

Abstract
Biallelic inactivation of MEN1 encoding menin in pancreatic neuroendocrine tumors (PNETs) associated with the multiple endocrine neoplasia type 1 (MEN1) syndrome is well established, but how menin loss/inactivation initiates tumorigenesis is not well understood. We show that menin activates the long noncoding RNA maternally expressed gene 3 (Meg3) by histone-H3 lysine-4 trimethylation and CpG hypomethylation at the Meg3 promoter CRE site, to allow binding of the transcription factor cAMP response element-binding protein. We found that Meg3 has tumor-suppressor activity in PNET cells because the overexpression of Meg3 in MIN6 cells (insulin-secreting mouse PNET cell line) blocked cell proliferation and delayed cell cycle progression. Gene expression microarray analysis showed that Meg3 overexpression in MIN6 mouse insulinoma cells down-regulated the expression of the protooncogene c-Met (hepatocyte growth factor receptor), and these cells showed significantly reduced cell migration/invasion. Compared with normal islets, mouse or human MEN1-associated PNETs expressed less MEG3 and more c-MET. Therefore, a tumor-suppressor long noncoding RNA (MEG3) and suppressed protooncogene (c-MET) combination could elicit menin's tumor-suppressor activity. Interestingly, MEG3 and c-MET expression was also altered in human sporadic insulinomas (insulin secreting PNETs) with hypermethylation at the MEG3 promoter CRE-site coinciding with reduced MEG3 expression. These data provide insights into the β-cell proliferation mechanisms that could retain their functional status. Furthermore, in MIN6 mouse insulinoma cells, DNA-demethylating drugs blocked cell proliferation and activated Meg3 expression. Our data suggest that the epigenetic activation of lncRNA MEG3 and/or inactivation of c-MET could be therapeutic for treating PNETs and insulinomas.
Unraveling the molecular mechanisms controlled by genes associated with hereditary tumor syndromes may offer insights into the pathogenesis of their sporadic counterpart tumors and other tumor types. Multiple endocrine neoplasia type 1 (MEN1) is a familial tumor syndrome caused by two inactivating hits to the tumor suppressor gene MEN1 that encodes the protein menin (1, 2). The first hit is inherited in the germline, and the second hit is tissue-specific-causing tumors, most notably in multiple endocrine tissues: parathyroids, anterior pituitary, and enteroendocrine-pancreas (3). These individual tumor types can also occur sporadically in patients who do not have the MEN1 syndrome (4, 5).
Targeted disruption of both copies of Men1 in mice leads to early embryonic lethality, whereas mice with the targeted disruption of a single Men1 allele develop the MEN1 syndrome with tumors that show biallelic Men1 inactivation in the parathyroids, anterior pituitary, and endocrine pancreas (6). Interestingly, a major difference between mouse and man is the occurrence of pancreatic endocrine tumors that are insulinoma in Men1+/− mice as opposed to the more common nonfunctioning pancreatic neuroendocrine tumors (PNETs) and gastrinoma in humans with MEN1 (5, 6). Therefore, investigating pathways downstream of menin could be relevant for understanding the pathogenesis of mouse and human insulinomas.
Tumors of the endocrine pancreas known as PNETs are classified as nonfunctioning or functioning based on hormone secretion and clinical symptoms. In human nonfunctioning PNETs, 30%–44% show somatic MEN1 mutations and 43% show DAXX/ATRX mutations (7, 8). The most commonly occurring functioning PNET is insulinoma that arises from pancreatic islet β-cells and continuously secretes insulin (9). In human sporadic functioning PNETs (insulinomas), 2%–19% show MEN1 mutations, 2% show ATRX mutations, and 30% show a recurrent YY1/T372R mutation (8, 10–12).
Biallelic inactivation of MEN1 in PNETs associated with the MEN1 syndrome is well established, but how menin loss/inactivation leads to tumorigenesis is not well understood. Understanding the mechanism of action of menin in pancreatic endocrine cells through its downstream targets could provide insights about MEN1-associated tumorigenesis. An obvious question that follows is whether dysregulation of the same targets by menin-independent mechanisms could also initiate tumor formation in non-MEN1-functioning PNETs (insulinomas) that usually lack MEN1 mutations.
Menin, located primarily in the nucleus, has been reported to participate in diverse biological functions through various interacting proteins (5). One of the intensively investigated associations of menin is in the miked lineage leukemia (MLL) protein complex that catalyzes the histone-H3 lysine-4 trimethyl mark (H3K4me3) in chromatin, a mark of active transcription (13, 14). We have previously shown by genome-wide chromatin immunoprecipitation (ChIP)- sequencing (ChIP-Seq) analysis that H3K4me3 at the Meg3 locus was specifically lost in menin-null mouse embryonic stem cells (mESCs) (15). And consequently, the expression of the long noncoding RNA (lncRNA) Meg3 was significantly reduced in menin-null mESCs (15). Whether lncRNAs play a role in MEN1 pathogenesis and for menin to elicit its tumor suppressor function was largely unknown until our recent findings from mESCs implicating the lncRNA MEG3 (15).
lncRNAs are polyadenylated RNA polymerase II-transcribed RNAs, 200 or more nucleotides in length but without obvious open reading frames to encode proteins (16). Maternally expressed gene 3 (MEG3), also known as gene-trap locus 2 (GTL2), is a monoallelically expressed lncRNA expressed from the maternal allele (online mendelian inheritance in man identification number 605636). Germline or somatic mutations in MEG3 have not been reported (online mendelian inheritance in man and COSMIC databases); however, the loss of MEG3 expression is found in various human tumors and tumor cell lines (17). Nevertheless, MEG3 target genes are not well defined, and the mechanisms of MEG3 regulation and function are not well understood.
In this study, we have characterized the epigenetic regulation of MEG3 by menin in pancreatic β-cells and identified the c-MET protooncogene as a MEG3 target gene. c-MET is the tyrosine kinase receptor for hepatocyte growth factor. MET amplification or overexpression is observed in a variety of tumors, and it is associated with proliferation, invasion, and metastasis (18), thus serving as an attractive therapeutic target for which several inhibitors are under clinical investigation (18). We show that MEG3 and c-MET levels are reciprocally correlated in human and mouse MEN1-associated PNETs and human sporadic insulinomas. Also, epigenetic inhibitors of DNA methylation could block the proliferation of mouse insulinoma cells and restore Meg3 expression. Thus, our data provide a strong basis for the investigation of modulating MEG3 and c-MET expression as a therapeutic option for PNETs, including insulinomas.
Materials and Methods
Primers and detailed methods
All the primers are listed in Supplemental Table 1. Detailed methods are provided in the Supplemental Methods.
Mouse and human tissues (pancreas)
Mouse experiments were conducted under the guidelines of the National Institutes of Health Animal Care and Use Committee, and an approved animal study protocol (number K070-MDB-12). Human tumor samples [four frozen MEN1 and three frozen non-MEN1 PNETs, one frozen MEN1 insulinoma and 23 archived formalin-fixed and paraffin-embedded (FFPE) sporadic insulinomas] were obtained with the informed consent from patients under National Institutes of Health Institutional Review Board-approved protocols (number NCT01005654). All five MEN1 patients carry germline mutations in MEN1. For immunohistochemistry (IHC) controls, FFPE pancreas sections of normal human pancreas (n = 7) were obtained (Abcam, IHC World, Origene, ProSci, US Biomax, Zyagen, and Dr Michael Emmert-Buck of National Cancer Institute). For RNA and DNA isolation, normal human pancreatic islet preparations (n = 6) were obtained (Lonza and the University of Alabama, Birmingham, Alabama).
Isolation of mouse pancreatic islets [wild type (WT) and tumor]
Mice (WT) were put to sleep and intraductal collagenase perfusion was performed following a standard protocol (19), and the islets were hand-picked under the dissecting microscope. Islet tumors were directly excised from the pancreas.
Meg3 cDNA cloning
Meg3 cDNA was amplified by RT-PCR using RNA from normal human islets, WT mouse islets, or RNA isolated from 5′-aza-2′-deoxycytidine (decitabine; Sigma)-treated MIN6-4N cells, with primers located at the beginning of exon 1 and the end of exon 10. PCR products were cloned into pcDNA3.1 (Invitrogen) and sequenced (Supplemental Figure 1).
Cell culture and stable cell lines
Mouse insulinoma cell lines, MIN6 (20) and MIN6-4N (21), were cultured in low-glucose DMEM supplemented with 15% fetal bovine serum and 1× antibiotic/antimycotic (Invitrogen, Gemini) at 37°C and 5% CO2. MIN6-4N is a tetraploid (4N) cell line that was isolated from MIN6 cells that have mixed ploidy (2N and 4N) (21). Stable cell lines were maintained in the above medium containing 500 μg/mL of G418 (Life Technologies).
Vec and M-27 are MIN6 stable cell lines containing vector and pcDNA3.1-Myc-His-Menin, respectively (22). Vec-4N and M-27-4N are MIN6-4N stable cell lines containing vector and pcDNA3.1-Myc-His-Menin, respectively, derived by single-cell isolation from Vec and M-27.
Vector or mouse Meg3-transfected stable cell lines were generated in MIN6-4N cells as described (22). Vec-3 and Meg-5 are stable cell lines of MIN6-4N-containing vector and pcDNA3.1-mMeg3-3, respectively. Vec-9 and Meg-14 are MIN6-4N stable cell lines containing vector and pcDNA3.1-mMeg3-1, respectively. mMeg3-3 was the most abundant cDNA clone (in WT mouse islets) that lacks exons 2b and 4, and mMeg3-1 is full length with all 10 exons (Supplemental Figure 2).
DNA, RNA isolation, and quantitative real time-PCR (QRT-PCR)
DNA and RNA were isolated using the DNeasy and RNeasy kits (QIAGEN). RNA samples were treated with deoxyribonuclease I (Ambion), and first-strand cDNA synthesis was performed with an oligodeoxythymidine primer and Superscript-III (Invitrogen). QRT-PCR was performed with the Brilliant SYBR Green quantitative PCR master mix (QIAGEN) and Mx3000p thermal cycler (Stratagene). Cycle threshold (Ct) values were normalized to mouse or human glyceraldehyde-3-phosphate dehydrogenase (Gapdh). Relative gene expression changes were calculated by the 2-Δct method, and the fold changes are plotted with respect to the controls.
DNA methylation analysis
The Meg3 promoter region near the transcriptional start site was amplified from untreated and bisulfite-treated DNA, cloned into the PCR2.1 vector (Invitrogen), sequenced, and analyzed to detect differences in methylation. A second independent method for analyzing methylation at the CRE site, methylation-specific PCR, was performed with the bisulfite-treated DNA.
Meg3 promoter cloning and luciferase assay
Mouse Meg3 promoter (−560 to +122) and human MEG3 promoter (−539 to +140) were amplified from mouse or human genomic DNA and cloned into the luciferase reporter vector pGL4.10 (Promega). Luciferase assays were performed in MIN6-4N cells.
Chromatin immunoprecipitation
Chromatin from Vec and M-27 cells was processed for the ChIP assay (Upstate/Millipore). ChIP-PCR was performed with a primer pair at the CRE site in the Meg3 promoter. PCR products were detected by agarose gel electrophoresis.
Microarray analysis and validation
Deoxyribonuclease-treated RNA from three independent cultures of the vector (Vec-3) and Meg3-transfected (Meg-5) stable clones were used for gene expression microarray analysis with Affymetrix Genechip mouse genome ST 1.0 arrays (Affymetrix). Microarray hybridization and data analysis were performed at the National Institute of Diabetes and Digestive and Kidney Diseases genomics core facility. Fifteen up- and 36 down-regulated genes (changed >2.5-fold, P < .05) were validated by QRT-PCR.
Cell proliferation and viability assays
For assessing the proliferation of stable cell lines, after 6 days in culture, cells were trypsinized and counted using cell counter slides (Nexcelcom). For the effect of DNA demethylating drugs 5′-aza-2′-deoxycytidine (decitabine; Sigma) and ascorbic acid (Sigma) and the effect of c-Met inhibitor PHA-665752 (Sigma), viable cells were assessed by the dimethylthiazoldiphenyltetra-zoliumbromide (MTT) assay (Promega).
Flow cytometry and cell cycle analysis
After 6 days in culture, cells were harvested with trypsin and incubated in Vindelov's propidium iodide buffer and analyzed by fluorescence-activated cell sorting (FACSCalibur; BD Biosciences). The raw data were subjected to ModFit analysis (BD Biosciences) to determine the percentages of cells in the G0/G1, S, and G2/M phases.
Apoptosis and cell migration/invasion assays
Stable cell lines were cultured in chamber slides to assess apoptosis by using the DeadEnd terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) fluorometric assay (Promega). Cell migration/invasion was assessed with the CytoSelect 24-well cell invasion assay kit (Cell Biolabs) that contained polycarbonate membrane inserts (8 μm pore size) (basement membrane, colorimetric format). Cells were seeded at 30 000 cells/well for 48 hours. The invasive cells in the membrane inserts were stained, the stain was extracted in an extracting solution, and the OD was measured at 560 nm (Molecular Devices) as an index of the relative number of migrating/invasive cells.
Immunofluorescence and IHC
FFPE pancreas sections were stained for menin and insulin using standard double-immunofluorescence procedures. For IHC, the FFPE pancreas sections were processed for insulin and c-MET staining as described (21). The c-MET antibody (1:1000) was validated in HeLa cells (American Type Culture Collection) (Supplemental Figure 3).
Statistical analysis
Data from at least three independent experiments were considered and presented as mean ± SEM. Differences between groups were compared by Student's t tests. The P values [P < 0.05 (*) or P < 0.005 (**) or P < 0.0001 (***)] were considered significant.
Results
Menin up-regulates the expression of Meg3 in MIN6 mouse insulinoma β-cells
Menin is a part of the multiprotein MLL complex that adds the H3K4me3 mark in chromatin (14). We have previously investigated mESCs with or without menin by H3K4me3-ChIP-sequence and gene expression microarray. Both methods identified MEG3 as the most significant target of menin (15). To investigate whether Meg3 acts downstream of menin in pancreatic β-cells, we studied the effect of menin on Meg3 expression in MIN6-cells. MIN6 is a mouse β-cell tumor (insulinoma) cell line (20) that responds to changes in menin concentration (22) (23). Transient overexpression or knockdown of menin in MIN6-cells did not alter Meg3 mRNA expression or Meg3 promoter activity (Supplemental Figure 4). These data suggested that menin might regulate Meg3 by affecting epigenetic modifications that require the stable presence of menin and additional cell divisions to acquire or remove such modifications, which cannot be achieved on episomal Meg3 promoter-containing plasmids. Therefore, exogenous menin-expressing MIN6 stable cell lines were analyzed for Meg3 expression (Figure 1A). MIN6 cell lines stably expressing menin (M-27 and M-27-4N) showed significantly increased Meg3 expression compared with vector-transfected stable cell lines (P < .0001) (Figure 1B). These data indicate that Meg3 is a downstream target of menin in MIN6 mouse insulinoma β-cells.
Figure 1.
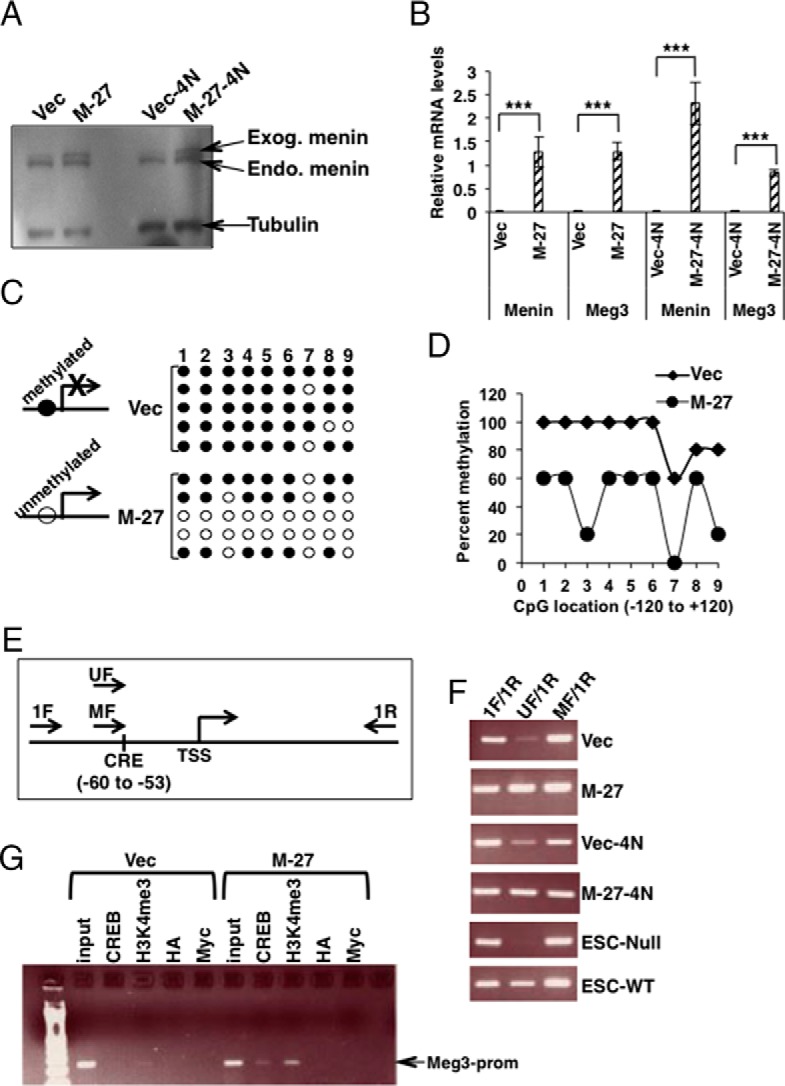
Epigenetic regulation of Meg3 by menin in MIN6 mouse insulinoma β-cells. A, Western blot detecting menin in whole-cell lysates prepared from MIN6 cell lines stably transfected with a menin-expressing plasmid [M-27 (mixed 2N+4N MIN6 cells) and M-27–4N (4N MIN6 cells)] or empty vector (Vec and Vec-4N). Tubulin was used as the loading control. Endo, endogenous menin; Exog, exogenous menin (stably transfected myc-his-tagged-menin). B, QRT-PCR assays showing significantly increased menin and Meg3 in RNA isolated from MIN6 stable cell lines shown in panel A (P < .0001). Gapdh was used as the internal control. C, Bisulfite sequencing assay of the promoter region (−120 to +120) of Meg3 using DNA isolated from MIN6 cells stably transfected with a menin-expressing plasmid (M-27) or empty vector (Vec). Reduced DNA methylation would allow Meg3 expression and increased methylation would block Meg3 expression. The numbers 1–9 indicate the nine differentially methylated CpGs in the −120 to +120 region of Meg3. Filled circles indicate methylated CpG and open circles indicate unmethylated CpG (see also Supplemental Figure 7). D, Data from panel C with significantly decreased methylation in the menin transfected MIN6 stable clone (M-27) compared with its vector control (Vec). CpG number 3 corresponds to the location of the conserved CRE site in the human and mouse Meg3 promoter. E, Schematic representation of the CRE site in the mouse Meg3 promoter and the primers for the methylation-specific PCR assay. Unmethylated (UF) and methylated (MF) primers are specific to the unmethylated and methylated CRE site, respectively. The 1F/1R primer pair spans 251 bp. The UF/1R or MF/1R primer pair spans 207 bp. TSS, transcriptional start site. F, Methylation-specific PCR assay using DNA from the indicated cell types. ESC-Null and ESC-WT are embryonic stem cells from the Men1−/− and WT mice, respectively. An increased amount of PCR product in the wells marked for UF/1R shows that exogenous menin expression in M-27 and M-27-4N unmethylates the CRE site, and the absence of the PCR product in the ESC-Null shows that, in the absence of menin, the CRE site is completely methylated. G, Agarose gel of ChIP-PCR assay with antibodies against the indicated antigens using chromatin from menin transfected MIN6 stable clone (M-27) compared with its vector control (Vec). Input corresponds to DNA isolated from chromatin used in the ChIP assay. Antihemagglutinin (HA) was used as a negative control. Anti-Myc was used to chromatin immunoprecipitate the transfected myc-his-tagged menin.
Reduced H3K4me3 has been shown to coincide with increased DNA methylation (24). Also, the Meg3 promoter region is regulated by changes in DNA methylation at CpG sites (17). Bisulfite sequencing of the −120 to +120 region of the mouse Meg3 promoter (which has nine differentially methylated CpGs) showed significantly reduced methylation in M-27 cells (P < .0001) (Figure 1, C and D). Interestingly, CpG number 3 is located in the cAMP responsive element (CRE) (5′-TGACGTCA-3′) in the Meg3 promoter that binds CRE-binding protein (CREB) (Figure 1E) (Supplemental Figure 5). The DNA methylation at the CpG in this CRE site was further analyzed by a second independent method, methylation-specific PCR (MSP) assay that could be used for the Meg3 promoter CRE-site-specific methylation analysis. The bisulfite-treated DNA from M-27 and M-27-4N cells showed an increased amount of PCR product with the ummethylated primer pair, indicating that the CpG in the CRE site was not methylated in the presence of menin, which also coincides with the expression of the Meg3 transcript (Figure 1F). The unmethylated PCR product was absent in the Men1-Null-embryonic stem cells (ESCs), showing that in the absence of menin, the CRE site is completely methylated, which coincides with the loss of the Meg3 transcript (Figure 1F).
In vitro methylation experiments have shown that CpG methylation at the conserved MEG3 CRE site disrupts CREB binding (25). However, it is not known whether this occurs in vivo. We tested this in our Vec vs M-27 cell culture model by a ChIP-PCR assay. A high occupancy of CREB and the presence of H3K4me3 at the Meg3 promoter region was observed in the M-27 cells compared with the Vec cells (Figure 1G).
Overall, these results suggest that in MIN6 mouse insulinoma β-cells, menin epigenetically regulates Meg3 expression by promoting the loss of DNA methylation due to the presence of menin-dependent H3K4me3 and CREB recruitment at the CRE site of the Meg3 promoter, thus leading to an increased expression of Meg3.
Meg3 overexpression affects cell proliferation and cell cycle progression in MIN6 mouse insulinoma β-cells
To evaluate whether Meg3 acts as a tumor suppressor in insulinoma cells, stable cell lines of MIN6 with vector or two different mouse Meg3 variants, mMeg3-3 (lacks exons 2b and 4) and mMeg3-1 (full length) were analyzed (Figure 2, A and B, and Supplemental Figure 6, A and B). Meg3-overexpressing cell lines showed significantly reduced cell proliferation compared with vector-transfected cell lines (P < .0001) (Figure 2C and Supplemental Figure 6C). Cell cycle analysis showed that when Meg3 was overexpressed, cell cycle progression was delayed, with a significant decrease in the percentage of cells in the G0/G1 phases (P < .05) and a significant increase in the percentage of cells in the G2/M phases (P < .005) (Figure 2D and Supplemental Figure 6D). These results suggest that Meg3 has a tumor suppressor function in MIN6 mouse insulinoma β-cells.
Figure 2.
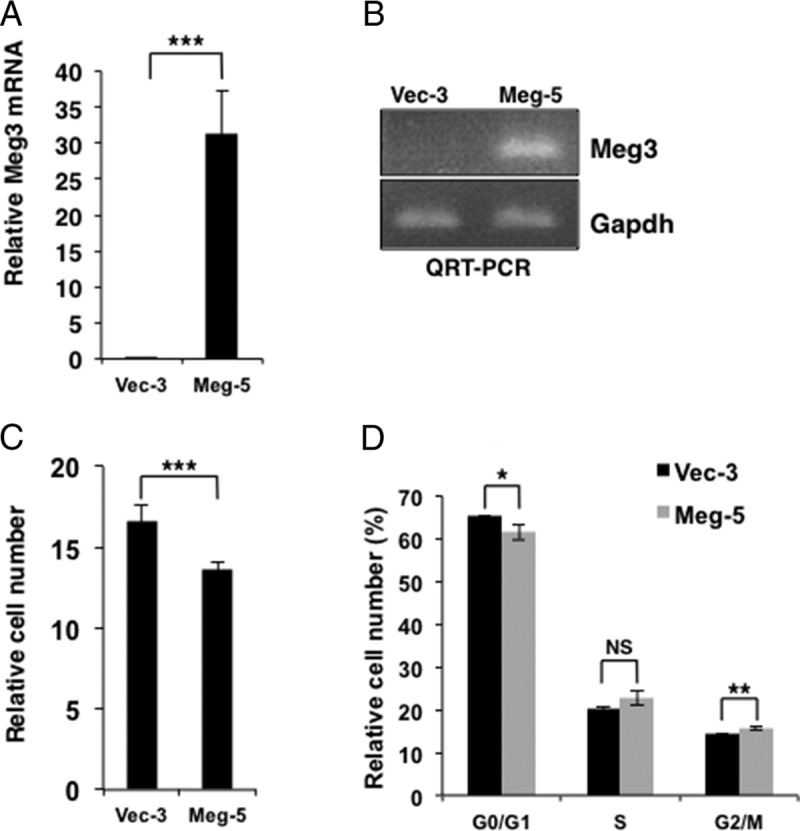
Meg3 reduces cell proliferation and delays the cell cycle in MIN6 mouse insulinoma β-cells. A, QRT-PCR assay showing significantly increased Meg3 in RNA isolated from MIN6-4N cell lines stably transfected with a mouse Meg3-expressing plasmid (cell line name is Meg-5) compared with cells stably transfected with vector (Vec-3) (P < .0001). Gapdh was used as the internal control. Meg-5 contains the transcript variant of Meg3 that lacks exons 2b and 4. B, Agarose gel showing Meg3 and Gapdh in representative RT-PCR reactions from panel A. Vector-transfected MIN6-4N cells (Vec-3) show very low levels of endogenous Meg3. C, Cell proliferation assay of Vec-3 and Meg-5 showing a significantly decreased cell number (P < .0001) in Meg 3-overexpressing MIN6 cells after 6 days in culture. D, Flow cytometry assay for cell cycle distribution of Vec-3 and Meg-5 showing a significantly decreased percentage of cells in the G0/G1 phases (P < .05) and a significantly increased percentage of cells in the G2/M phases (P < .005) in Meg 3-overexpressing MIN6-4N cells after 6 days in culture. NS, not significant. See also Supplemental Figure 6 for data from the Meg-9 stable cell line with the full-length transcript of Meg3.
Meg3 regulated genes in MIN6 mouse insulinoma β-cells
To determine Meg3 target genes, a microarray gene expression analysis was performed with RNA from the Meg-5 and Vec-3 cells. Using a 2.5-fold change and a value of P < .05 as the cutoff, 36 genes were down-regulated and 15 genes were up-regulated when Meg3 was overexpressed (Table 1). These genes were further validated by QRT-PCR (Table 1). Functional classification using the Database for Annotation, Visualization, and Integrated Discovery (26) showed an enrichment of genes associated with cell adhesion and apoptosis. Interestingly, the c-Met protooncogene was one of the top hits in the down-regulated genes. Increased expression of c-MET has been reported in many human primary and metastatic tumors (18). Thus, a microarray analysis suggested that Meg3 could suppress the expression of the protooncogene c-Met and regulate the expression of genes involved in cell adhesion and apoptosis. Meg-5 cells and Meg-14 cells showed reduced cell migration/invasion and modestly increased apoptosis compared with their vector-transfected control cells (Figure 3, A–C). We also assessed the expression of c-Met in Men1-Null-ESCs that showed reduced Meg3 expression and found that c-Met mRNA was significantly increased in the Men1-Null-ESCs (Figure 3D).
Table 1.
Genes Regulated by Meg3 in MIN6-4N Cells (Meg-5 vs Vec-3, P < .05)
| Gene Symbol | Gene Name | Microarray Fold Change | QRT-PCR Fold Change |
|---|---|---|---|
| Down-regulated | |||
| A630095E13Rik | RIKEN cDNA A630095E13 | −6.57 | −11.55 |
| Met | Met protooncogene | −5.48 | −8.37 |
| Pon3 | Paraoxonase 3 | −4.39 | −6.91 |
| Ceacam10 | Carcinoembryonic antigen-related cell adhesion molecule | −3.98 | −3.82 |
| Tnfsf10 | TNF (ligand) superfamily, member 10 | −3.92 | −4.18 |
| Hoxb9 | Homeobox B9 | −3.85 | −17.95 |
| Rab3c | Member RAS oncogene family | −3.74 | −5.41 |
| Gm501 | Predicted gene 501 | −3.61 | −10.45 |
| BC028528 | cDNA sequence BC028528 | −3.55 | 3.55 |
| Cldn1 | Claudin 1 | −3.53 | −4.39 |
| Pcdhb2 | Protocadherin-β-2 | −3.52 | −3.24 |
| Nrk | Nik-related kinase | −3.48 | −3.65 |
| Casp4 | Caspase-4 | −3.41 | −3.40 |
| Prkd3 | Protein kinase D3 | −3.32 | −4.53 |
| Cd9 | CD9 antigen | −3.31 | −2.72 |
| 4930550L24Rik | RIKEN cDNA 4930550L24 | −3.27 | −3.00 |
| Fam129a | Family with sequence similarity 129, member A | −3.23 | −6.10 |
| Gatm | Glycine amidinotransferase | −3.08 | −3.06 |
| Kctd12b | Potassium channel tetramerisation domain containing 12b | −3.05 | −4.05 |
| Hectd2 | HECT domain containing 2 | −3.02 | −5.06 |
| Klhl1 | Kelch-like 1 (Drosophila) | −2.95 | −3.10 |
| Rnasel | Ribonuclease L | −2.89 | −5.79 |
| Tfpi | Tissue factor pathway inhibitor | −2.82 | −4.56 |
| Egln3 | EGL 9 homolog 3 (Caenorhabditis elegans) | −2.82 | −4.44 |
| Mgam | Maltase-glucoamylase | −2.80 | −29.26 |
| Phldb2 | Pleckstrin homology-like domain, family B, member 2 | −2.77 | −3.10 |
| Pcdhb3 | Protocadherin-β3 | −2.71 | 2.82 |
| Fn1 | Fibronectin 1 | −2.69 | −5.01 |
| Cldn11 | Claudin 11 | −2.60 | −3.41 |
| 2610018G03Rik | RIKEN cDNA 2610018G03 | −2.55 | −3.61 |
| Samd9l | Sterile α motif domain containing 9-like | −2.50 | −8.33 |
| Casp1 | Caspase-1 | −2.50 | 1.07 |
| H2-K1 | Histocompatibility 2, K1 | −2.49 | −3.75 |
| Cd82 | CD82 antigen | −2.46 | −2.65 |
| Tm4sf4 | Transmembrane 4, superfamily member 4 | −2.46 | −2.82 |
| Pcdhb5 | Protocadherin-β5 | −2.45 | −3.00 |
| Up-regulated | |||
| Anxa2 | Annexin A2 | 2.46 | 8.75 |
| Mmd2 | Monocyte to macrophage differentiation-associated 2 | 2.46 | 13.25 |
| Enpp3 | Ectonucleotide pyrophosphatase/phosphodiesterase 3 | 2.51 | 9.79 |
| Dach2 | Dachshund 2 | 2.57 | 7.504 |
| Rhox2e | Reproductive homeobox 2E | 2.63 | 4.05 |
| Ptprm | Protein tyrosine phosphatase, receptor type, M | 2.64 | 11.74 |
| Matn2 | Matrilin 2 | 2.65 | 9.69 |
| Moxd1 | Monooxygenase, DBH-like 1 | 2.79 | 10.46 |
| Sp110 | Sp110 nuclear body protein | 2.82 | 7.65 |
| Dgkb | Diacylglycerol kinase, β | 3.22 | 11.86 |
| Meg3 | Maternally expressed 3 | 3.25 | 8127.9 |
| Cdh6 | Cadherin 6 | 3.54 | 8.83 |
| Coch | Coagulation factor C homolog | 3.60 | 16.30 |
| Sntg1 | Syntrophin, γ1 | 4.16 | 34.49 |
| Car8 | Carbonic anhydrase 8 | 4.50 | 31.17 |
Figure 3.

Effect of Meg3 on cell migration/invasion and apoptosis and the effect of menin on c-Met. A, Cell migration/invasion assay showing decreased cell migration (represented by OD at 560 nm) in exogenous Meg3-expressing MIN6 stable cell lines (Meg-5 and Meg-14) compared with their respective vector controls (Vec-3 and Vec-9) (P < .005). B and C, Apoptosis assessed by TUNEL staining showing more apoptotic cells in exogenous Meg3-expressing MIN6 stable cell lines (Meg-5 and Meg-14) compared with their respective vector controls (Vec-3 and Vec-9) cultured in chamber slides. Representative green fluorescence images are shown (magnification, ×100), with TUNEL-positive cells indicated by white arrows (B). Nuclei were stained with 4′,6′-diamino-2-phenylindole (DAPI; blue) to show the location of the cells in the field. Five different fields were observed per cell line to count TUNEL-positive cells (C). D, QRT-PCR assay for c-Met using RNA isolated from ESC-Null and ESC-WT embryonic stem cells from Men1−/− and WT mice, respectively, showing significantly increased c-Met in ESC-null cells (P < .05). Gapdh was used as the internal control. E, QRT-PCR assay for c-Met using RNA isolated from the menin transfected MIN6 stable clone M-27-4N compared, with its vector control Vec-4N showing significantly reduced c-Met in M-27-4N cells (P < .0001). Gapdh was used as the internal control. Inset, Western blot for menin (upper bands): endogenous (Endo) menin in Vec-4N, and endogenous and exogenous (Exog) myc-his-tagged menin in M-27-4N. Tubulin was used as a loading control (lower band). F, Cell viability detected by an MTT assay in Vec-4N and M-27-4N before and after 24 hours of treatment with the indicated concentrations of the c-Met inhibitor PHA-665752. Significantly reduced number of cells were observed in the Vec-4N cells at 5 μM concentration of PHA-665752 (P < .0001), but no significant (NS) reduction in cell number was observed in the M-27-4N cells.
Having observed that Meg3 could down-regulate c-Met, we asked the question whether the MIN6-cells stably expressing menin that show Meg3 up-regulation could have altered c-Met expression. QRT-PCR analysis showed significantly reduced c-Met expression in the MIN6 cell line that stably expresses menin compared with its vector control (P < .0001) (Figure 3E). This result suggests that menin-Meg3 action can regulate c-Met in MIN6 mouse insulinoma β-cells. In addition, MIN6-cells stably expressing menin were less sensitive to the cytotoxic effects of a c-Met inhibitor (Figure 3F). Note that MIN6-cells show very little endogenous Meg3 and very high c-Met expression that makes them respond to c-Met inhibitor treatment. Whether the depletion of c-Met can reverse the effects of the menin-null conditions remain to be determined.
Expression of Meg3 and its target c-Met in mouse and human islet cells
To evaluate the physiological role of Meg3 in pancreatic endocrine cell tumorigenesis, Meg3 mRNA levels were examined in pancreatic islets of 22-month-old WT and Men1+/− mice. Menin loss was clearly seen in the insulin-positive larger hyperplastic islet and islet tumor in the Men1+/− pancreas sections (Figure 4A). These large islets showed significantly decreased Meg3 mRNA compared with WT islets (P < .05) (Figure 4B). Also, the islet tumor of Men1+/− mice showed very strong staining for c-Met protein compared with the normal-looking smaller islets in the same pancreas section (Figure 4C). The relevance of Meg3 and c-Met regulation was further evaluated in frozen samples of human MEN1 and non-MEN1 islet tumors (PNETs). Four of five MEN1 PNETs and all three non-MEN1 PNETs showed significantly decreased MEG3 mRNA (P < .0001) (Figure 4D). Significantly increased c-MET protein expression was observed in all five MEN1 PNETs and in two of the three non-MEN1 PNETs (P < .005) (Figure 4E).
Figure 4.

Expression of Meg3 and its target c-Met in mouse and human islet cells. A, Immunofluorescence detection of menin and insulin in the pancreas section of a 22-month-old Men1+/− mouse. Left panel, Menin (green) in small normal-looking islets (yellow arrows) and loss of menin expression in a large hyperplastic islet (blue arrow) and an islet tumor (white arrow). Right panel, Insulin in the normal-looking islets (yellow arrows), the large hyperplastic islet (blue arrow), and the islet tumor (white arrow) (magnification, ×50). B, QRT-PCR assay showing significantly reduced Meg3 in RNA isolated from pancreatic islet tumors of Men1+/− mice (n = 7) compared with islets of WT mice (n = 6) (P < .005). The age of mice was 22 months. Gapdh was used as the internal control. C, Immunohistochemical detection of insulin and c-Met in 22-month-old Men1+/− mouse pancreas section; (1 and 2, ×100 and ×400 magnification, respectively), showing insulin-stained normal-looking islets; (3 and 4, ×100 and ×400 magnifications, respectively), showing no staining for c-Met in the normal-looking islets. Islets are marked with a black dotted outline; (5 and 6, ×400 and ×1000 magnification, respectively), showing insulin-stained islet tumor (red arrow); (7 and 8, ×400 and ×1000 magnification, respectively), showing positive staining for c-Met in the islet tumor (red arrow). The black dashed line marks the boundary of the islet tumor and nonislet tissue (green arrow). Blue is hematoxylin counterstain in the nucleus. D and E, QRT-PCR assay for MEG3 and c-MET using RNA isolated from frozen specimens of human MEN1 and non-MEN1 pancreatic islet tumors and normal control islets. FP2, FP4, FP9, and FP10 are MEN1 PNETs; FP5 is a MEN1 insulinoma; FP1, FP6, and FP8 are sporadic insulinomas. Tumors show significantly decreased MEG3 expression (P < .0001) except in tumor FP10 (E) and significantly increased c-MET expression (P < .005) except in tumor FP1 (F). Gapdh was used as the internal control.
These data show the physiological relevance of menin, the lncRNA MEG3, and the protooncogene c-MET in pancreatic islets cells and in MEN1 and non-MEN1-associated islet-cell tumors.
Reciprocal correlation of MEG3 and c-MET in human sporadic insulinomas
To understand the importance of MEG3 and c-MET in human β-cell tumors (insulinomas) that usually lack MEN1 mutations, we studied 23 archived FFPE human sporadic insulinoma samples. Normal human islets were used as controls (n = 6). Significantly decreased expression of MEG3 mRNA was observed in most of the insulinomas compared with the normal islets (P < .005) (Figure 5A). CRE-MSP showed increased methylation at the CpG in the CRE site, from 34% to 58% in insulinomas, compared with the normal islets (Figure 5, B and C). IHC for insulin and c-MET showed that 13 of 15 insulinomas examined had strong staining for c-MET (Figure 5D). These results suggest that targets downstream of menin are pathologically relevant in sporadic insulinomas.
Figure 5.

Reduced MEG3 and increased c-MET expression in human insulinomas. A, QRT-PCR assay for MEG3 expression using RNA isolated from 23 FFPE sporadic insulinoma specimens and six normal islet preps showing significantly decreased expression of MEG3 in insulinomas (P < .005). Gapdh was used the internal control. B and C, Methylation specific PCR assay of DNA isolated from the specimens in panel A showing increased methylation at the CRE site in the human MEG3 promoter in the insulinomas (34% in normal islets increased to 58% in insulinomas). Meth, methylated; unmeth, unmethylated; D, Immunohistochemical detection of insulin and c-MET (representative images of one normal and one tumor) (1 and 2, ×400 and 1000 magnification, respectively), showing an insulin-stained normal islet (3 and 4, ×400 and ×1000 magnification, respectively), showing no staining with the c-MET antibody in the normal islet. Normal islet is outlined with a black dashed line (5 and 6, ×1000 magnification), showing positive staining for insulin and c-MET, respectively. Blue is hematoxylin counterstain in the nucleus.
DNA-demethylating drugs reduce mouse insulinoma cell proliferation and restore Meg3 expression
Treatment of various cell types with DNA-demethylating drugs such as decitabine (5′-aza-2′-deoxycytidine) or ascorbic acid have been shown to activate the expression of genes silenced by promoter CpG methylation including MEG3 (27–29). To assess whether pharmacological DNA-demethylating agents could demethylate the promoter region of Meg3 and restore Meg3 expression in insulinoma β-cells, MIN6-cells were treated with the DNA-demethylating drugs, decitabine or ascorbic acid. Significantly increased cell death was observed with increasing concentrations of the two DNA-demethylating agents within 24–48 hours of treatment (P < .0001) (Figure 6, A–C). QRT-PCR analysis showed an increased expression of Meg3, with an increasing concentration of the DNA-demethylating agents (Figure 6D) and the presence of the abundant isoform of Meg3 (Meg3-3) and two other isoforms Meg3-1 and Meg3-2 (Supplemental Figure 1). Thus, inhibition of DNA methylation caused cell death and allowed the expression of endogenous Meg3. DNA methylation inhibitors have been shown to cause changes in DNA methylation at differentially methylated regions and promoters of imprinted genes such as MEG3 (28). Therefore, our observations could have therapeutic implications for the treatment of human insulinomas by targeting Meg3 promoter methylation and methylation at other relevant genes.
Figure 6.

DNA-demethylating drugs reduce mouse insulinoma cell proliferation and restore Meg3 expression, A and B, Cell proliferation detected by an MTT assay as an index of cell viability of MIN6-4N cells after treatment with the indicated concentrations of DNA-demethylating agents, 5′-aza-2′-deoxycytidine (DZ) and ascorbic acid (AA). An MTT assay was performed at the indicated time points. Significantly reduced number of cells was observed after 48–96 hours in all concentrations of DZ, and in 1 mM AA (P < .0001). C, Bright-field microscopy images of MIN6-4N cells showing cell death when treated with increasing concentrations of DZ and AA at the indicated time points. D, QRT-PCR assay for Meg3 using RNA isolated from MIN6-4N cells in panels A and B, showing that Meg3 expression was restored after treatment with 1 μM DZ (72 h) and 1 mM AA (96 h) (P < .0001).
Discussion
Molecular mechanisms controlled by genes associated with hereditary tumor syndromes could also play a significant role in the pathogenesis of their sporadic counterpart tumors. We investigated this concept in relation to menin and the regulation of its target gene MEG3 in MEN1-associated PNETs and sporadic functioning PNETs (insulinomas). MEG3 encodes a lncRNA, and investigations to elucidate how lncRNAs participate in development and disease have been of immense interest. Our studies therefore also provide insights about the regulation and functionality of a lncRNA.
Using exogenous menin or Meg3-expressing MIN6 stable cell lines, we demonstrate that menin regulates Meg3 by an epigenetic mechanism, and Meg3 could inhibit cell proliferation and cell cycle. From microarray analysis, we found that Meg3 could act as a tumor suppressor in MIN6 mouse insulinoma cells by affecting genes associated with cell adhesion and apoptosis and by targeting the expression of the protooncogene c-Met. Interestingly, c-Met was also down-regulated in exogenous menin-expressing MIN6-cells. We provide evidence for the physiological relevance of the menin-MEG3-c-MET axis: low MEG3 and high c-MET expression in mouse and human MEN1 or non-MEN1-associated islet tumors. In MIN6 mouse insulinoma cells, DNA-demethylating drugs, decitabine and ascorbic acid, caused cell death and rescued the expression of endogenous Meg3, a very important result with therapeutic implications. It is important to determine the overall contribution of factors downstream of menin or downstream of menin's targets in the insulinoma phenotype of MIN6 cells. Whether the loss of Meg3 alone or in combination with loss or gain of other factors that have been identified to act downstream of menin in MIN6 cells are sufficient to cause β-cell tumors remains to be determined.
Evidence from in vitro methylation and reporter assays had previously shown that the Meg3 CRE site CpG could be methylated to interfere with CREB binding and abolish promoter activity (25). We now show in vivo evidence whereby menin up-regulates Meg3 expression by H3K4me3 and reduced DNA methylation at the CpG in the CRE site in the Meg3 promoter and by CREB recruitment. Whether methylation of a CpG located within a transcription factor binding motif affects DNA binding in vivo has not yet been investigated for most transcription factors (30). Our data underscore the importance of this mechanism for the menin-dependent recruitment of the transcription factor CREB at the Meg3 promoter. However, menin-independent mechanisms in sporadic non-MEN1 tumors that can silence Meg3 by acquiring DNA methylation at the CRE site or other CpGs remain to be determined.
Both human and mouse Meg3 has various alternatively spliced isoforms (17, 31). We found that in MIN6 mouse insulinoma cells, which normally express very low levels of Meg3 mRNA, after decitabine treatment, mMeg3-1, mMeg3-2, and mMeg3-3 transcripts were expressed. It will be of interest to determine whether alternative splicing is another mechanism to regulate MEG3 in tumors.
microRNAs elicit their action by seed sequence base pairing with the target RNA region (32). How lncRNAs act is not well understood. In the cytoplasm they have been shown to affect mRNA stability or translation (33). lncRNAs function in the nucleus by targeting chromatin-modifying proteins such as the polycomb group-repressive proteins, thus effecting gene transcription. The PRC2 accessory subunit JARID2 was shown to interact with MEG3 to recruit and assemble PRC2 at a subset of target genes in pluripotent stem cells (34). From microarray analysis of MIN6 mouse insulinoma cells, we found that Meg3 altered the expression of c-Met and other genes associated with cell adhesion and apoptosis. It is possible that MEG3 could participate in recruiting and assembling similar factors at target genes in β-cells and other endocrine cells.
Conventional Meg3 (Gtl2) knockout mouse models show parent-of-origin-specific defects. Maternal deletion caused perinatal death and skeletal muscle defects; however, paternal deletion caused perinatal death or no effect, and homozygous deletion did not have any effect (35, 36). Therefore, to circumvent the variety of phenotypes observed in conventional Meg3 knockout mice, conditional Meg3 inactivation in tissues affected by menin loss in MEN1 such as the parathyroids, anterior pituitary, and endocrine pancreas will be informative to study the pathological role of Meg3 in endocrine tumorigenesis.
Mouse models with c-Met loss in β-cells have been well studied. Mice with targeted inactivation of c-Met in β-cells have defective insulin secretion, reduced islet size, enhanced β-cell death, and accelerated onset of diabetes (37–39). Also, the hepatocyte growth factor/c-Met axis has been shown to be critical for β-cell survival and β-cell proliferation in situations of diminished β-cell mass (40). Furthermore, the β-cells of human type 2 diabetes patients who present with β-cell loss show epigenetic regulation of the microRNA cluster located near MEG3 (41). These studies support our data about MEG3 and c-MET from the analysis of functioning PNETs (insulinoma), thus providing information about the pathways important to proliferate β-cells that retain their functional properties and provide valuable insights into the mechanisms of β-cell proliferation. It will be important to determine the direct consequence of modulating MEG3 and/or c-MET in human islets (normal and tumor), specifically β-cells, for which viable pure preparations of normal human islet cell types that match the tumor will be desirable.
Although MEN1 mutations are observed in 30%–40% of sporadic parathyroid and pancreatic endocrine tumors, significant MEN1 mutations are not observed in sporadic pituitary tumors (42). Given that MEG3 mRNA reduction occurs in 40% of sporadic pituitary tumors (43), our data about MEG3 as a downstream effector of menin help to understand how a downstream target of menin can be causative in the absence of MEN1 mutations in pituitary cells. Similarly, our current observation of MEG3 mRNA reduction in non-MEN1 sporadic insulinomas shows how MEG3 could be regulated in a menin-independent manner. Interestingly, other genes mutated in insulinomas and PNETs (YY1 and DAXX/ATRX) have been also shown to regulate either MEG3 or c-MET. YY1 was recruited to a region 400 bp upstream of MEG3 exon 1 and to a region immediately downstream of the transcriptional start site (44). DAXX and ATRX together form a repression complex, and DAXX has been shown to suppress c-MET transcription by binding to the c-MET promoter (45, 46). Furthermore, consistent with our data, increased c-MET expression was also observed in primary tumors and their metastatic lesions using a tissue microarray of sporadic nonfunctioning PNETs (47). Thus, the MEG3/c-MET pathway is not only physiologically relevant in MEN1 and non-MEN1 tumors that carry MEN1 mutations, but it is also a relevant pathway downstream of the other genes (YY1 and DAXX/ATRX) mutated in insulinomas and nonfunctioning PNETs, underscoring the importance of our findings. Our data also highlight the importance of different paths to the same target genes in diseases with similar pathology.
Our data demonstrate menin-dependent or menin-independent mechanisms of cellular transformation of pancreatic neuroendocrine cells that act by inhibiting the expression of a tumor suppressor gene (MEG3) and enhancing the expression of an oncogene (c-MET). These observations provide a strong basis for the investigation of MEG3 activation and c-MET suppression as novel therapeutic approaches for the treatment of pancreatic neuroendocrine tumors and possibly for the prevention of metastasis.
Database submission
The microarray data files have been submitted to the Gene Expression Omnibus (accession number GSE57716).
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Dr Phillip McCoy (National Heart, Lung, and Blood Institute flow cytometry core, Bethesda, Maryland) for cell cycle analysis; Dr Oksana Gavrilova [mouse metabolism core, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)] for islet isolation technical assistance; Dr Qiang Du and Dr Michael R. Emmert-Buck, (National Cancer Institute) for immunohistochemistry technical assistance; Dr Gregory Germino (NIDDK) for the use of microscopes; Dr Shruti Desai (NIDDK) of our laboratory for helpful discussions; and Dr Lee S. Weinstein (NIDDK) for his support and guidance.
This work was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases (Project 1ZIADK07503503, to SK.A.) and the National Cancer Institute (Project 1ZIABC01127503, to E.K.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ChIP
- chromatin immunoprecipitation
- CRE
- cAMP responsive element
- CREB
- CRE-binding protein
- ESC
- embryonic stem cell
- FFPE
- formalin-fixed and paraffin-embedded
- Gapdh
- glyceraldehyde-3-phosphate dehydrogenase
- H3K4me3
- histone-H3 lysine-4 trimethyl mark
- IHC
- immunohistochemistry
- lncRNA
- long noncoding RNA
- Meg3
- maternally expressed gene 3
- MEN1
- multiple endocrine neoplasia type 1
- mESC
- mouse ESC
- MTT
- dimethylthiazoldiphenyltetra-zoliumbromide
- PNET
- pancreatic neuroendocrine tumor
- QRT-PCR
- quantitative real time-PCR
- TUNEL
- terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling
- WT
- wild type.
References
- 1. Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276(5311):404–407. [DOI] [PubMed] [Google Scholar]
- 2. Lemmens I, VandeVen WJM, Kas K, Zhang CX, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Hum Mol Genet. 1997;6(7):1177–1183. [DOI] [PubMed] [Google Scholar]
- 3. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990–3011. [DOI] [PubMed] [Google Scholar]
- 4. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab. 2010;24(3):355–370. [DOI] [PubMed] [Google Scholar]
- 5. Agarwal SK. Multiple endocrine neoplasia type 1. Front Horm Res. 2013;41:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piret SE, Thakker RV. Mouse models for inherited endocrine and metabolic disorders. J Endocrinol. 2011;211(3):211–230. [DOI] [PubMed] [Google Scholar]
- 7. Jiao YC, Shi CJ, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corbo V, Dalai I, Scardoni M, et al. MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer. 2010;17(3):771–783. [DOI] [PubMed] [Google Scholar]
- 9. Shin JJ, Gorden P, Libutti SK. Insulinoma: pathophysiology, localization and management. Future Oncol. 2010;6(2):229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhuang Z, Vortmeyer AO, Pack S, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 1997;57(21):4682–4686. [PubMed] [Google Scholar]
- 11. Cao YN, Gao ZB, Li L, et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun. 2013;4:2810. [DOI] [PubMed] [Google Scholar]
- 12. Gortz B, Roth J, Krahenmann A, et al. Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol. 1999;154(2):429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339(2):240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milne TA, Hughes CM, Lloyd R, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci USA. 2005;102(3):749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agarwal SK, Jothi R. Genome-wide characterization of menin-dependent H3K4me3 reveals a specific role for menin in the regulation of genes implicated in MEN1-like tumors. PLoS One. 2012;7(5):e37952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. 2012;48(3):R45–R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. [Erratum (2012) 12(9):637] 12(2):89–103. [DOI] [PubMed] [Google Scholar]
- 19. O'Dowd JF. The isolation and purification of rodent pancreatic islets of Langerhans. Methods Mol Biol. 2009;560:37–42. [DOI] [PubMed] [Google Scholar]
- 20. Miyazaki JI, Araki K, Yamato E, et al. Establishment of a pancreatic β-cell line that retains glucose-inducible insulin-secretion-special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127(1):126–132. [DOI] [PubMed] [Google Scholar]
- 21. Desai SS, Modali SD, Parekh VI, Kebebew E, Agarwal SK. GSK-3β protein phosphorylates and stabilizes HLXB9 protein in insulinoma cells to form a targetable mechanism of controlling insulinoma cell proliferation. J Biol Chem. 2014;289(9):5386–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi K, Parekh VI, Roy S, Desai SS, Agarwal SK. The embryonic transcription factor Hlxb9 is a menin interacting partner that controls pancreatic β-cell proliferation and the expression of insulin regulators. Endocr Relat Cancer. 2013;20(1):111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA. 2005;102(41):14659–14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balasubramanian D, Akhtar-Zaidi B, Song LY, et al. H3K4me3 inversely correlates with DNA methylation at a large class of non-CpG-island-containing start sites. Genome Med. 2012;4(5):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao J, Zhang X, Zhou Y, Ansell PJ, Klibanski A. Cyclic AMP stimulates MEG3 gene expression in cells through a cAMP-response element (CRE) in the MEG3 proximal promoter region. Int J Biochem Cell Biol. 2006;38(10):1808–1820. [DOI] [PubMed] [Google Scholar]
- 26. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 27. Zhao J, Dahle D, Zhou YL, Zhang X, Klibanski A. Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J Clin Endocrinol Metab. 2005;90(4):2179–2186. [DOI] [PubMed] [Google Scholar]
- 28. Stadtfeld M, Apostolou E, Ferrari F, et al. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nat Genet. 2012;44(4):398–405, S391–S392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wesemann DR, Portuguese AJ, Magee JM, et al. Reprogramming IgH isotype-switched B cells to functional-grade induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109(34):13745–13750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. 2013;288(48):34287–34294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schuster-Gossler K, Bilinski P, Sado T, Ferguson-Smith A, Gossler A. The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an RNA. Dev Dyn. 1998;212(2):214–228. [DOI] [PubMed] [Google Scholar]
- 32. Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149(3):515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20(3):300–307. [DOI] [PubMed] [Google Scholar]
- 34. Kaneko S, Bonasio R, Saldana-Meyer R, et al. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell. 2014;53(2):290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Y, Cheunsuchon P, Nakayama Y, et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development. 2010;137(16):2643–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takahashi N, Okamoto A, Kobayashi R, et al. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum Mol Genet. 2009;18(10):1879–1888. [DOI] [PubMed] [Google Scholar]
- 37. Roccisana J, Reddy V, Vasavada RC, et al. Targeted inactivation of hepatocyte growth factor receptor c-met in β-cells leads to defective insulin secretion and GLUT-2 downregulation without alteration of β-cell mass. Diabetes. 2005;54(7):2090–2102. [DOI] [PubMed] [Google Scholar]
- 38. Dai CS, Huh CG, Thorgeirsson SS, Liu YH. β-Cell-specific ablation of the hepatocyte growth factor receptor results in reduced islet size, impaired insulin secretion, and glucose intolerance. Am J Pathol. 2005;167(2):429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mellado-Gil J, Rosa TC, Demirci C, et al. Disruption of hepatocyte growth factor/c-Met signaling enhances pancreatic β-cell death and accelerates the onset of diabetes. Diabetes. 2011;60(2):525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alvarez-Perez JC, Ernst S, Demirci C, et al. Hepatocyte growth factor/c-Met signaling is required for β-cell regeneration. Diabetes. 2014;63(1):216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kameswaran V, Bramswig NC, McKenna LB, et al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab. 2014;19(1):135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Agarwal SK, Ozawa A, Mateo CM, Marx SJ. The MEN1 gene and pituitary tumours. Horm Res. 2009;71(suppl 2):131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou Y, Zhang X, Klibanski A. Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol Cell Endocrinol. 2014;386(1–2):16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McMurray EN, Schmidt JV. Identification of imprinting regulators at the Meg3 differentially methylated region. Genomics. 2012;100(3):184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishov AM, Vladimirova OV, Maul GG. Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J Cell Sci. 2004;117(17):3807–3820. [DOI] [PubMed] [Google Scholar]
- 46. Morozov VM, Massoll NA, Vladimirova OV, Maul GG, Ishov AM. Regulation of c-met expression by transcription repressor Daxx. Oncogene. 2008;27(15):2177–2186. [DOI] [PubMed] [Google Scholar]
- 47. Hansel DE, Rahman A, House M, et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res. 2004;10(18 Pt 1):6152–6158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.