

Abstract
In the course of detecting nuclear transcription factors by electrophoretic mobility shift assay using digoxigenin (DIG)-labeled probes, we encountered a problem with a considerable nonspecific shift band in negative control lanes from which protein extracts were omitted. This nonspecific shift band can interfere with the detection of the desired target protein. Purification of the DIG-labeled probes by removing unincorporated DIG-labeled nucleotides did not resolve the problem. However, the introduction of an additional step of heating at 95°C for 5 min and subsequent re-annealing after DIG-labeled probe synthesis eliminated these nonspecific shift bands and allowed accurate analysis of the target protein.
Keywords: Non-radioactive labeling, Digoxigenin, Electrophoretic mobility shift assay
The use of non-radioisotopic (non-RI)1 nucleic acid probes has increased extensively in the past two decades. Advantages of the non-RI labeling systems over RI labeled probes include economy, stability and safety, and have led to applications in various techniques. One of the most successful labeling and detection systems is the DIG system [1–7], based on the hapten digoxigenin (DIG) (derived from Digitalis) and anti-DIG antibody conjugates [6, 7]. In the course of analysis of nuclear transcription factors by electrophoretic mobility shift assay (EMSA) using DIG-labeled probes, we encountered a problem with a considerable nonspecific shift band in a negative control lane from which protein extract had been omitted. This nonspecific shift band interfered with the detection of the desired target protein and disturbed accurate analysis. Here, we report on a method to eliminate the nonspecific shift band in EMSA using the DIG system.
EMSAs were carried out using a DIG gel shift kit (Roche Diagnostic Corporation, Indianapolis, IN, USA) according to the manufacturer’s instructions. The oligonucleotide containing the consensus binding sequence for the nuclear factor-kappa B (NF-κB) (sense, 5′-AGTTGAGGGGACTTTCCCAGGC-3′; antisense, 5′-GCCTGGGAAAGTCCCCTCAACT-3′) was DIG-labeled by terminal transferase (TdT) using a 3′-end labeling kit after annealing. The reaction mix included 4 μL of 5× concentrated buffer (1 M potassium cacodylate, 0.125 M Tris-HCl, 1.25 mg/mL bovine serum albumin; pH 6.6), 4 μL of 25 mM CoCl2, 1 μL of 1 mM Dig-ddUTP, 100 ng of double-stranded oligonucleotide, 400 units of TdT and double-distilled H2O to obtain a final reaction volume of 20 μL. The labeling reaction was carried out by incubating at 37°C for 15 min and then stopped with 2 μL of a 0.2 M ethylenediaminetetraacetic acid (EDTA) (pH 8.0). Then we added double-distilled H2O to obtain a final probe concentration of 4 ng/μl. We confirmed that the labeling reaction produced the expected amount of labeled oligonucleotides by dot-blot. Nuclear extracts were prepared as described previously [8]. Nuclear extracts were added to 20 μL (final volume) of reaction buffer (20 mM Hepes, pH 7.6, 30 mM KCl, 10 mM (NH4)2SO4, 1 mM dithiothreitol(DTT), 1 mM EDTA, 0.2% (v/v) Tween 20, 1 μg poly (dI-dC) · poly(dI-dC) and 0.1 μg poly L-lysine) together with the DIG-labeled probes (~0.8 ng) and the reaction mixture was incubated at room temperature for 15 min. Protein-DNA complexes were then separated on a 5 % (w/v) native polyacrylamide gel run in 0.5× TBE buffer and electrically transferred to a nylon membrane (Roche). The DIG-label was detected by chemiluminescence and film exposure employing alkaline phosphatase-conjugated anti-DIG antibody (Roche) and CSPD (Roche).
The problem we encountered involving a band in negative control lanes with no protein extracts is illustrated in Fig. 1, lane 1. A band of equal intensity and migration position was also observed in all lanes containing protein extracts (Fig. 1, lane 2, 3). The non-specificity of these observed bands was demonstrated by a lack of competition with a 200-fold excess of cold oligonucleotides (Fig. 1, lane 3). This nonspecific shift band could interfere with detection of a specific band, particularly when a target band might have similar mobility (e.g. [9]).
Fig. 1.
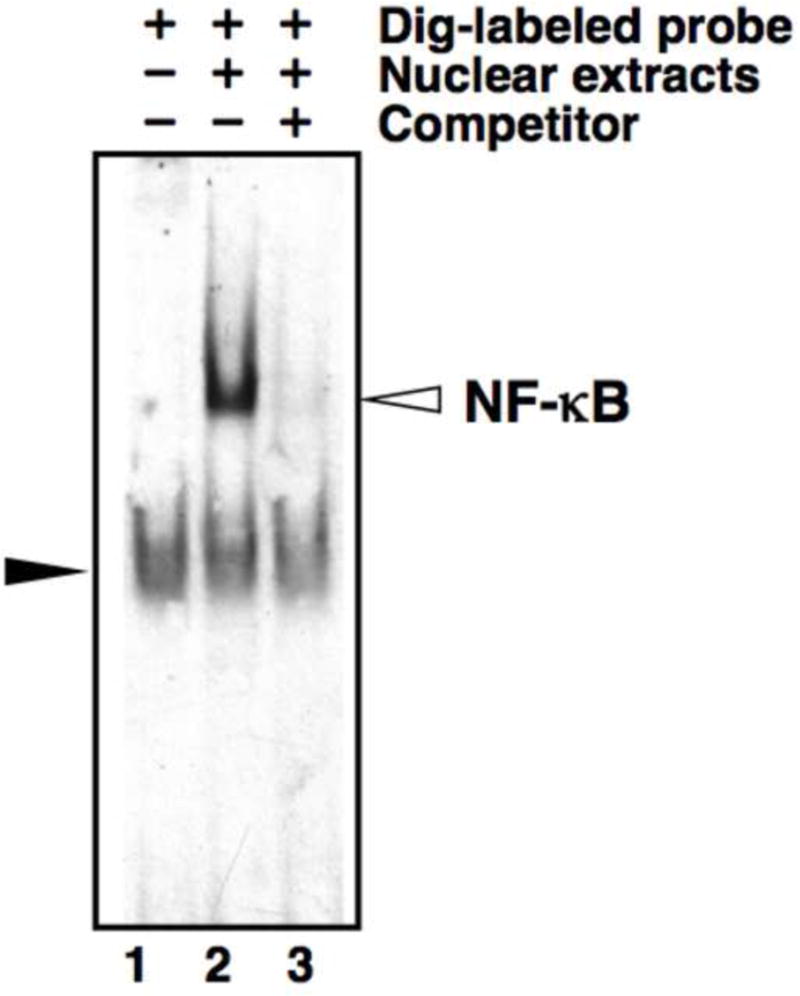
Nonspecific shift bands in EMSA using DIG-labeled probes. 1 μg of total nuclear extract from WEHI-231 cells was used for EMSA with DIG-labeled double-stranded NF-κB consensus oligonucleotide probes. Lane 1: negative control, WEHI-231 nuclear extract omitted. Lane 2: WEHI-231 nuclear extract included. Lane 3: WEHI-231 nuclear extract plus 200-fold excess of unlabeled NF-κB consensus oligonucleotide (specific competitor). The open arrow indicates the position of NF-κB band; the closed arrow indicates the nonspecific shift band.
In the standard protocol of the DIG system, it is considered unnecessary to purify the DIG-labeled probes after the labeling reaction, because the unincorporated free DIG-labeled nucleotides do not bind to the membrane and do not cause background signal, unlike RI-labeled probes [5]. Just to be sure, the unincorporated free DIG-labeled nucleotides were separated from the DIG-labeled probes using Amicon Ultra Centrifugal Filters 10K (Millipore Corporation, Billerica, MA, USA), but that did not resolve the problem. Additionally, we used fresh reagents of the highest quality, and avoided denaturation of the DIG-labeled probes that might result from overheating during electrophoresis by performing the electrophoresis at 4°C, and used fresh oligonucleotides of the highest grade available. However, the problem was not resolved, although the intensity of the nonspecific shift band decreased somewhat by using the highest-grade oligonucleotides compared to the signal seen when using oligonucleotides purified just by desalting.
On examining the molecular weight of the nonspecific shift band, we noticed that it was about 50 kDa, which is near the molecular weight of TdT (Fig. 2A, lane 1). The molecular weight of TdT is about 58 kDa [10], but the TdT that we used was a recombinant enzyme with a molecular weight of about 45 kDa (according to the manufacturer, Roche). We speculated that TdT is involved in the nonspecific shift band. To test this hypothesis, we heated the DIG-labeled probes at 95°C for 5 min and subsequently cooled the probes slowly to 15°C to re-anneal. We then used this probe as is for EMSA. The addition of this simple manipulation resulted in the complete elimination of the nonspecific shift band in the negative control lane (Fig. 2B, lane 2), thereby eliminating also the interference with the detection of the desired target protein (Fig. 2B, lane 4).
Fig. 2.
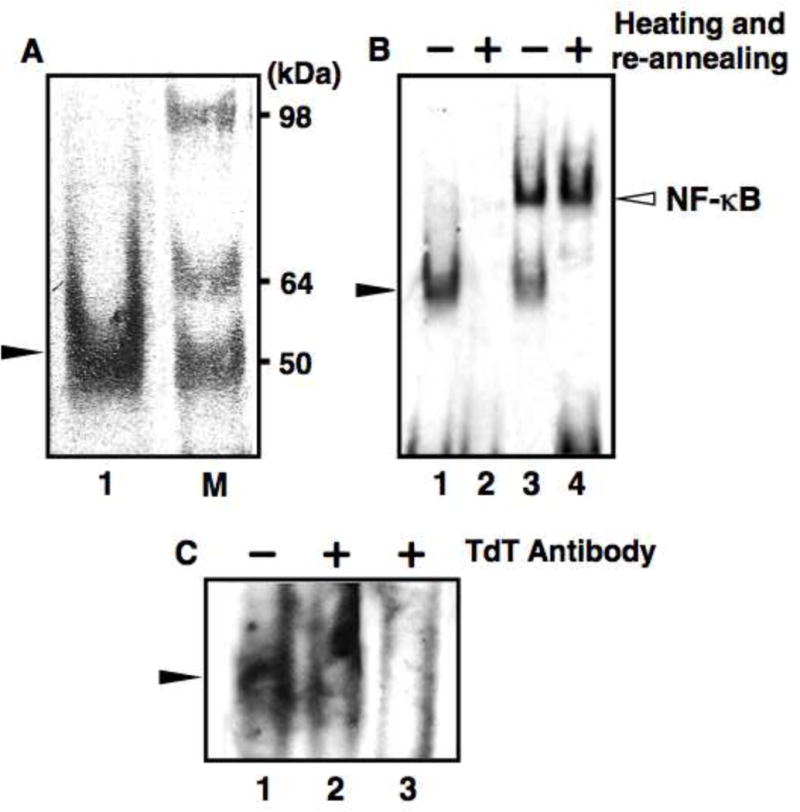
Elimination of nonspecific shift bands in EMSA using DIG-labeled probes. Non-specific bands were eliminated by heating at 95°C for 5 min and subsequent re-annealing of DIG-labeled probes. (A) Approximate molecular weight (MW) of the nonspecific shift band. EMSA was performed with a sample from which nuclear extracts were omitted (lane 1). SeeBlue marker (Invitrogen, Carlsbad, CA, USA) was used as a reference to determine the approximate MW (lane M). (B) EMSA with DIG-labeled double-stranded NF-κB consensus oligonucleotide probes that were unheated (lane 1, 3) or heated at 95°C for 5 min and re-annealed (lane 2, 4). Lane 1, 2: Negative control, WEHI-231 nuclear extracts were omitted. Lane 3, 4: WEHI-231 nuclear extracts (1 μg) were included. (C) Identification of the protein bound to DIG-labeled probes. Lane 1: no TdT antibody (Santa Cruz Biotechnologies, Santa Cruz, CA, USA) added. Lanes 2 and 3: addition of 1 μg and 3 μg of TdT antibody respectively. The open arrow indicates the position of NF-κB band; the closed arrow indicates the nonspecific shift band.
It has been reported that TdT can bind to double stranded DNA and retard the band corresponding to double-stranded DNA in EMSA [11]. We surmised that the nonspecific shift band was caused by binding of TdT to double stranded DNA (DIG-labeled probes). In order to verify this, we used TdT antibody (Fig. 2C). The addition of increasing amounts of TdT antibody decreased the nonspecific shift band intensity; 3 μg of TdT antibody almost completely disrupted the band, suggesting that this band was formed by binding of TdT with the DIG-labeled probes.
A likely explanation for the cause of the nonspecific shift band that was eliminated following the heating step is as follows: It is conceivable that TdT remained bound to a part of the double-stranded DIG-labeled probes for some reason, and that it thereby contributed to the nonspecific shift bands we observed, by the difference in mobility from the free double-stranded DIG-labeled probes to which TdT was not bound. The same problems with the nonspecific shift band in negative control lanes occurred also for DIG-labeled probes with various DNA sequences and chain length (data not shown), which suggests that this problem occurs for some reason that is not dependent on the DNA sequence. According to the Manufacturer’s instructions for the DIG-system (e.g. [5]), the labeling reaction of DNA is stopped by addition of EDTA. This is a non-denaturing method of stopping catalytic activity that makes use of the property of TdT whereby catalytic activity is dramatically enhanced by the presence of divalent metal ions [12]. However, restart of catalysis could happen, if conditions recover that allow the enhancement of catalysis. In enzyme catalysis, the enzyme temporarily binds to a substrate and separates from a product through intermediate enzyme/substrate complex and enzyme/product complex. Even under the divalent metal ion-absent condition following the chelation by EDTA, catalysis might have proceeded at very slow rate and the intermediate might have been observed as the nonspecific shift band. A portion of the DIG-labeled oligonucleotides appears to have separated from the enzyme (TdT) as products normally, because the desired target proteins were observed along with the nonspecific band in EMSA (Fig. 1, lane 2). We have no definite information on the reasons why TdT remains bound to the DIG-labeled probes. A further analysis could include purification of the labeled probe by agarose gel electrophoresis and recombine that with fresh terminal transferase to evaluate the conditions for complex formation. However, while TdT may not be able to separate completely from the products under certain conditions, this appears to be corrected by thermal denaturation, accomplished by heating at 95°C.
TdT is routinely used to add one or more deoxynucleotides to the 3′-termini of single-stranded [13] or double-stranded DNA [14] and to label oligodeoxyribonucleotides with modified nucleotides (e.g. biotin- [15], fluorescein- [16], and digoxigenin- [3] labeled nucleotides). In order to label nucleic acid, TdT is used not only for EMSA, but also for Southern blot, Northern blot, dotblot, slotblot, in situ hybridization and so on, extensively. Therefore, not only in EMSA but also in these methods, similar problems involving TdT remaining bound to the labeled probes might happen under some conditions. In most cases, experimental data with a problem, such as nonspecific bands in negative control lanes in EMSA, would not likely be published. Therefore, these issues may not come to light, even if there are widespread problems. However, it also is possible that not a few researchers have had similar problems. In any case, our results highlight the utility of adding a simple step of heating the DIG-labeled probes once and re-annealing them, in cases in which nonspecific bands or signals have been observed.
Acknowledgments
We thank Dr. Jennifer J. Schlezinger for providing the WEHI-231 nuclear extracts and suggestions. Support by NIH-SRP P42ES007381 to JJS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used: RI, radioisotopic; DIG, digoxigenin; EMSA, electrophoretic mobility shift assay; TdT, terminal transferase; EDTA, ethylenediaminetetraacetic acid; DTT, dithiothreitol; NF-κB, nuclear factor-kappa B.
References
- 1.Kessler C. The digoxigenin system: principle and applications of the novel nonradioactive DNA labeling and detection system. BioTechnology Int. 1990;1990:183–194. [Google Scholar]
- 2.Höltke HJ, Kessler C. Nonradioactive labeling of RNA transcripts in vitro with the hapten digoxigenin (DIG); hybridization and ELISA-based detection. Nucleic Acids Res. 1990;18:5843–5851. doi: 10.1093/nar/18.19.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmitz GG, Walter T, Seibl R, Kessler C. Nonradioactive labeling of oligonucleotides in vitro with the hapten digoxigenin by tailing with terminal transferase. Anal Biochem. 1991;192:222–231. doi: 10.1016/0003-2697(91)90212-c. [DOI] [PubMed] [Google Scholar]
- 4.Höltke HJ, Sagner G, Kessler C, Schmitz G. Sensitive chemiluminescent detection of digoxigenin-labeled nucleic acids: a fast and simple protocol and its applications. BioTechniques. 1992;12:104–114. [PubMed] [Google Scholar]
- 5.Höltke HJ, Ankenbauer W, Mühlegger K, Rein R, Sagner G, Seibl R, Walter T. The digoxigenin (DIG) system for non-radioactive labelling and detection of nucleic acids–an overview. Cell Mol Biol. 1995;41:883–905. [PubMed] [Google Scholar]
- 6.Kessler C. The digoxigenin system: anti-digoxigenin technology – a survey on the concept and realization of a novel bioanalytic indicator system. Mol Cell Probes. 1991;5:161–205. doi: 10.1016/0890-8508(91)90041-h. [DOI] [PubMed] [Google Scholar]
- 7.Seibl R, Höltke HJ, Rüger R, Meindl A, Zachau HG, Rasshofer R, Roggendorf M, Wolf H, Arnold N, Wienberg J, Kessler C. Nonradioactive labeling and detection of nucleic acids: III. Applications of the digoxigenin system. Biol Chem Hoppe-Seyler. 1990;371:939–951. doi: 10.1515/bchm3.1990.371.2.939. [DOI] [PubMed] [Google Scholar]
- 8.Schlezinger JJ, Jensen BA, Mann KK, Ryu HY, Sherr DH. Peroxisome proliferator-activated receptor gamma-mediated NF-kappa B activation and apoptosis in pre-B cells. J Immunol. 2002;162:6831–6841. doi: 10.4049/jimmunol.169.12.6831. [DOI] [PubMed] [Google Scholar]
- 9.Goldstone HMH, Tokunaga S, Schlezinger JJ, Goldstone JV, Stegeman JJ. EZR1: a novel family of highly expressed retroelements induced by TCDD and regulated by a NF-κB-like factor in embryos of zebrafish (Danio rerio) Zebrafish. 2012;9:15–25. doi: 10.1089/zeb.2011.0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollum FJ, Brown M. A high molecular weight form of terminal deoxynucleotidyl transferase. Nature. 1979;278:191–192. doi: 10.1038/278191a0. [DOI] [PubMed] [Google Scholar]
- 11.Robbins DJ, Coleman MS. Initiator role of double stranded DNA in terminal transferase catalyzed polymerization reactions. Nucleic Acids Res. 1988;16:2943–2957. doi: 10.1093/nar/16.7.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robbert LR. Terminal deoxynucleotidyltransferase. In: Boyer PD, editor. The Enzymes. 3. Academic Press; New York: 1973. pp. 105–118. [Google Scholar]
- 13.Hayes FN, Mitchell VE, Ratliff RL, Schwartz AW, Williams DL. Incorporation efficiency of small oligo-5′-nucleotide initiators in the terminal deoxyribonucleotide transferase reaction. Biochemistry. 1966;5:3625–3629. doi: 10.1021/bi00875a035. [DOI] [PubMed] [Google Scholar]
- 14.Roychoudhury R, Jay E, Wu R. Terminal labeling and addition of homopolymer tracts to duplex DNA fragments by terminal deoxynucleotidyl transferase. Nucleic Acids Res. 1976;3:863–877. doi: 10.1093/nar/3.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A, Tchen P, Roullet F, Cohen J. Nonradioactive labeling of synthetic oligonucleotide probes with terminal deoxynucleotidyl transferase. Anal Biochem. 1988;169:376–382. doi: 10.1016/0003-2697(88)90299-0. [DOI] [PubMed] [Google Scholar]
- 16.Igloi GL, Schiefermayr E. Enzymatic addition of fluorescein- or biotin-riboUTP to oligonucleotides results in primers suitable for DNA sequencing and PCR. BioTechniques. 1993;15:486–497. [PubMed] [Google Scholar]