E54A) were a generous gift from Heinz Gut [22]. B3GLCT construct was obtained from RIKEN (Clone ID: G830038O03) and subcloned into pSecTag2 HygroC.
siRNA experiments
siRNA targeting POFUT2 was the same as used previously [4] and Stealth siRNA (Life Technologies) targeting β3GlcT were obtained (sense: 5′-CCUCCUUCAUCAGCUGGCUAAACAA, antisense: 5′-UUGUUUAGCCAGCUGAUGAAGGAGG). siRNA and plasmid DNA encoding proteins of interest were co-transfected using Lipofectamine 2000 (Life Technologies) in HEK293T cells according to the manufacturer’s protocol. The cells were then allowed to grow in OptiMEM for 48 h before harvesting media and cell fractions. Equal fractions of media and cell lysate were analyzed by western blotting. Imaging and quantitation was performed using an Odyssey 9120 infrared imaging system (Li-Cor).
Azide-alkyne cycloaddition (click reaction)
Reactions were performed as described before [19]. Media and cell lysate fractions were collected from cells metabolically labeled with 200 μM 6AF and tagged with Biotin (0.1 mM Azido-Biotin (CC Tools), 0.1 mM Tris-(benzyltriazolylmethyl)amine catalyst (AnaSpec), 1 mM copper sulfate, 2 mM sodium ascorbate) at room temperature for 1 h.
EndoH and PNGaseF digests
HEK293T cells were transiently transfected with ADAMTS13-V5-His6 or ADAMTSL1-myc-His6 and labeled with 200 μM 6AF for 48 h. Media samples were purified using Ni-NTA agarose (Qiagen) and lysates were immunopurified using anti-V5 (Invitrogen) or anti-myc (9E10). Samples were “clicked” as described above and treated with PNGaseF or EndoH as described [34].
Ribosome experiments
HEK293T cells transiently transfected with N-3XFlag-ADAMTS13 plasmid and labeled with 200 μM 6AF for 24 h, then treated with 100 μM cycloheximide (Sigma) for 15 min at 37°C. The cells were then lysed in 50 mM NH4HCl, 12 mM MgCl2, 50 mM Tris acetate and 0.5% NP-40. The lysates were passed through a 27.5 gauge syringe twice to thoroughly free any adherent ribosomes and debris is removed by centrifuging at 12,000 g (20 min at 4°C). The lysate was “clicked” (as described above) and loaded on a 34% sucrose cushion containing 50 mM NH4HCl, 12 mM MgCl2, 50 mM Tris acetate and centrifuged at 100,000 g for 3 h. The pellet was rinsed 3X with lysis buffer, resuspended in sample buffer, and analysed by western blotting (anti-Flag (Sigma), Streptavidin IRDye 800 (Rockland Immunochemicals)), and imaging performed with the Odyssey system.
Analysis of cell-associated ADAMTS13
HEK293T cells were transiently transfected with N-3XFlag-ADAMTS13 plasmid and allowed to grow for 48 h before being lysed in RIPA buffer containing 100 mM iodoacetamide (Sigma). The lysates were incubated on ice for 30 min to allow alkylation before insolubles were cleared by centrifuging at 20,000 g for 10 min. Following immunoprecipitation (Flag-M2 agarose beads, Sigma) and elution (3X Flag peptide, Sigma), the eluate was divided and run on reducing and non-reducing gels and detected by western blot analysis. Relevant bands were subjected to in-gel protease digestions and mass spectral analysis as described [8]. Semi-quantitative analysis of different glycoforms of each peptide was performed by generating Extracted Ion Chromatograms (EIC) as described [35].
Co-immunoprecipitation experiments
Lec13 CHO cells were grown in the presence or absence of 1 mM fucose and transiently transfected with plasmids encoding the indicated TSR-containing protein. 24 h after transfection, the cells were lysed and the TSR-containing protein was immunoprecipitated with antibodies against the respective tags. The eluates were analyzed on a reducing gel and probed with the antibodies indicated.
In-vitro unfolding assay
TSP1-TSR3 was generated in BL21 cells as described [2]. Fuc-TSR was synthesized by incubating TSP1-TSR3 (10 μM) with 200 μM GDP-fucose and POFUT2 in buffer containing 50 mM HEPES pH 6.8 overnight at 37°C. The fucosylated TSR was re-purified from the reaction mixture using reverse phase HPLC and used in a similar reaction with recombinant B3GLCT and UDP-glucose to generate GlcFuc-TSR. TSRs were incubated with 0.6 M guanidine hydrochloride and 5 mM DTT at room temperature. At specified time points, aliquots of the unfolding reaction were acid-trapped and analyzed by reverse phase HPLC as described in [36]. Folded and unfolded species were quantified by determining the area under the curve (AUC) for each species. For quantitation, the AUC for each peak was measured using curve symmetry, the background AUC subtracted, and calculated as a percentage of the starting material (total TSR at 0 min).
In-vitro folding assay
TSP1-TSR3 was denatured in 100 mM Tris pH 7.5, 6 M GnHCl and 10 mM DTT at room temperature for 1.5 h. Denaturants were removed using a Sephadex G-25 column (Pharmacia). The desalted protein was then allowed to refold in the presence of 25 mM GSSG, 50 mM GSH (Sigma) and combinations of equimolar recombinant POFUT2 and 200 μM GDP-fucose (final). At specified time points, aliquots of the reaction were taken and acid-trapped in 4% Trifluoroacetic acid (final). The samples were then analyzed by reverse phase HPLC as described in [36]. POFUT2 (wt and E54A) were expressed and purified from HEK293T cells as described in [22]. Quantitation was determined using AUC as described above.
Supplementary Material
Acknowledgments
We thank Dr. Pamela Stanley for providing the Lec13 CHO cell lines, Dr. Suneel Apte for the ADAMTSL2 construct, Dr. Wali Karzai and his lab members for guidance on ribosome purifications, Dr. Ari Helenius for the anti-puromycin antibody, Dr. Heinz Gut for hPOFUT2 constructs, Drs. Erwin London and Deborah Brown for helpful discussions, and all the members of the Haltiwanger lab for their input. This work was supported by NIH grants CA12307101.
Abbreviations
- 6AF
6-alkynylfucose
- B3GLCT
β3-glucosyltransferase
- DSP
Dithiobis [succinimidyl propionate]
- EIC
Extracted Ion Chromatogram
- EndoH
Endoglycosidase H
- ERGIC
ER-Golgi Intermediate Compartment
- LC
Liquid Chromatography
- MS
Mass spectrometry
- PFC
Protein Folding Complex
- PNGaseF
Peptide N-Glycanase F
- POFUT
Protein O-Fucosyltransferase
- TSP
Thrombospondin
- TSR
Thrombospondin Type I Repeat
Footnotes
Author contributions: DV and RSH designed the experiments and wrote the paper, and DV and HT performed the experiments. SSJ did preliminary experiments and EM provided valuable ideas and reagents. The authors declare no conflicts of interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. The Journal of biological chemistry. 2006;281:9393–9399. doi: 10.1074/jbc.M511975200. [DOI] [PubMed] [Google Scholar]
- 2.Luo Y, Nita-Lazar A, Haltiwanger RS. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. The Journal of biological chemistry. 2006;281:9385–9392. doi: 10.1074/jbc.M511974200. [DOI] [PubMed] [Google Scholar]
- 3.Luther KB, Haltiwanger RS. Role of unusual O-glycans in intercellular signaling. The international journal of biochemistry & cell biology. 2009;41:1011–1024. doi: 10.1016/j.biocel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ricketts LM, Dlugosz M, Luther KB, Haltiwanger RS, Majerus EM. O-fucosylation is required for ADAMTS13 secretion. The Journal of biological chemistry. 2007;282:17014–17023. doi: 10.1074/jbc.M700317200. [DOI] [PubMed] [Google Scholar]
- 5.Wang LW, Dlugosz M, Somerville RP, Raed M, Haltiwanger RS, Apte SS. O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. The Journal of biological chemistry. 2007;282:17024–17031. doi: 10.1074/jbc.M701065200. [DOI] [PubMed] [Google Scholar]
- 6.Kozma K, Keusch JJ, Hegemann B, Luther KB, Klein D, Hess D, Haltiwanger RS, Hofsteenge J. Identification and characterization of abeta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats. The Journal of biological chemistry. 2006;281:36742–36751. doi: 10.1074/jbc.M605912200. [DOI] [PubMed] [Google Scholar]
- 7.Du J, Takeuchi H, Leonhard-Melief C, Shroyer KR, Dlugosz M, Haltiwanger RS, Holdener BC. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Developmental biology. 2010;346:25–38. doi: 10.1016/j.ydbio.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonhard-Melief C, Haltiwanger RS. O-fucosylation of thrombospondin type 1 repeats. Methods in enzymology. 2010;480:401–416. doi: 10.1016/S0076-6879(10)80018-7. [DOI] [PubMed] [Google Scholar]
- 9.Sato T, Sato M, Kiyohara K, Sogabe M, Shikanai T, Kikuchi N, Togayachi A, Ishida H, Ito H, Kameyama A, et al. Molecular cloning and characterization of a novel human beta1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology. 2006;16:1194–1206. doi: 10.1093/glycob/cwl035. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez de Peredo A, Klein D, Macek B, Hess D, Peter-Katalinic J, Hofsteenge J. C-mannosylation and o-fucosylation of thrombospondin type 1 repeats. Molecular & cellular proteomics : MCP. 2002;1:11–18. doi: 10.1074/mcp.m100011-mcp200. [DOI] [PubMed] [Google Scholar]
- 11.Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, Peter-Katalinic J. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. The Journal of biological chemistry. 2001;276:6485–6498. doi: 10.1074/jbc.M008073200. [DOI] [PubMed] [Google Scholar]
- 12.Wang LW, Leonhard-Melief C, Haltiwanger RS, Apte SS. Post-translational modification of thrombospondin type-1 repeats in ADAMTS-like 1/punctin-1 by C-mannosylation of tryptophan. The Journal of biological chemistry. 2009;284:30004–30015. doi: 10.1074/jbc.M109.038059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maillette de Buy Wenniger-Prick LJ, Hennekam RC. The Peters’ plus syndrome: a review. Annales de genetique. 2002;45:97–103. doi: 10.1016/s0003-3995(02)01120-6. [DOI] [PubMed] [Google Scholar]
- 14.Hess D, Keusch JJ, Oberstein SA, Hennekam RC, Hofsteenge J. Peters Plus syndrome is a new congenital disorder of glycosylation and involves defective Omicron-glycosylation of thrombospondin type 1 repeats. The Journal of biological chemistry. 2008;283:7354–7360. doi: 10.1074/jbc.M710251200. [DOI] [PubMed] [Google Scholar]
- 15.Lesnik Oberstein SA, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, Breuning MH, Hennekam RC. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. American journal of human genetics. 2006;79:562–566. doi: 10.1086/507567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weh E, Reis L, Tyler R, Bick D, Rhead W, Wallace S, McGregor T, Dills S, Chao MC, Murray J, et al. Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clinical genetics. 2013;86:142–148. doi: 10.1111/cge.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu L, Hou X, Shi S, Korner C, Stanley P. Slc35c2 promotes Notch1 fucosylation and is required for optimal Notch signaling in mammalian cells. The Journal of biological chemistry. 2010;285:36245–36254. doi: 10.1074/jbc.M110.126003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishikawa HO, Ayukawa T, Nakayama M, Higashi S, Kamiyama S, Nishihara S, Aoki K, Ishida N, Sanai Y, Matsuno K. Two pathways for importing GDP-fucose into the endoplasmic reticulum lumen function redundantly in the O-fucosylation of Notch in Drosophila. The Journal of biological chemistry. 2010;285:4122–4129. doi: 10.1074/jbc.M109.016964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Shareffi E, Chaubard JL, Leonhard-Melief C, Wang SK, Wong CH, Haltiwanger RS. 6-alkynyl fucose is a bioorthogonal analog for O-fucosylation of epidermal growth factor-like repeats and thrombospondin type-1 repeats by protein O-fucosyltransferases 1 and 2. Glycobiology. 2013;23:188–198. doi: 10.1093/glycob/cws140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azzam ME, Algranati ID. Mechanism of puromycin action: fate of ribosomes after release of nascent protein chains from polysomes. Proceedings of the National Academy of Sciences of the United States of America. 1973;70:3866–3869. doi: 10.1073/pnas.70.12.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ripka J, Adamany A, Stanley P. Two Chinese hamster ovary glycosylation mutants affected in the conversion of GDP-mannose to GDP-fucose. Archives of biochemistry and biophysics. 1986;249:533–545. doi: 10.1016/0003-9861(86)90031-7. [DOI] [PubMed] [Google Scholar]
- 22.Chen CI, Keusch JJ, Klein D, Hess D, Hofsteenge J, Gut H. Structure of human POFUT2: insights into thrombospondin type 1 repeat fold and O-fucosylation. The EMBO journal. 2012;31:3183–3197. doi: 10.1038/emboj.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosuri P, Alegre-Cebollada J, Feng J, Kaplan A, Ingles-Prieto A, Badilla CL, Stockwell BR, Sanchez-Ruiz JM, Holmgren A, Fernandez JM. Protein folding drives disulfide formation. Cell. 2012;151:794–806. doi: 10.1016/j.cell.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 25.Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, Bauer F, Flori E, Prost-Squarcioni C, Krakow D, et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nature genetics. 2008;40:1119–1123. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashikov A, Routier F, Fuhlrott J, Helmus Y, Wild M, Gerardy-Schahn R, Bakker H. The human solute carrier gene SLC35B4 encodes a bifunctional nucleotide sugar transporter with specificity for UDP-xylose and UDP-N-acetylglucosamine. The Journal of biological chemistry. 2005;280:27230–27235. doi: 10.1074/jbc.M504783200. [DOI] [PubMed] [Google Scholar]
- 27.Luhn K, Wild MK, Eckhardt M, Gerardy-Schahn R, Vestweber D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nature genetics. 2001;28:69–72. doi: 10.1038/ng0501-69. [DOI] [PubMed] [Google Scholar]
- 28.Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:913–917. doi: 10.1073/pnas.91.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ritter C, Helenius A. Recognition of local glycoprotein misfolding by the ER folding sensor UDP-glucose:glycoprotein glucosyltransferase. Nature structural biology. 2000;7:278–280. doi: 10.1038/74035. [DOI] [PubMed] [Google Scholar]
- 30.Tan K, Duquette M, Liu JH, Zhang R, Joachimiak A, Wang JH, Lawler J. The structures of the thrombospondin-1 N-terminal domain and its complex with a synthetic pentameric heparin. Structure. 2006;14:33–42. doi: 10.1016/j.str.2005.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanson SR, Culyba EK, Hsu TL, Wong CH, Kelly JW, Powers ET. The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3131–3136. doi: 10.1073/pnas.0810318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh JC, Kodjabachian L, Rebbert ML, Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB, Nathans J. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398:431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- 33.Koo BH, Le Goff C, Jungers KA, Vasanji A, O’Flaherty J, Weyman CM, Apte SS. ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein that is widely expressed during mouse embryogenesis and is regulated during skeletal myogenesis. Matrix biology : journal of the International Society for Matrix Biology. 2007;26:431–441. doi: 10.1016/j.matbio.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Luo Y, Haltiwanger RS. O-fucosylation of notch occurs in the endoplasmic reticulum. The Journal of biological chemistry. 2005;280:11289–11294. doi: 10.1074/jbc.M414574200. [DOI] [PubMed] [Google Scholar]
- 35.Rana NA, Nita-Lazar A, Takeuchi H, Kakuda S, Luther KB, Haltiwanger RS. O-glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. The Journal of biological chemistry. 2011;286:31623–31637. doi: 10.1074/jbc.M111.268243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang JY, Li L, Lai PH. A major kinetic trap for the oxidative folding of human epidermal growth factor. The Journal of biological chemistry. 2001;276:4845–4852. doi: 10.1074/jbc.M005160200. [DOI] [PubMed] [Google Scholar]
- 37.Rana NA, Haltiwanger RS. Fringe benefits: functional and structural impacts of O-glycosylation on the extracellular domain of Notch receptors. Current opinion in structural biology. 2011;21:583–589. doi: 10.1016/j.sbi.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.