

Abstract
Bioluminescence imaging with luciferase-luciferin pairs is a popular method for visualizing biological processes in vivo. Unfortunately, most luciferins are difficult to access and remain prohibitively expensive for some imaging applications. Here we report cost-effective and efficient syntheses of D-luciferin and 6′-aminoluciferin, two widely used bioluminescent substrates. Our approach employs inexpensive anilines and Appel's salt to generate the luciferin cores in a single pot. Additionally, the syntheses are scalable and can provide multi-gram quantities of both substrates. The streamlined production and improved accessibility of luciferin reagents will bolster in vivo imaging efforts.
Bioluminescence imaging is among the most powerful techniques for visualizing cells and other biological features.1,2 At the heart of this technology are enzymes (luciferases) that catalyze the oxidation of small molecule substrates (luciferins), releasing visible light in the process (Figure 1).2 Bioluminescent photons can penetrate tissues— even in intact rodents—making this technique well suited for imaging in vivo and other complex environments. Indeed, bioluminescence has been widely used to monitor cell proliferation and migration, gene expression patterns, and enzyme activities in a variety of preclinical models.1,2
Figure 1.
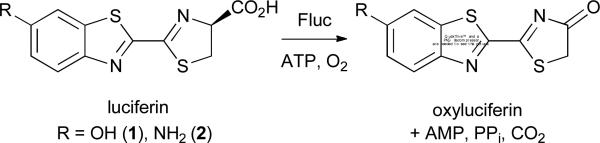
Firefly luciferase (Fluc) catalyzes the oxidation of D-luciferin (1), its native substrate, and various analogs, including the 6′-amino compound (2). Light is produced during the enzymatic reactions.
The most prevalent luciferase-luciferin pair in biological imaging originates from the firefly. Firefly luciferase (Fluc) catalyzes the oxidation of D-luciferin (1), producing yellow-green light at room temperature (Figure 1).3 Fluc can be expressed in many cell and tissue types, and when D-luciferin is administered, photons are produced. Fluc can also catalyze light emission with a variety of D-luciferin analogs,4 including the 6′-amino variant (2)5-7 and related cyclic amino analogs,8,9 heterocyclic derivatives,4,10-12 and luciferins with extended pi systems.13 Some of these molecules emit different colours of light and are thus useful for multi-spectral imaging. Other analogs are more cell permeant than D-luciferin,14 making them attractive for sensitive imaging applications. “Caged” forms of luciferin can also be used in conjunction with Fluc to measure enzyme activities15 and monitor cell-cell interactions in vivo.16
Given their broad utility and critical roles in biological imaging, it is surprising that luciferins remain difficult to access in an expedient and cost-effective manner.17,18 The first synthesis of D-luciferin reported by White in 1961 was seven steps and provided the compound in only 6% overall yield.19-21 Iterative improvements to this route have been reported over the past several decades, although the basic strategy remains the same: generate a 2-carboxy-substituted benzothiazole, replace the carboxylate with a cyano group, and condense the resulting cyanobenzothiazole with D-cysteine.17,22-24 While reliable, these routes remain low yielding and unnecessarily long. Functionalized cyanobenzothiazoles can be purchased directly from commercial suppliers and condensed with cysteine to prepare luciferins in a single step. However, these reagents are expensive and not practical for studies requiring multi-gram quantities of a light-emitting substrate.
We recently reported an alternative method to synthesize D-luciferin.10 This approach relies on benzothiazole formation from aniline starting materials and the dithiazolium chloride salt 3 (also known as Appel's salt, Scheme 1). Appel's salt condenses readily with arylamines, and the resulting iminodithiazoles can be opened with a variety of nucleophiles, generating cyanothioformamides; these moieties can then be cyclized to afford benzothiazole products (Scheme 1).25-27 Appel's salt can also be prepared in bulk quantities (>500 g from simple addition of sulphur monochloride to chloroacetonitrile) and stored indefinitely, making it attractive for large-scale work. To prepare D-luciferin via this approach, we treated p-anisidine (4) with Appel's salt and fragmented the resulting dithiazole with DBU. The product, thioformamide 6, was then cyclized using modern C-H activation chemistry to afford cyanobenzothiazole 7. Subsequent protecting group removal and cysteine condensation ultimately provided the desired luciferin 1. Altogether, this route provided D-luciferin in just 5 steps and 42% overall yield, marked improvements over previous syntheses. We were also able to use this procedure to access unique luciferin analoguess. For example, when ortho-substituted anilines (e.g., o-NH2 and o-OH) were used in place of 4, benzimidazole and benzoxazole luciferins were isolated.10,12
Scheme 1.

Retrosynthetic analysis of D-luciferin (1) and its 6′ amino analogue (2). Both molecules can be accessed from aniline starting materials and Appel's salt (3).
Despite the initial successes and notable advances of our published route, limitations remained. The synthesis employed expensive metal reagents and large volumes of solvent. Several time-intensive purification steps were also required. These features precluded the easy preparation of large quantities (>10 g) of 1. Here we report an improved, streamlined synthesis of D-luciferin (1) that requires only two steps from inexpensive starting materials. This chemistry is also applicable to synthesizing D-luciferin analogs, including aminoluciferin (2).
In early attempts to optimize our published synthesis, we focused on the dithiazole fragmentation procedure (step 2, Scheme 2). This reaction employed DBU, a reagent that is notoriously difficult to remove from reaction mixtures via extraction or flash column chromatography. Indeed, in our hands, multiple chromatographic separations were required to isolate 5 from residual DBU upon scale up; this purification scheme quickly became impractical. Since DBU functions to break apart the dithiazole, we reasoned that other, more tractable nucleophiles could be used in its place. Initial attempts with triphenyl phosphine and sodium sulphide, though, proved unsuccessful. Triphenyl phosphine (and the corresponding oxide) was similarly difficult to remove from the reaction mixture. Sodium sulphide, by contrast, was easy to remove, but converted 5 to an N-aryl dithiooxamide (S1, Scheme S1) instead of the desired formamide 6. We attributed this result to sulphide being a strong, yet sterically unencumbered nucleophile, capable of additional reactivity with cyanothioformamides. Fortunately, a less nucleophilic anion— thisosulfate (S2O 2−3)—provided 6 in excellent yield (Scheme 3) with no dithiooxamide observed. Compound 6 also precipitated from the reaction mixture and could be collected by filtration, eliminating the need for column chromatography. We further discovered that both the formation and fragmentation of 5 (with thiosulfate) could be performed in a single pot, eliminating another onerous purification step (Scheme 3). With 6 in hand, we completed the synthesis of D-luciferin (1) using our previously published method.10 Collectively, this route improved the overall yield of D-luciferin (60%) and shortened the time involved by eliminating one synthetic step entirely, along with two chromatographic separations.
Scheme 2.

Previously reported synthesis of D-luciferin.10
Scheme 3.

Synthesis of D-luciferin (1) using an initial one-pot condensation/fragmentation procedure.
In addition to the fragmentation step, we recognized that formation of cyanobenzothiazole 7 in the original synthesis (step 3, Scheme 2) was not optimal. This step required expensive metal reagents, non-ideal solvents, excess TBAB, and dilute conditions.28 Based on work from Rees,29-31 we reasoned that 7 might be attainable directly from 5 via thermolysis. This would eliminate the need for metal-mediated cyclization entirely and shorten the overall synthesis (Scheme 4, lower). However, initial attempts to cyclize 5 in refluxing DMF resulted in degradation or afforded thioformamide 6 as the major product. The cyclization yield improved dramatically at higher temperatures using sulfolane and sealed tubes. Under these conditions, we also observed trace amounts of deprotected phenol 8. This prompted us to explore whether 5 could be cyclized and deprotected in a single pot (Scheme 4, lower). Indeed, upon heating 5 in sulfolane and subsequent treatment with pyr•HCl, 8 was isolated in moderate yield (61%), along with the protected phenol 7 (20%). Re-subjecting 7 to pyr•HCl increased the total yield of 8. Excitingly, the cyclization/deprotection sequence could also be coupled with the first synthetic step: formation of imine 5 (as in Scheme 3). When 4 was treated with Appel's salt, followed by rigorous heating and pyr•HCl, 8 and 7 were isolated in 51 and 21 percent yields, respectively. (Scheme 4, upper). Subsequent condensation of 8 with D-cysteine provided luciferin 1. Thus, D-luciferin (1) can be prepared from inexpensive starting materials in only two steps and 44% overall yield. To our knowledge, this represents the shortest synthesis of D-luciferin from simple anilines to date. Moreover, these conditions are scalable (> 20 g of 8 can be routinely produced in a single reaction step), and the route is compatible with a variety of phenol protecting groups.
Scheme 4.

Synthesis of D-luciferin (1) via Appel's salt condensation and thermolysis. The iminodithiazole adduct 5 can be isolated following treatment of p-anisidine (4) with Appel's salt (lower) or carried on directly into the cyclization and deprotection steps (upper).
We next attempted to apply the improved route to the synthesis of aminoluciferin 2. This analogue and related variants are attractive for bioluminescence imaging owing to their ease of derivatization and, in some cases, enhanced cell permeability.4,6,14 Initial attempts to access 2 from p-phenylenediamine, though, were unsuccessful and afforded a complex mixture of products. We therefore moved forward with nitroaniline 9 and condensed this material with Appel's salt 3 (Scheme 5A). Attempts to generate benzothiazole 11 via thermal cyclization of 10 were low yielding, likely due to the electron-deficient nature of the aromatic ring. Benzothiazole 11 was accessible on gram-scale, though, using a route similar to the one shown in Scheme 3. Aniline 9 was treated with Appel's salt (3), followed by sodium thiosulfate (to fragment the dithiazole intermediate) to provide 10. Subsequent Pd-mediated cyclization generated 11 in 74% yield (Scheme 5C). Reduction of the nitro group followed by cysteine condensation ultimately afforded aminoluciferin 2.
Scheme 5.

Synthesis of aminoluciferin 2. (A) Cyanobenzothiazole intermediate 11 was accessed via palladium-mediated cyclization (top) or metal-free cyclization (bottom). (B) A reduction-condensation sequence was used to prepare 2.
We were able to further improve the aminoluciferin synthesis drawing inspiration from our earlier work. We previously demonstrated that ortho-nucleophiles can trap Appel's salt adducts to afford cyclized products.10 Thus, we reasoned that installing a thiol ortho to the amine in 9 would enable more facile access to 6′-aminoluciferin (2, Scheme 5). Attempts to prepare aminothiol 14 from the corresponding ortho-chloro aniline via nucleophilic aromatic substitution were not successful. However, 14 could be generated in situ by incubating commercially available 13 with hydrazine (Scheme 5B). Subsequent addition of Appel's salt 3 provided 11 in 62% yield. Subsequent nitro group reduction and cyclization provided 2 in 54% overall yield. Notably, this is the shortest and highest yielding synthesis of 6′-aminoluciferin (2) to date.
In summary, we developed an improved method to access two key luciferin molecules for bioluminescence imaging. The approach builds off earlier work using anilines and readily accessible Appel's salt to generate luciferin cores. The processes eliminate costly reagents and time-intensive purifications, improving both the speed and efficiency of luciferin construction. The syntheses are also scalable and have used to produce bulk quantities of the light-emitting substrates. The methods reported here will streamline the production of both known and novel luciferins, and thus drive the continued expansion of the bioluminescent toolkit.
Supplementary Material
Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01-GM107630 to J.A.P.). D.C.M. was supported by an Allergan Graduate Fellowship, and W.B.P. was supported by the National Science Foundation via the BEST IGERT program (DGE-1144901). We also thank the Chamberlin, Overman, Jarvo, Rychnovsky, and Nowick labs at UCI for reagents, along with members of the Prescher lab for helpful discussions.
Footnotes
Electronic supplementary information (ESI) available.
Notes and references
- 1.Paley MA, Prescher JA. MedChemComm. 2014;5:255. doi: 10.1039/C3MD00288H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prescher JA, Contag CH. Curr. Op. Chem. Bio. 2010;14:80. doi: 10.1016/j.cbpa.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Wood KV. Photochem. Photobiol. 1995;62:662. [Google Scholar]
- 4.Adams ST, Jr., Miller SC. Curr. Op. Chem. Bio. 2014;21:112. doi: 10.1016/j.cbpa.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White EH, Worther H, Seliger HH, Mcelroy WD. J. Am. Chem. Soc. 1966;88:2015. [Google Scholar]
- 6.Shinde R, Perkins J, Contag CH. Biochemistry. 2006;45:11103. doi: 10.1021/bi060475o. [DOI] [PubMed] [Google Scholar]
- 7.Viviani VR, Neves DR, Amaral DT, Prado RA, Matsuhashi T, Hirano T. Biochemistry. 2014;53:5208. doi: 10.1021/bi500160m. [DOI] [PubMed] [Google Scholar]
- 8.Reddy GR, Thompson WC, Miller SC. J. Am. Chem. Soc. 2010;132:13586. doi: 10.1021/ja104525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mofford DM, Reddy RR, Miller SC. J. Am. Chem. Soc. 2014;136:13277. doi: 10.1021/ja505795s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCutcheon DC, Paley MA, Steinhardt RC, Prescher JA. J. Am. Chem. Soc. 2012;134:7604. doi: 10.1021/ja301493d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conley NR, Dragulescu-Andrasi A, Rao J, Moerner WE. Angew. Chem. Int. Ed. Engl. 2012;51:3350. doi: 10.1002/anie.201105653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodroofe CC, Meisenheimer PL, Klaubert DH, Kovic Y, Rosenberg JC, Behney CE, Southworth TL, Branchini BR. Biochemistry. 2012;51:9807. doi: 10.1021/bi301411d. [DOI] [PubMed] [Google Scholar]
- 13.Jathoul AP, Grounds H, Anderson JC, Pule MA. Angew. Chem. Int. Ed. Engl. 2014 doi: 10.1002/anie.201405955. DOI: 10.1002/anie.201405955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans MS, Chaurette JP, Adams ST, Jr., Reddy GR, Paley MA, Aronin N, Prescher JA, Miller SC. Nat. Methods. 2014;11:393. doi: 10.1038/nmeth.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Chen LZ, Du LP, Li MY. Chem Soc Rev. 2013;42:662. doi: 10.1039/c2cs35249d. [DOI] [PubMed] [Google Scholar]
- 16.Sellmyer MA, Bronsart L, Imoto H, Contag CH, Wandless TJ, Prescher JA. Proc. Natl. Acad. Sci. U. S. A. 2013;110:8567. doi: 10.1073/pnas.1218336110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meroni G, Rajabi M, Santaniello E. ARKIVOC. 2009:265. [Google Scholar]
- 18.Fraga H. Photochem. Photobiol. Sci. 2008;7:146. doi: 10.1039/b719181b. [DOI] [PubMed] [Google Scholar]
- 19.White EH, McCapra F, Field GF, McElroy WD. J. Am. Chem. Soc. 1961;83:2402. [Google Scholar]
- 20.White EH, McCapra F, Field GF. J. Am. Chem. Soc. 1963;85:337. [Google Scholar]
- 21.White EH, Rapaport E, Seliger, H H, Hopkins TA. Bioorg. Chem. 1971;1:92. [Google Scholar]
- 22.Seto S, Ogura K, Nishiyama Y. Bull. Chem. Soc. Jpn. 1963;36:331. [Google Scholar]
- 23.Wurfel H, Weiss D, Beckert R, Guther A. J. Sulfur Chem. 2012;33:9. [Google Scholar]
- 24.Ciuffreda P, Casati S, Meroni G, Santaniello E. Tetrahedron. 2013;69:5893. [Google Scholar]
- 25.Appel R, Janssen H, Siray M, Knoch F. Eur. J. Inorg. Chem. 1985;118:1632. [Google Scholar]
- 26.Kim K. Sulfur Reports. 1998;21:147. [Google Scholar]
- 27.Cuadro AM, Alvarezbuilla J. Tetrahedron. 1994;50:10037. [Google Scholar]
- 28.Inamoto K, Hasegawa C, Hiroya K, Doi T. Org. Lett. 2008;10:5147. doi: 10.1021/ol802033p. [DOI] [PubMed] [Google Scholar]
- 29.Rees CW. J. Heterocycl. Chem. 1992;29:639. [Google Scholar]
- 30.Rakitin OA, Rees CW, Vlasova OG. Tetrahedron Lett. 1996;37:4589. [Google Scholar]
- 31.Besson T, Rees CW. J. Chem. Soc., Perkin Trans. 1995;1:1659. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.