

SUMMARY
N-terminal acetylation is among the most common protein modifications in eukaryotes and is mediated by evolutionarily conserved N-terminal acetyltransferases (NATs). NatD is among the most selective NATs; its only known substrates are histones H4 and H2A, containing the N-terminal sequence SGRGK in humans. Here we characterize the molecular basis for substrate-specific acetylation by NatD by reporting its crystal structure bound to cognate substrates and performing related biochemical studies. A novel N-terminal segment wraps around the catalytic core domain to make stabilizing interactions, and the α1-α2 and β6-β7 loops adopt novel conformations to properly orient the histone N termini in the binding site. Ser1 and Arg3 of the histone make extensive contacts to highly conserved NatD residues in the substrate binding pocket, and flanking glycine residues also appear to contribute to substrate-specific binding by NatD, together defining a Ser-Gly-Arg-Gly recognition sequence. These studies have implications for understanding substrate-specific acetylation by NAT enzymes.
INTRODUCTION
N-terminal acetylation is one of the most widespread protein modifications in eukaryotes, which occurs in over 60% and 80% of the yeast and human proteomes, respectively (Starheim et al., 2012). The functional consequence of N-terminal acetylation is diverse. For example, this modification has been shown to play a role in the N-end rule for protein degradation through E3 ligases Ubr1 and Doa10 (Hwang et al., 2010); to be important for E2-mediated activation of an E3 neddylation enzyme (Scott et al., 2011); to regulate cell survival pathways through regulation by Bcl-xL (Yi et al., 2011); to inhibit the targeting of proteins to the endoplasmic reticulum (Forte et al., 2011); and to facilitate heterochromatin formation in yeast (Arnaudo et al., 2013; Yang et al., 2013). N-terminal acetylation is believed to be an irreversible process, as no N-terminal deacetylase has been identified to date (Starheim et al., 2012). Several studies have also connected the aberrant activity of N-terminal acetyltransferases (NATs) to diseases such as cancer, suggesting that these enzymes may be attractive targets for drug development (Kalvik and Arnesen, 2013).
In eukaryotes, there are five evolutionarily conserved enzymes responsible for N-terminal acetylation, termed NatA-NatE. NatA-NatC contain at least two subunits, an auxiliary and catalytic subunit, whereas NatD and NatE can both be active as monomeric enzymes (Starheim et al., 2012). The NATs vary in their substrate specificity. NatA has the broadest substrate profile of the NATs and acetylates substrates that have small N-terminal side chains (Ala, Cys, Gly, Ser, Thr, or Val) (Polevoda and Sherman, 2003). These N-terminal residues are exposed after cleavage of the initiator methionine by the action of methionine aminopeptidases (Huang et al., 1987). The other NATs acetylate methionine-containing N termini with specificity further dictated by the identity of at least the second residue. NatB acetylates proteins that have an N-terminal methionine followed by Asp, Asn, Glu, or Gln as their second residue (Van Damme et al., 2012); and NatC and NatE acetylate proteins that have an N-terminal methionine followed by a hydrophobic second residue (Evjenth et al., 2009; Polevoda and Sherman, 2003). NatF, which is present only in higher eukaryotes, has a wider variety of substrates and acetylates N-terminal methionines followed by Ala, Leu, or Lys residues (Van Damme et al., 2011). NATs A, B, C, and E have been found to associate with the ribosome; it is believed that N-terminal acetylation is largely a cotranslational process wherein the NATs acetylate the N terminus as it emerges from the ribosomal peptide exit channel (Starheim et al., 2012).
NatD (also known as Nat4, Naa40p, or Patt1) activity was first observed in the yeast Saccharomyces cerevisiae when a NatA yeast knockout strain was noted to harbor N-terminal acetylated histones H2A and H4, which contain an N-terminal serine residue that is typically N-terminally acetylated by NatA (Mullen et al., 1989). NatD from S. cerevisiae was subsequently cloned (Song et al., 2003), and the human homolog was later characterized (Hole et al., 2011). The only NatD substrates known to date are histones H2A and H4, making it the most substrate-selective NAT enzyme so far characterized. Both the human and yeast orthologs were found to be associated with ribosomal fractions from whole cell lysates. Interestingly, the human ortholog localized to the nucleus in addition to the cytoplasm, suggesting that NatD may act both post- and cotranslationally (Hole et al., 2011; Polevoda et al., 2009).
The biological role of N-terminal H4 acetylation has only recently come to light. Schiza et al. demonstrated that the presence of an N-terminal acetyl group on H4 upregulates rDNA expression through crosstalk with H4 Arg-3 methylation. The H4 N-terminal acetyl group inhibits asymmetric dimethylation of H4 Arg-3 (H4R3me2a) by the Hmt1 arginine methyltransferase (Schiza et al., 2013). When H4R3me2a is found at the rDNA locus, transcription is repressed. In this way, the presence of an acetyl group on H4 may act as a sensor for cell growth. NatD has also been implicated in disease. NatD is downregulated in hepatocellular carcinoma, and overexpression of NatD in the cancerous cells enhances apoptosis (Liu et al., 2009). NatD liver-specific knockout male mice have decreased body mass and are protected from age-associated hepatic steatosis (Liu et al., 2012).
In this study, we set out to understand the molecular basis for H4- and H2A-specific acetylation by NatD. To do so, we crystallized NatD in binary complex with acetyl coenzyme A (CoA), and in ternary complex with CoA and an N-terminal histone H4/H2A substrate peptide, and performed biochemical and enzymatic characterization of the enzyme.
RESULTS
Overall Structure of NatD
An alignment of NatD sequences from various organisms reveals a ~150-residue region (residues 55–216 in human) of high sequence conservation, which we hypothesized mapped to the acetyltransferase domain (Figure 1). In order to prepare recombinant NatD protein variants for biochemical and structural studies, we bacterially overexpressed several NatD protein variants derived from Schizosaccharomyces pombe or human. This led to the identification of S. pombe NatD (SpNatD, residues 13–204) and human NatD (hNatD, residues 17–220) that formed cocrystals with substrate(s) suitable for X-ray structure determinations. Crystals were prepared of SpNatD and hNatD in the presence of acetyl-CoA, and of hNatD in the presence of CoA and an N-terminal histone H4/H2A pentapeptide (SGRGK). Selenomethionine-labeled SpNatD was prepared to determine the SpNatD/Ac-CoA structure using single-wavelength anomalous diffraction, and this structure was used to determine the hNatD/Ac-CoA and hNatD/CoA/peptide structures using molecular replacement. The structures were refined to high quality and to medium/high resolution (Table 1). The overall SpNatD and hNatD structures are essentially superimposable, with a root-mean-square deviation (rmsd) of 1.08 Å (Figure 2A). The remainder of this article describes the human NatD structures, NatD/Ac-CoA (resolution = 1.78 Å, Rfree/Rwork=20.6%/16.9%) and NatD/CoA/peptide (resolution = 2.5 Å (Rfree/Rwork=22.3%/17.5%).
Figure 1. Sequence Alignment of NatD Orthologs.

The sequence alignment contains the following NatD orthologs: human (Homo sapiens), fission yeast (Schizosaccharomyces pombe), fruit fly (Drosophila melanogaster), spikemoss (Selaginella moellendorffii), and sea anemone (Nematostella vectensis). The blue boxes represent conserved patches of sequence alignment. Residues in red are highly conserved, and residues in white with a red background are strictly conserved. Above the sequence alignment are indicated amino acid numbering for H. sapiens and secondary structure elements. Amino acid residues are indicated that make contacts to the substrate peptide (●), show mutational sensitivity (+), or are proposed to play catalytic roles (*).
Table 1.
Data Statistics for NatD Crystal Structures
| Selenomethionine SpNatD | hNatD/acetyl-CoA | hNatD/CoA/H4-H2A Peptide | |
|---|---|---|---|
| PDB ID | 4AU3 | 4U9V | 4U9W |
| Crystal Parameters | |||
| Space group | C2 | C2 | P21 |
| Cell dimensions | |||
| a, b, c, (Å) | 114.701, 42.629, 85.417 | 88.256, 44.064, 50.356 | 52.901, 93.288, 100.329 |
| α, β, γ (°) | 90.00, 98.68, 90.00 | 90.00, 95.44, 90.00 | 90.00, 96.69, 90.00 |
| Data Collectiona | |||
| Resolution (Å) | 50–1.85 (1.92–1.85) | 50–1.78 (1.84–1.78) | 50–2.50 (2.59–2.50) |
| Unique reflections | 34807 | 18519 | 33496 |
| Rmerge | 0.067 (0.595) | 0.074 (0.471) | 0.102 (0.552) |
| I/σI | 32.6 (6.1) | 22.6 (1.7) | 12.3 (2.3) |
| Completeness (%) | 99.4 (98.7) | 98.5 (87.5) | 98.8 (97.6) |
| Redundancy | 17.4 (16.7) | 8.1 (4.2) | 3.8 (3.7) |
| Refinement | |||
| Rwork/Rfree (%) | 17.9/23.8 | 16.9/20.6 | 17.5/22.3 |
| No. of atoms | |||
| Protein | 3132 | 1582 | 6348 |
| Acetyl-CoA/CoA | 96 | 51 | 192 |
| Peptide | N/A | N/A | 125 |
| Solvent | 234 | 166 | 457 |
| B factors (Å2) | |||
| Protein | 64.63 | 23.99 | 26.61 |
| Acetyl-CoA/CoA | 62.97 | 21.59 | 24.92 |
| Peptide | N/A | N/A | 25.85 |
| Solvent | 77.18 | 32.57 | 27.23 |
| Root-mean-square deviation | |||
| Bond lengths (Å) | 0.007 | 0.009 | 0.010 |
| Bond | 1.078 | 1.126 | 1.206 |
| angles (°) | |||
| Ramachandran statistics (%) | |||
| Favored | 100 | 97.44 | 98.72 |
| Allowed | 0 | 2.56 | 1.15 |
| Outlier | 0 | 0 | 0.13 |
Values in parentheses are for the highest resolution shell.
Figure 2. Overall Structure of NatD Complexes.

(A) Superposition of the SpNatD/acetyl-CoA (violet), hNatD/acetyl-CoA (brown), and hNatD/CoA/H4-H2A peptide (cyan) complexes. CoA is shown as sticks, and the H4-H2A peptide is omitted for clarity.
(B) Overall structure of hNatD/CoA/H4-H2A peptide with structurally unique elements of NatD relative to other NATs highlighted in yellow. CoA is shown in orange and H4 is shown in magenta.
The overall NatD structure has a mixed α/β GCN5-like fold (Figure 2B) similar to the other reported NAT structures (Liszczak et al., 2011; Liszczak et al., 2013; Liszczak and Marmorstein, 2013). However, there are notable differences between NatD and the other NAT structures, NatA and NatE (Figure 3A). In particular, differences are noted in the loops forming the peptide binding site, and NatD contains a unique N-terminal segment.
Figure 3. Unique Structural Features of NatD.

(A) Superposition of hNatD (cyan), SpNatA (orange), and hNatE (green) complexes. The H4-H2A peptide is shown in magenta. Only the CoA from the hNatD/CoA/H4-H2A peptide structure is shown for clarity.
(B) Close-up view of the substrate binding groove of NatD in comparison with NatA and NatE. The color coding is as in Figure 3A. NatA substrate (SASE) and NatE substrate (MLGP) are shown as orange and green sticks, respectively.
(C) View of the interaction of the NatD N-terminal segment (yellow cartoon representation) with the catalytic core domain (cyan surface representation).
(D) Detailed interactions between the N-terminal segment and catalytic core domain of NatD. Residues from the N terminus that mediate interactions are labeled in black, and residues from the core domain are colored in dark blue and labeled in white. Met-162 is omitted for clarity.
A striking divergent feature of NatD is its loops flanking the substrate binding site, β6-β7 and α1-α2. A characteristic feature of the peptide binding site of the NAT proteins is that these loops appear to be used to prevent internal lysines from inserting into the active site. This is in contrast to the more open loop configuration of lysine acetyltransferases such as Gcn5 (Rojas et al., 1999) and Hat1 (Li et al., 2014). In the case of NatA and NatE, a β6-β7 hairpin loop serves this function. In NatD, the corresponding β6-β7 hairpin loop is flipped away from the substrate binding site, and instead, the binding pocket is blocked by an extended α1-α2 loop on the opposite side (Figure 3B). As discussed below, this difference appears to be critically important for substrate-specific binding by NatD.
The novel NatD N-terminal segment (residues 24–51) adopts a helix-loop-strand topology that wraps around the catalytic core domain, making complementary interactions with a hydrophobic patch, which appears to be important for stability of the protein (Figure 3C). The interface between the N-terminal segment and a cleft on a surface of the catalytic core domain is largely hydrophobic. Specifically, residues Pro-38, Leu-39, Phe-42, Phe-45, and Tyr-48 of the N-terminal segment make van der Waals contacts to residues Leu-53, Ile-57, Cys-59, Trp-107, Leu-109, Phe-124, Phe-126, Leu-151, Phe-154, Ile-158, Leu-161, and Met-162 of the core domain (Figure 3D). Consistent with the importance of these contacts for stability of the catalytic core domain, N-terminally truncated proteins beginning at residue 47 were not stable through purification, yet constructs beginning at residue 37 were (data not shown). The region from residues 37–48 contains the key hydrophobic residues that interact with the hydrophobic surface cleft of the core domain. Notably, the residues that line this hydrophobic cleft are replaced with more hydrophilic residues in the other NAT proteins, which explains why they lack the extended N terminus. We have designated the α helix and β strand in this novel N-terminal region α0 and β0, respectively, in order to keep secondary structure nomenclature consistent with the other NAT proteins (Liszczak et al., 2011; Liszczak et al., 2013; Liszczak and Marmorstein, 2013).
Substrate Recognition by NatD
The structure of NatD in complex with CoA and the N terminus of histones H4/H2A provides important insights into substrate binding. The overall structure of the protein from the ternary complex is similar to the binary complex, with an rmsd of 0.313 Å. The density for the peptide is well defined, and we were able to unambiguously place all atoms expect for the Lys5p side chain. The binding pocket for the peptide is highly acidic, which is in line with the basicity of the histone tails (Figure 4A). The ternary complex reveals extensive interactions with the substrate peptide and NatD (Figure 4B). Notably, contacts occur along the entire length of the peptide; each amide nitrogen and oxygen is hydrogen bonded, except for the Arg3p amide oxygen. There are direct hydrogen bonds to the backbone amide groups on the peptide from residues Tyr-138, Thr-174, Ser-197, and Tyr-211 of NatD. In addition, there are water-mediated contacts between residues Glu-100, Tyr-136, and Thr-174 to the backbone amides of the peptide. The aliphatic region of Trp-90 interacts with Gly2p and Gly4p of the peptide via van der Waals contacts. Ile-213 makes van der Waals contacts with the backbone of Lys5p.
Figure 4. Peptide Binding Site of NatD.

(A) Electrostatic surface of the NatD peptide binding site with the peptide shown in magenta stick figure. Residues from NatD are labeled in black and residues from the peptide are labeled in yellow with their corresponding one-letter codes and numerical positions in the peptide. The side chain of Lys5p was disordered and not modeled into the crystal structure.
(B) Detailed interactions between NatD and the H4-H2A peptide. NatD is shown as a transparent cyan surface, and residues that interact with the peptide are yellow. Hydrogen bonds are shown as dashed lines and waters are shown as red spheres.
(C) Close-up view of the NatD active site highlighting interactions made by Ser1p. Acetyl-CoA was modeled into the figure by aligning the binary and ternary NatD structures.
The residues along the peptide path are poised to confer specificity for recognition by the enzyme. Ser1p sits in a pocket, which would not accommodate residues with larger side chains, as they would be occluded by Met-81, Tyr-85, and Glu-139. There are two water molecules in an extended network of hydrogen bonds that fill the other side of the pocket (Figure 4C). In addition to the size restrictive pocket, the side chain of Ser1p makes hydrogen bonds to Tyr-85 and Glu-139. Arg3p makes numerous contacts with NatD as well (Figure 4B). The aliphatic region of the Arg3p side chain sits on top of Tyr-136 in an extensive van derWaals interaction. The two terminal nitrogens in the guanidino group of the side chain make hydrogen bonds with the NatD side chains Asp-127, Glu-129, and Tyr-138. The glycines at positions 2 and 4 also appear to be playing important roles in peptide substrate recognition by NatD. It is notable that Gly2p is in a conformation with torsion angles that are unfavorable for any residues other than glycine in the Ramachandran plot (Phi: 69.59, Psi: −171.02). In addition, both Gly2p and Gly4p are in grooves that are sterically tailored for glycines. Gly2p lies in a narrow tunnel, which connects Ser1p with the rest of the peptide and is in close contact with Lys-97 and Glu-100. Gly4p makes intimate contacts with Trp-90, Thr-174, and Tyr-211. It appears that residues with side chains in position 2 or 4 would thus lead to unfavorable clashes (Figure 4B). The majority of the residues making contacts with the peptide are highly conserved among NatD homologs (Figure 1).
To address the contribution of specific residues in substrate recognition, we prepared point mutants to alanine for residues of interest and assayed them for acetyltransferase activity against a 19-residue peptide of the H4 N terminus (Figure 5A). All of the mutants assayed were purified to homogeneity in the same manner as the wild-type enzyme and displayed the same gel filtration elution profile, suggesting that the mutants did not perturb overall protein folding. Individual mutations of residues that contribute to the intimate Gly4p interaction (T174A and Y211A) show wild-type catalytic efficiency (Figures 5A and 5B). In contrast, mutations of residues that contact Arg3p (Y136A, Y138A, and D127A/E129A) show significantly decreased catalytic efficiency (1.6- to 18-fold) (Figures 5A and 5B), highlighting the particular importance of this residue for substrate-specific binding. The W90A and Y85A mutants also exhibited markedly decreased catalytic efficiency (2.8- and 5.8-fold, respectively). The W90A mutant lacks the stabilizing van der Waals contacts with the peptide. The decrease in activity in the Y85A mutant is likely due to the absence of hydrophobic stabilization provided by Tyr-85 in the α1-α2 loop that forms a part of the cavity for histone recognition, in addition to its hydrogen bond to Ser1p (Figures 5A and 5B). Glu-100 is a highly conserved residue that may act to position α1 and α2 through an interaction with Lys-97, another conserved residue. This is supported by the observation that an E100A mutation exhibits a 5-fold reduction in catalytic efficiency. Notably, Lys-97 mutants were not stable through purification (data not shown).
Figure 5. Mutational Analysis of NatD.

(A) Catalytic efficiency of selected NatD mutants. Mutations that have negligible effect are in blue, while those that decrease the catalytic efficiency of the enzyme are in pink.
(B) Residues targeted for mutagenesis are mapped onto the NatD structure. The color scheme is the same as Figure 5A.
(C) The catalytic efficiency of wild-type NatD toward N-terminal histone H4 peptides of varying length. Data are represented as mean ± SEM. Each mutant and peptide of varying length was assayed in triplicate.
NatD Catalysis
The structure of NatD suggests several potential catalytic residues. Classically, acetyltransferases in the GNAT family use a general base residue to deprotonate the lysine side chain or N terminus, which carries out a nucleophilic attack on the acetyl group of acetyl-CoA (Yuan and Marmorstein, 2013). Interestingly, the type and relative location of residue that is used to carry out this function can differ between acetyltransferase enzymes (Liszczak et al., 2011; Liszczak and Marmorstein, 2013; Vetting et al., 2005). In the NatD structure, both Tyr-136 and Glu-139 are positioned to potentially play a role as important catalytic residues (Figures 4B and 4C). Although Tyr-136 is not close enough to deprotonate the N terminus directly, there is an ordered water molecule bridging its phenol oxygen and the peptide N terminus. This water molecule is positioned similarly to one that appears to be involved in catalysis in NatE, which uses a tyrosine as one of its general bases (Liszczak et al., 2011). In order to distinguish between the two residues, we mutated Tyr-136 to phenylalanine and Glu-139 to glutamine and tested the activity of these mutants. While the catalytic efficiency of the enzyme is modestly affected by the Y136F mutation, it is effectively abolished in the E139Q mutant, demonstrating that the catalytic residue of NatD is likely Glu-139 (Figure 5A). This is in agreement with a previous study (Liu et al., 2009) and consistent with the functional importance of Glu-139, as it is strictly conserved among NatD proteins (Figure 1). As Glu-139 is not positioned to directly deprotonate the N terminus, it may not act as a general base but instead increase the nucleophilicity of the N terminus through an induced dipole effect from the carboxylic acid side. Glu-139 may also be important for orienting the amino group properly across from the acetyl-CoA. This is similar to Glu-24 in NatA, which is not directly positioned to deprotonate the N terminus but is essential for catalysis (Liszczak et al., 2013).
Our results are in contrast to previous reports on the catalytic requirements of NatD. A previous modeling study predicted that Cys-137 could also act as an important residue in the catalytic mechanism of NatD (Jedrzejewski and Kazmierkiewicz, 2013), in a mechanism similar to MYST family acetyltransferases (Yan et al., 2002). However, the cysteine is not in a suitable position in the structure to act as an acetyl group acceptor, and while the C137A mutant does show a decrease in activity, it still retains much of its catalytic power (Figure 5A). Previous studies also suggested that NatD requires a longer N-terminal tail than other NATs in order to recognize its histone H4 and H2A substrates. In particular, in an in vivo assay in S. cerevisiae, it was found that chimeric cytochrome c with H4 N termini showed complete acetylation with chimeric proteins containing 50 N-terminal H4 residues, and only incomplete acetylation with chimeric proteins containing 30 N-terminal H4 residues (Polevoda et al., 2009). However, when we tested synthetic H4 peptides of varying length (5, 8, and 19 residues) we did not see a significant difference in catalytic efficiency (Figure 5C). This result is in agreement with the crystal structure, suggesting that the determinants for NatD-specific binding and acetylation are contained within the first four N-terminal residues. It is also consistent with a previous study, which showed that NatD substrate specificity is not affected by residues past the fifth position in a peptide when the enzyme was tested against a library of oligopeptides (Hole et al., 2011).
DISCUSSION
The structures and biochemical activities of two of the six NAT proteins have recently been characterized, NatE (Liszczak et al., 2011) and NatA (Liszczak et al., 2013). Here, we report the molecular characterization of one of the most substrate-selective NAT enzymes, NatD, which has been reported to acetylate the N terminus of only two substrates (histones H4 and H2A) that harbor the identical sequence of SGRGK in humans. We find that the overall fold of NatD is similar to other NATs; however, there are structural differences that appear to be critical for maintaining protein integrity and cognate substrate binding. A novel N-terminal segment wraps around the catalytic core domain to make numerous stabilizing interactions, and deletion analysis suggests that this N-terminal segment is required for maintaining the stability of the catalytic core domain. We note that several NAT proteins (NatA, NatB, and NatC) require auxiliary subunits for activity; the NatA structure reveals that the Naa15p auxiliary subunit wraps around the Naa10p catalytic subunit to increase stability of the catalytic subunit to induce a conformation of Naa10p that is compatible for catalysis (Liszczak et al., 2013). In this regard, the N-terminal segment of NatD may play an analogous role as the NatA auxiliary subunit.
The N-terminal sequence requirements of NatD are different from other NATs. In particular, most other NATs show the greatest specificity for residue 1, with reduced preference for residue 2 and significantly reduced preference for residues 3 on (Starheim et al., 2012). This is borne out by the crystal structures of NatA and NatE bound to cognate peptides, showing the most extensive side chain interactions with residue 1 followed by residue 2 of the cognate substrate (Liszczak et al., 2011; Liszczak et al., 2013). NatD, in contrast, has sequence requirements for the first 4 residues of its substrate peptide (Hole et al., 2011). Unique structural features of NatD appear to play direct roles in substrate-specific binding by NatD. In particular, the β6-β7 loop of NatD is oriented away from the peptide substrate binding site relative to the corresponding loops in the other NATs. Instead, the extended α1-α2 loop of NatD flips toward the peptide binding site and in so doing tilts the path of the NatD peptide by about 75° relative to the paths of the NatA and NatE substrates (Figure 3B). An important consequence of this is that Arg3p is forced into a unique and complementary pocket of NatD that is not present in the other NATs and particularly not in NatA, which also acetylates N termini that contain a serine in position 1 (Figure 6A). Indeed, both NatA and NatE contain a conserved tyrosine (Tyr-138 in both proteins) that would sterically block peptides from being oriented toward the β6-β7 loop as in NatD. This may be a mechanism by which NatA and NatE ensure a broader substrate profile, as orienting the peptide away from the enzyme surface prevents significant interaction beyond the first two residues of the N terminus.
Figure 6. Comparison between Substrate Recognition of NatD and NatA.
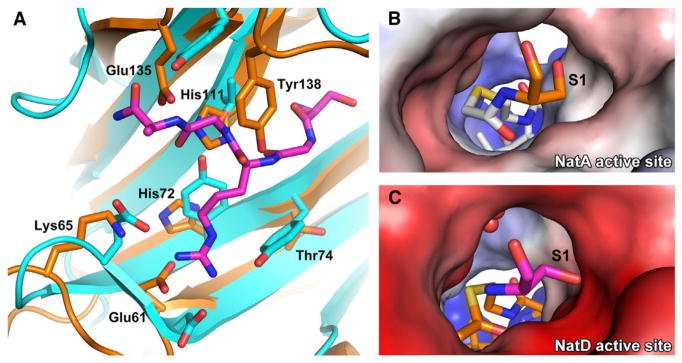
(A) Overlay of the peptide binding site of NatA (orange) and NatD (cyan with magenta histone substrate).
(B) Electrostatic potential surface of the NatA active site with a bound covalently linked bisubstrate inhibitor. The N-terminal serine of the substrate peptide is shown in orange, and the acetyl-CoA moiety of the bisubstrate inhibitor is in white.
(C) Electrostatic potential surface of the NatD active site. The N-terminal serine of the substrate peptide is shown in magenta, and acetyl-CoA is in orange. Waters are shown as red spheres. Acetyl-CoA was modeled into the figure by aligning the binary and ternary NatD structures.
The Arg3p pocket also raises the intriguing question of whether Arg3 methylation may inhibit H4 acetylation. H4 N-terminal acetylation inhibits Arg3 asymmetric dimethylation in yeast (Schiza et al., 2013), and based on our structural data, it may be the case that Arg3p methylation also inhibits N-terminal acetylation. If Arg3p were methylated, and particularly asymmetrically dimethylated, we predict that it would disrupt the hydrogen bond network in its binding pocket (Figure 4B) and therefore occlude substrate binding and acetylation.
Similarly to NatA, the peptide binding pocket of NatD can only accommodate residues with small side chains as position 1 in the peptide. However, unlike NatA, the pocket is more tailored to accommodate a serine. Tyr-85 and Glu-139 are oriented to have their side chains precisely where the side chain of the N-terminal residue will lie in the pocket (Figure 4C). Whereas the active site of NatA lacks significant electrostatic potential (Figure 6B), the presence of these polar and charged residues in the active site likely makes it unfavorable for residues with small aliphatic side chains to be the N-terminal residue in the peptide (Figure 6C). In addition, the active site pocket of NatD is even more constricted than NatA, particularly due to the placement of Glu-139. This helps explain the preference for serine over threonine, a residue that NatA can acetylate. Gly2p and Gly4p of the H4/H2A peptide also appear to contribute to substrate-specific binding by NatD. Gly2p contains unusual Phi/Psi angles, which are only favorably accommodated by a glycine residue, and it does not appear that a residue with a side chain would be sterically accommodated in positions 2 or 4 of a substrate. hNatD therefore appears to be specific for a Ser-Gly-Arg-Gly sequence in its cognate substrate. Notably, S. cerevisiae H2A has a divergent sequence of SGGKG. It was shown that heterologous expression of human NatD in an S. cerevisiae NatD knockout strain restored N-terminal acetylation of yeast H4 but not yeast H2A in vivo (Hole et al., 2011). Therefore, the substrate specificities of NatD correspond to the sequences of H2A and H4 in the organism, and yeast NatD has likely evolved to accommodate both N termini.
Given the fairly short consensus sequence of NatD, we wondered why NatD is specific for histones H4 and H2A. We first performed a Uniprot search to query what other proteins share an N-terminal SGRGK sequence. To our surprise, the only proteins containing this N-terminal sequence in the human proteome are H2A, H4, and H2A.X. If we decrease our specificity by removing Lys-5 (which does not appear to make sequence-specific interactions in the structure), there is only one other protein with an N terminus of SGRG, an SWI/SNF-related chromatin regulator called SMARCD2 (Ring et al., 1998). There is no evidence that SMARCD2 is acetylated by NatD, but our studies predict that this would be the case. There are an additional 17 proteins in the human proteome with an N terminus of SGR that may also be substrates for NatD. Importantly, this method does not account for N termini generated through proteolytic cleavage, nor for any hitherto undiscovered N termini that diverge from the sequence of SGRG but may still act as substrates for NatD. However, it still appears that the N-terminal proteome of potential NatD substrates is limited relative to other NAT proteins. Further studies will have to be carried out to establish if the pool of NatD client proteins is indeed broader than previously appreciated.
EXPERIMENTAL PROCEDURES
Homo sapiens NatD (hNatD) Expression and Purification
The full-length hNatD gene (encoding residues 1–237) was a gift from Dr Thomas Arnesen from the University of Bergen. Several N-terminal and C-terminal truncation constructs were engineered into a modified PCDF vector containing an N-terminal tobacco etch virus (TEV) protease-cleavable His tag. All constructs were transformed into Rosetta (DE3)pLysS competent Escherichia coli cells, which were grown to an OD600 of 0.7–0.9 and induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 16°C for ~16 hr. All subsequent purification steps were carried out at 4°C. Cells were isolated by centrifugation and lysed by sonication in lysis buffer containing 25 mM Tris, pH 8.0, 1 M NaCl, 10 mM β-mercaptoethanol (βME) and 10 μg/ml phenylmethanesulfonylfluoride. The lysate was clarified by centrifugation and passed over nickel resin (Thermo Scientific), which was subsequently washed with >20 column volumes of lysis buffer supplemented with 25 mM imidazole. The protein was eluted in lysis buffer supplemented with 300 mM imidazole. His tagged TEV protease was added to the eluent containing the target protein for the duration of a 14 hr dialysis into dialysis buffer containing 25 mM Tris, pH 8.5, 200 mM NaCl, 10 mM βME. This solution was passed through an additional nickel column to remove TEV protease as well as any uncut NatD. The resin was then washed with approximately seven column volumes of dialysis buffer supplemented with 25 mM imidazole, which was pooled with the initial flow-through. This solution was dialyzed into ion exchange buffer containing 25 mM Tris, pH 8.5, 50 mM NaCl and 1 mM dithiothreitol (DTT) and loaded onto a 5 ml HiTrap Q ion exchange column (GE Healthcare). The protein was eluted in the same buffer with a salt gradient (50–750 mM NaCl) over the course of 20 column volumes. Peak fractions were pooled and dialyzed into sizing buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl, and 1 mM DTT for ~16 hours. This was concentrated to a volume of 500 μl (10 kDa concentrator; Amicon Ultra, Millipore), and loaded onto and run on an s75prep gel filtration column (GE Healthcare) in sizing buffer. Peak fractions were concentrated to 10 mg/ml as measured by UV280 for crystallization trials.
S. pombe NatD (SpNatD) Expression and Purification
The full-length SpNatD gene (encoding residues 1–204) was cloned from the S. pombe genome (ATCC), and several N-terminal truncation constructs were engineered into a modified pETDUET vector containing a TEV protease-cleavable His tag. All constructs were transformed into Rosetta (DE3) pLysS competent E. coli cells, which were grown to an OD600 of 0.7–0.9 and induced with 0.5 mM IPTG at 16°C for ~16 hr. These constructs all expressed as insoluble inclusion bodies in Rosetta (DE3)pLysS E. coli cells. To improve solubility, a number of constructs of the SpNatD gene were subcloned into a modified pETDUET vector containing an N-terminal SUMO tag. These constructs were transformed and induced with IPTG in the same way and were soluble in E. coli cells. All subsequent studies were carried out at 4°C and are identical to the purification protocol used for purifying the human protein, with the following changes. After the nickel column, His tagged Ulp1 protease was used in place of TEV protease to cleave the SUMO tag. The ion exchange buffer contained 25 mM HEPES, pH 7.0, 50 mM NaCl, and 1 mM DTT, and a 5 ml HiTrap SP ion-exchange column (GE Healthcare) was used. The selenomethionine derivative was produced by expressing the SUMO tagged SpNatD (residues 13–204) in minimal media (Molecular Dimensions) with 50 μg/l selenomethionine (Sigma) purified in the same manner as above.
NatD Crystallization and Data Collection
hNatD (8 mg/ml) was incubated with acetyl-CoA (Sigma-Aldrich) at a 1:3 molar ratio. Crystals were obtained with hanging-drop vapor diffusion in a drop containing a 1:1 mixture of protein to a well solution containing 20% PEG 3350, 200 mM sodium citrate tribasic dihydrate, 100 mM citric acid, pH 4.0. Diffraction-quality crystals grew in 2–3 days. The ternary complex was crystallized using an hNatD construct containing residues 17–220 at 10 mg/ml. The protein was incubated with CoA, and the C-terminally amidated substrate peptide (SGRGK, GenScript) at a 1:3:5 molar ratio of protein:CoA:peptide. Crystals were obtained through hanging-drop vapor diffusion at 20°C at a 1:1 ratio of protein to a well solution containing 20% PEG 3350 and 200 mM sodium malonate, pH 6.0. Crystals grew after 1 day. SpNatD was crystallized at a concentration of 10 mg/ml using a construct that contained residues 13-204. The protein was mixed with acetyl-CoA at a molar ratio of 1:3 protein to acetyl-CoA. Crystals were obtained with hanging-drop vapor diffusion at 20°C at a 1:1 ratio of protein to well solution containing 20% PEG 3350 and 200 mM potassium formate. Crystals grew after 1 day. Selenomethionine crystals of SpNatD were generated in the same manner. All crystals were cryoprotected by transferring them to their respective well solutions supplemented with 20% glycerol before being flash frozen in liquid nitrogen. Data for the hNatD ternary complex were collected at a home source using a MicroMax-007 HF rotating anode (Rigaku). Data for all other crystals were collected at beamline X29A at the National Synchrotron Light Source (Brookhaven National Laboratory). All data sets were processed using HKL2000 (Otwinowski and Minor, 1997).
Structure Determination and Refinement
Initially, a number of full-length and truncated search models based on known structures of NATs (Liszczak et al., 2011; Liszczak et al., 2013; Liszczak and Marmorstein, 2013) were used in an attempt to solve either hNatD or SpNatD structure through molecular replacement (McCoy et al., 2007). However, these attempts were unsuccessful in generating a suitable starting model for either hNatD or SpNatD. A data set was collected on the selenomethionine-labeled SpNatD crystals at the selenium peak wavelength (0.9788 Å), heavy atoms were placed in the asymmetric unit, and initial phases were generated using single-wavelength anomalous diffraction phasing using SOLVE in the Phenix suite (Adams et al., 2011). The SpNatD structure was initially built into the resulting density by AutoBuild in the Phenix suite. The rest of the structures described were determined using molecular replacement. The selenomethionine-labeled SpNatD structure was used as a search model for the wild-type SpNatD crystals. However, these crystals did not yield a model of sufficient quality, so only the selenomethionine data set was refined fully. The program CHAINSAW (Stein, 2008) was used to generate a search model from the SpNatD structure for the binary hNatD structure, and the final refined hNatD structure was used as a search model for the ternary hNatD crystals. For all of the structures, the ligands acetyl-CoA/CoA and the substrate peptide for the ternary complex were built into the Fo − Fc electron density map. All of the structures were refined iteratively by using Phenix.Refine and manual model building in the molecular graphics program Coot (Emsley et al., 2010). The SpNatD/acetyl-CoA complex crystallized as a dimer with the N terminus of one monomer in the active site of another. The N-terminal serine of the enzyme is acetylated, and CoA is bound to the enzyme. This is likely an artifact of crystallization, wherein the subunit packing positions the N terminus of SpNatD (SRRMK), which superficially resembles the natural substrate, in the active site. The final model and structure factors for all of the structures were submitted to the Protein Data Bank (PDB). Refinement statistics and PDB IDs can be found in Table 1.
Acetyltransferase Assays
Acetyltransferase assays were carried out in 25 mM HEPES pH 7.0, 100 mM NaCl, and 1 mM DTT. The substrate peptide used in the assay for the mutant enzymes corresponds to the first 19 residues of human H4 (NH2-SGRGKGGKGLGKGGAKRHR-COOH; GenScript). In the assay, a saturating amount (400 μM) of radiolabeled [14C]acetyl-CoA (4 mCi/mmol; PerkinElmer Life Sciences) and varying concentrations of the substrate peptide (6.25–200 μM) were incubated with 75 nM hNatD (500 nM for E139A and E139Q mutants) in 75 μl of reaction volume for 20–180 min at 37°C. To quench the reaction, 20 μl of the reaction mixture was added to negatively charged P81 paper disks (Whatman), and the paper disks were immediately placed in wash buffer (10 mM HEPES, pH 7.5). The paper disks were washed three times, at 5 min per wash, to remove unreacted acetyl-CoA. The papers were then dried with acetone and added to 4 ml of scintillation fluid, and the signal was measured with a Packard Tri-Carb 1500 liquid scintillation analyzer. Background control reactions were performed in the absence of enzyme or in the absence of substrate peptide to ensure that any possible signal due to autoacetylation was negligible. Each reaction was performed in triplicate. Complications from product inhibition at high peptide concentrations kept us from generating full catalytic curves. However, by keeping the substrate concentration far below Km, we were able to plot velocity versus [substrate] and use the slope of the resulting line to obtain kcat/Km values for the mutants based on the equation: v = (kcat/Km)[E][S]. The counts per minute were converted to molar units using a standard curve of known [14C]acetyl-CoA concentrations in scintillation fluid. The assay was performed in the same manner for substrate peptides corresponding to the first five and eight residues of human H4 (SGRGK and SGRGKGGK, respectively) with wild-type hNatD.
Highlights.
NatD is among the most selective N-terminal acetyltransferases (NATs)
NatD-specific structural features accommodate its high substrate selectivity
NatD substrate site is tailored for a Ser-Gly-Arg-Gly histone H4/H2A N terminus
Studies have implications for understanding substrate-specific acetylation by NATs
Acknowledgments
This work was supported by NIH grant R01 GM060293 awarded to R.M. and NIH grant T32 GM071339 awarded to G.P.L. and R.S.M. We acknowledge support of the Proteomics core facility at the Wistar Institute (NIH P30 CA010815) and the University of Pennsylvania DNA Sequencing Facility at the Perelman School of Medicine, University of Pennsylvania (NIH P30 CA016520). We thank Petra Van Damme for critical reading of the manuscript. The coordinates of the structures have been deposited in the PDB under accession numbers 4UA3 (selenomethionine-labeled SpNatD/Ac-CoA), 4U9V (hNatD/Ac-CoA) and 4U9W (hNatD/Ac-CoA/Peptide).
Footnotes
AUTHOR CONTRIBUTIONS
R.S.M. and G.P.L. performed the experiments described in the manuscript. R.S.M. prepared manuscript figures and text. R.M. designed and supervised experiments by R.S.M. and G.P.L. and prepared manuscript text. All authors read and approved the submitted manuscript.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. The Phenix software for automated determination of macromolecular structures. Methods. 2011;55:94–106. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaudo N, Fernandez IS, McLaughlin SH, Peak-Chew SY, Rhodes D, Martino F. The N-terminal acetylation of Sir3 stabilizes its binding to the nucleosome core particle. Nat Struct Mol Biol. 2013;20:1119–1121. doi: 10.1038/nsmb.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evjenth R, Hole K, Karlsen OA, Ziegler M, Arnesen T, Lillehaug JR. Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J Biol Chem. 2009;284:31122–31129. doi: 10.1074/jbc.M109.001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte GM, Pool MR, Stirling CJ. N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 2011;9:e1001073. doi: 10.1371/journal.pbio.1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole K, Van Damme P, Dalva M, Aksnes H, Glomnes N, Varhaug JE, Lillehaug JR, Gevaert K, Arnesen T. The human N-alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS One. 2011;6:e24713. doi: 10.1371/journal.pone.0024713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Elliott RC, Liu PS, Koduri RK, Weickmann JL, Lee JH, Blair LC, Ghosh-Dastidar P, Bradshaw RA, Bryan KM, et al. Specificity of cotranslational amino-terminal processing of proteins in yeast. Biochemistry. 1987;26:8242–8246. doi: 10.1021/bi00399a033. [DOI] [PubMed] [Google Scholar]
- Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejewski RP, Kazmierkiewicz R. Structure of Patt1 human proapoptotic histone acetyltransferase. J Mol Model. 2013;19:5533–5538. doi: 10.1007/s00894-013-2043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvik TV, Arnesen T. Protein N-terminal acetyltransferases in cancer. Oncogene. 2013;32:269–276. doi: 10.1038/onc.2012.82. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang L, Liu T, Chai C, Fang Q, Wu H, Agudelo Garcia PA, Han Z, Zong S, Yu Y, et al. Hat2p recognizes the histone H3 tail to specify the acetylation of the newly synthesized H3/H4 heterodimer by the Hat1p/Hat2p complex. Genes Dev. 2014;28:1217–1227. doi: 10.1101/gad.240531.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Marmorstein R. Implications for the evolution of eukaryotic amino-terminal acetyltransferase (NAT) enzymes from the structure of an archaeal ortholog. Proc Natl Acad Sci USA. 2013;110:14652–14657. doi: 10.1073/pnas.1310365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Arnesen T, Marmorstein R. Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J Biol Chem. 2011;286:37002–37010. doi: 10.1074/jbc.M111.282863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Goldberg JM, Foyn H, Petersson EJ, Arnesen T, Marmorstein R. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat Struct Mol Biol. 2013;20:1098–1105. doi: 10.1038/nsmb.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Liu Y, Wang H, Ge X, Jin Q, Ding G, Hu Y, Zhou B, Chen Z, Zhang B, et al. Patt1, a novel protein acetyltransferase that is highly expressed in liver and downregulated in hepatocellular carcinoma, enhances apoptosis of hepatoma cells. Int J Biochem Cell Biol. 2009;41:2528–2537. doi: 10.1016/j.biocel.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhou D, Zhang F, Tu Y, Xia Y, Wang H, Zhou B, Zhang Y, Wu J, Gao X, et al. Liver Patt1 deficiency protects male mice from age-associated but not high-fat diet-induced hepatic steatosis. J Lipid Res. 2012;53:358–367. doi: 10.1194/jlr.M019257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Kayne PS, Moerschell RP, Tsunasawa S, Gribskov M, Colavito-Shepanski M, Grunstein M, Sherman F, Sternglanz R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989;8:2067–2075. doi: 10.1002/j.1460-2075.1989.tb03615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Sherman F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J Mol Biol. 2003;325:595–622. doi: 10.1016/s0022-2836(02)01269-x. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Hoskins J, Sherman F. Properties of Nat4, an N(alpha)-acetyltransferase of Saccharomyces cerevisiae that modifies N termini of histones H2A and H4. Mol Cell Biol. 2009;29:2913–2924. doi: 10.1128/MCB.00147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring HZ, Vameghi-Meyers V, Wang W, Crabtree GR, Francke U. Five SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin (SMARC) genes are dispersed in the human genome. Genomics. 1998;51:140–143. doi: 10.1006/geno.1998.5343. [DOI] [PubMed] [Google Scholar]
- Rojas JR, Trievel RC, Zhou J, Mo Y, Li X, Berger SL, Allis CD, Marmorstein R. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature. 1999;401:93–98. doi: 10.1038/43487. [DOI] [PubMed] [Google Scholar]
- Schiza V, Molina-Serrano D, Kyriakou D, Hadjiantoniou A, Kirmizis A. N-alpha-terminal acetylation of histone H4 regulates arginine methylation and ribosomal DNA silencing. PLoS Genet. 2013;9:e1003805. doi: 10.1371/journal.pgen.1003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song OK, Wang X, Waterborg JH, Sternglanz R. An Nalpha-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J Biol Chem. 2003;278:38109–38112. doi: 10.1074/jbc.C300355200. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Gevaert K, Arnesen T. Protein N-terminal acetyl-transferases: when the start matters. Trends Biochem Sci. 2012;37:152–161. doi: 10.1016/j.tibs.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J Appl Crystallogr. 2008;41:641–643. [Google Scholar]
- Van Damme P, Hole K, Pimenta-Marques A, Helsens K, Vandekerckhove J, Martinho RG, Gevaert K, Arnesen T. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 2011;7:e1002169. doi: 10.1371/journal.pgen.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Lasa M, Polevoda B, Gazquez C, Elosegui-Artola A, Kim DS, De Juan-Pardo E, Demeyer K, Hole K, Larrea E, et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc Natl Acad Sci USA. 2012;109:12449–12454. doi: 10.1073/pnas.1210303109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting MW, de S, Carvalho LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Yan Y, Harper S, Speicher DW, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat Struct Biol. 2002;9:862–869. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- Yang D, Fang Q, Wang M, Ren R, Wang H, He M, Sun Y, Yang N, Xu RM. Nalpha-acetylated Sir3 stabilizes the conformation of a nucleosome-binding loop in the BAH domain. Nat Struct Mol Biol. 2013;20:1116–1118. doi: 10.1038/nsmb.2637. [DOI] [PubMed] [Google Scholar]
- Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Vander Heiden MG, Yang R, Li F, Locasale JW, et al. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–620. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Marmorstein R. Histone acetyltransferases: rising ancient counterparts to protein kinases. Biopolymers. 2013;99:98–111. doi: 10.1002/bip.22128. [DOI] [PMC free article] [PubMed] [Google Scholar]