

Abstract
Context:
Aldosterone and PTH are implicated in the pathogenesis of cardiovascular and skeletal diseases. An expanding body of evidence supports a bidirectional and positive physiologic relationship between aldosterone and PTH. Large population-based studies confirming this relationship, and whether it may be targeted as a potential method to mitigate the clinical consequences associated with excess aldosterone and PTH, are needed.
Objective:
We hypothesized that higher aldosterone levels would associate with higher PTH, and that the use of renin-angiotensin-aldosterone system (RAAS) inhibitors would predict lower PTH in a large, multi-ethnic, community-based cohort.
Design, Setting, Participants:
We conducted cross-sectional analyses of participants in the Multi-Ethnic Study of Atherosclerosis without apparent primary hyperparathyroidism or chronic kidney disease (n = 5668). We evaluated associations of RAAS inhibitor use with PTH concentration among 1888 treated hypertensive participants. We also tested associations of serum aldosterone concentration with PTH concentration among 1547 participants with these measurements.
Outcome:
Serum PTH concentration.
Results:
Higher aldosterone associated with higher PTH (β = 0.19 pg/ml per 1 ng/dl of aldosterone, P < .0001), and this finding was most pronounced among those with a primary hyperaldosteronism-like phenotype. There was a stepwise increment in PTH when comparing untreated normotensives, hypertensives using RAAS inhibitors, untreated hypertensives, and treated hypertensives using non-RAAS inhibitors (40.8, 45.0, 46.2, 47.1 pg/ml, respectively). The use of any RAAS inhibitor independently associated with lower PTH (β = −2.327 pg/ml per use of RAAS inhibitor, P = .006), when compared with the use of any non-RAAS inhibitor medication.
Conclusions:
Higher serum aldosterone concentration is associated with higher serum PTH concentration, and the use of RAAS inhibitors is associated with lower PTH concentration. These results extend prior evidence from observational and intervention studies suggesting a potentially important and modifiable relationship between the RAAS and PTH in humans.
The renin-angiotensin-aldosterone system (RAAS), an established mediator of cardiovascular disease (1–3), has also been associated with skeletal disease (4, 5). Conversely, high levels of PTH have been associated with adverse cardiovascular outcomes including hypertension (6), cardiovascular dysfunction (7, 8), and cardiovascular mortality (9, 10) in addition to the established effects on bone and mineral metabolism (11, 12). Growing evidence points to a bidirectional physiologic relationship between the RAAS and PTH (13–17) that has the potential to become a “vicious cycle” in pathophysiologic states such as primary hyperparathyroidism and primary hyperaldosteronism, where cardiovascular and skeletal health implications are significant.
Observational studies in patients with primary hyperaldosteronism have associated elevations in aldosterone with higher PTH levels (18, 19) and with reduced bone mineral density (5, 20, 21), both of which improved following treatment of hyperaldosteronism (5, 18–20). Studies in primary hyperparathyroidism have also linked elevated PTH levels with higher aldosterone (22). These small observational studies have suggested an important relationship between PTH and the RAAS under pathophysiologic conditions. In healthy populations without primary hyperaldosteronism, controlled intervention studies have demonstrated that angiotensin II can acutely stimulate PTH and angiotensin-converting enzyme (ACE) inhibitors can acutely lower PTH (23, 24), and that chronic mineralocorticoid receptor blockade can lower PTH via interactions with the mineralocortcoid receptor that is expressed in the parathyroid gland (24).
We hypothesized that the use of RAAS inhibitors ie, medications that lower aldosterone levels such as ACE inhibitors and angiotensin receptor blockers (ARB), would associate with lower PTH levels in a population of individuals without primary hyperparathyroidism, and independent of glomerular filtration rate or other predictors of PTH in a large, multiethnic, community-based cohort. Further, we hypothesized that higher levels of aldosterone would associate with higher PTH levels. Assessment of these hypotheses in a large cohort is relevant to determine whether RAAS inhibition may serve as a potential medical therapy to lower PTH and impart both cardiovascular and skeletal health benefits.
Materials and Methods
Study population
The Multi-Ethnic Study of Atherosclerosis (MESA) is a prospective, multicenter cohort study of 6814 community-dwelling adults 45–84 years of age. From 2000–2002, subjects without heart failure were recruited at six centers (New York, New York; Baltimore, Maryland; Forsyth County, North Carolina; Chicago, Illinois; St Paul, Minnesota; Los Angeles, Californa) and were re-evaluated in followup at roughly 1.5, 3, and 4.5 years after enrollment (25). Additional details of study design and recruitment procedures have been described previously (26). The study was approved by institutional review boards at all sites, and all participants provided informed consent.
Demographic variables, biochemical values including PTH, and reported medication use were determined on all subjects at baseline study visit 1 (conducted over 24 months between 2000 and 2002). Serum aldosterone and plasma renin activity (PRA) were measured at followup study visits 2 (conducted over 18 months during 2002–2004) and 3 (conducted over 18 months during 2004–2005) in a random subset of 1960 participants. Of the 6814 participants, we excluded those with missing PTH measurements. In addition, given that PTH was our outcome of interest, we excluded participants who might have unrecognized primary hyperparathyroidism, defined as a serum calcium level greater than 10.2 mg/dl and a PTH level greater than 65 mg/dl (6, 27), and those with evidence of renal impairment (estimated glomerular filtration rate [eGFR] <60 ml/min/1.73 m2) (28) (Supplemental Figure 1). Of the remaining 5668 participants, we specifically analyzed the relation between RAAS inhibitor use and PTH in the subset of hypertensive individuals using antihypertensive medications (n = 1888), because hypertension status was an independent predictor of elevated PTH (see statistical analyses below). Among these individuals, 1547 had serum aldosterone and PRA measurements to analyze the relation between aldosterone and PTH (Supplemental Figure 1).
Demographic and laboratory measurements
With the exception of aldosterone and PRA, all demographic, historical, and laboratory measurements were made at the baseline examination in 2000–2002. Participants completed self-administered questionnaires and received standardized interviews to evaluate demographics, medical history, medication, and substance use (29). Diabetes status was determined by American Diabetes Association criteria (30) based on fasting blood glucose cutoffs of 100 mg/dl and 126 mg/dl for impaired fasting glucose and diabetes mellitus, respectively. Antihypertensive medication use was categorized by drug class; RAAS inhibitor use constituted any reported use of an ACEi and/or ARB. Non-RAAS inhibitor medication classes included calcium channel blockers, diuretics, β-blockers, and other infrequently used medications. Although β-blockers, and to some extent diuretics, may affect the dynamics of renin or the RAAS, they are not conventionally considered RAAS inhibitors or medications that modulate aldosterone secretion in the same context as ACEi and ARBs, and were therefore categorized as non-RAAS inhibitors. Mineralocorticoid receptor antagonists were not individually distinguished from other potassium-sparing diuretics in MESA, and were therefore jointly categorized in the non-RAAS inhibitor group; however, the overall number of individuals using these medications was only 158.
Serum samples were collected in the morning after an overnight fast and were stored at −80°C and thawed for analysis (25). eGFR was estimated from serum creatinine using the Chronic Kidney Disease Epidemiology Collaboration equation (CKD-EPI) (31), and mean annual 25-hydroxyvitamin D [25(OH)D] concentrations were estimated as previously described (32). Intact PTH concentration was measured at the University of Washington using the Beckman-Coulter DxI automated two-site immunoassay (Beckman-Coulter). Between-lot variations in PTH results were adjusted using repeated measurements of aliquoted serum samples from 20 normal controls. After adjustment, interassay coefficient of variation (CV) was 6.1% at 30.1 pg/ml and 3.4% at 94.5 pg/ml (6, 33), comparable to other validated PTH assays (34). Aldosterone was measured by RIA (Diasorin) (intra-assay CVs between 6.30% and 8.87%), and PRA was determined using a RIA of generated angiotensin I (Diasorin) (intra-assay CV between 6.89% and 18.38%) (25).
Statistical analysis
RAAS inhibitor use and PTH
To evaluate the effect of RAAS inhibitor medication use on PTH, we first compared mean PTH levels of normotensive vs untreated hypertensive individuals to confirm and determine the magnitude of the known association between hypertension and PTH within our study population. To disaggregate the effect of antihypertensive drug class from the known association between hypertension and PTH (35–38), we then limited the study population to those on at least one antihypertensive medication when analyzing the relationship between RAAS inhibitor use and PTH (n = 1888). Serum PTH level was the continuous dependent outcome variable. RAAS inhibitor use (yes/no) was the predictor of interest. Unpaired t tests were employed to compare mean PTH levels between normotensive and untreated hypertensive individuals and between individuals on RAAS inhibitor and non-RAAS inhibitor antihypertensive medications. We used linear regression to evaluate the adjusted independent relationship between RAAS inhibitor use and PTH. We performed a sensitivity analysis to exclude the possibility of antihypertensive drug combination interactions by further restricting the population to include only those participants taking a single antihypertensive drug (n = 1164). Regression models were adjusted for age, race, sex, body-mass index (BMI), eGFR, and 25(OH)D (Supplemental Table 1). We conducted extended multivariate models with additional terms (diabetes status, smoking history, level of education, physical activity level, waist-to-hip ratio, study site, serum calcium, serum phosphate, urine calcium-to-creatinine ratio, urine phosphate-to-creatinine ratio, and individual antihypertensive drug classes including diuretics, β-blockers, and calcium channel blockers); however, these additional adjustments did not affect the results.
Aldosterone and PTH
Serum aldosterone was evaluated as a continuous variable, and using quintiles, to permit assessment of mean PTH levels across categories of aldosterone concentrations. We used multivariable linear regression to estimate the continuous association between serum aldosterone and PTH, and we calculated the adjusted mean PTH level for each quintile of aldosterone. Regression models were adjusted for age, race, sex, BMI, eGFR, and 25(OH)D. We conducted extended multivariable models with additional terms (diabetes status, smoking history, level of education, physical activity level, waist-to-hip ratio, study site, serum calcium, serum phosphate, urine calcium-to-creatinine ratio, urine phosphate-to-creatinine ratio); however, these additional adjustments did not affect the results. We did not include serum or urinary sodium or potassium in our models because they are strong predictors of aldosterone and would result in collinearity; however, addition of these variables did not change the results or effect estimates. We did not apply any transformations to the serum aldosterone concentrations, given that linear modeling best approximated the association with PTH; addition of nonlinear polynomial terms did not improve the model fit (Supplemental Figure 2). Because our exclusion criterion for primary hyperparathyroidism was relatively permissive (serum calcium >10.2 mg/dl and PTH ≥65 pg/ml), we considered the possibility that some high PTH values in our sample might still represent milder forms of primary hyperparathyroidism, a pathophysiologic state that could obscure a physiologic association between PTH and aldosterone. To address this possibility, we repeated the above analyses in a more restricted population, excluding those participants with serum calcium greater than 10.2 mg/dl and PTH at least 30 pg/ml (n = 56 additional participants excluded).
All analyses were performed using SAS version 9.3 (SAS Institute). P < .05 was considered statistically significant.
Exploratory analyses: potential clinical implications of the aldosterone-PTH relationship
Aldosterone phenotype and PTH
To investigate potential physiologic mechanisms underlying the aldosterone-PTH relationship, we performed an exploratory analysis to evaluate the relationship between distinct hyperaldosteronism-like (HA-like) phenotypes with PTH. To maximize the sample size needed to capture these HA-like phenotypes, we removed all aforementioned exclusions for renal dysfunction and hyperparathyroidism, leaving a sample of 1694 subjects with RAAS measurements available. We defined three nonoverlapping HA-like phenotypes within this study population: 1) “primary HA-like” phenotype with apparent hyporeninemic hyperaldosteronism (aldosterone >15 ng/dl and PRA <0.6 ng/ml · h), 2) “secondary HA-like” phenotype with apparent hyperreninemic hyperaldosteronism (aldosterone >15 ng/dl and PRA >2.0 ng/ml · h), and 3) a normal phenotype with neither primary nor secondary HA-like features (aldosterone <12 ng/dl and PRA 1–2 ng/ml · h). Unpaired t tests were used to compare mean PTH and other variables between these groups.
Aldosterone, PTH, and hypertension prevalence
To explore whether a positive relationship between aldosterone and PTH could adversely affect cardiovascular health (vicious cycle), we assessed categories of aldosterone and PTH levels with the prevalence of hypertension. We categorized levels of these hormones as high (>75th percentile) or low (<25th percentile) for this analysis. We tested the interaction between aldosterone and PTH in predicting the prevalence of hypertension in an interaction model adjusted for age, race, sex, BMI, eGFR, and 25(OH)D.
Results
Study population
The mean age of the overall study population was 60.8 years, and 52.6% were women. Mean eGFR was 82.0 ml/min/1.73 m2 and mean 25(OH)D was 25.3 ± 10.9 ng/ml, with a corresponding mean PTH level of 43.2 pg/ml ± 17.8 pg/ml. The distribution of age, sex, BMI, and other baseline demographic characteristics was largely constant across aldosterone quintiles, although higher quintiles of aldosterone contained a greater proportion of caucasian and Hispanic participants compared with lower quintiles (Table 1). The highest quintile of aldosterone was also characterized by a higher proportion of hypertensive participants than lower quintiles. PTH levels were relatively constant at lower aldosterone quintiles but increased with the highest aldosterone quintile. There were no differences in renal function or serum calcium. RAAS inhibitors were the most frequently used antihypertensive medication (Table 1), and 62% of treated hypertensives were on only one single antihypertensive medication (Supplemental Table 2).
Table 1.
Demographic and Biochemical Characteristics by Aldosterone Quintile
| Variable | Aldo Quintile 1, <8.58 ng/dl | Aldo Quintile 2, 8.58–11.58 ng/dl | Aldo Quintile 3, 11.59–14.91 ng/dl | Aldo Quintile 4, 14.92–19.66 ng/dl | Aldo Quintile 5, >19.66 ng/dl |
|---|---|---|---|---|---|
| N | 309 | 310 | 309 | 310 | 309 |
| Age, y | 61.2 (9.4) | 61.8 (9.8) | 60.2 (9.1) | 61.3 (9.1) | 59.6 (9.5) |
| Female, n (%) | 150 (48.5%) | 164 (52.9%) | 159 (51.5%) | 138 (44.5%) | 156 (50.5%) |
| Race/ethnicity, n (%) | |||||
| White | 115 (37.2%) | 118 (38.1%) | 115 (37.2%) | 131 (42.3%) | 134 (43.4%) |
| Chinese American | 31 (10.0%) | 47 (15.2%) | 48 (15.5%) | 49 (15.8%) | 31 (10.0%) |
| African American | 91 (29.5%) | 67 (21.6%) | 56 (18.1%) | 52 (16.8%) | 46 (14.9%) |
| Hispanic | 72 (23.3%) | 78 (25.2%) | 90 (29.1%) | 78 (25.2%) | 98 (31.7%) |
| BMI, kg/m2 | 27.9 (5.5) | 28.1 (5.3) | 28.0 (4.9) | 27.7 (4.6) | 28.3 (5.2) |
| Fasting glucose, mg/dl | 98.4 (34.2) | 96.1 (26.1) | 95.0 (27.3) | 98.8 (32.3) | 97.3 (28.0) |
| Hypertension, n (%) | 111 (35.9%) | 123 (39.7%) | 120 (38.8%) | 123 (39.7%) | 163 (52.8%) |
| Antihypertensive medications, n (%) | 93 (30.1%) | 91 (29.4%) | 89 (28.9%) | 95 (30.7%) | 133 (43.0%) |
| RAAS inhibitor | 51 (16.5%) | 49 (15.8%) | 46 (14.9%) | 46 (14.8%) | 51 (16.5%) |
| Diuretic | 21 (6.8%) | 23 (7.4%) | 24 (7.8%) | 31 (10%) | 62 (20.1%) |
| Calcium channel blocker | 23 (7.4%) | 34 (11.0%) | 21 (6.8%) | 27 (8.7%) | 48 (15.5%) |
| β-blocker | 18 (5.8%) | 18 (5.8%) | 26 (8.4%) | 23 (7.4%) | 38 (12.3%) |
| Smoking status, n (%) | |||||
| Never smoker | 130 (42.1%) | 160 (51.61%) | 175 (56.6%) | 171 (55.3%) | 134 (43.4%) |
| Former | 131 (42.4%) | 107 (34.52%) | 96 (31.1%) | 102 (33.0%) | 128 (41.4%) |
| Current | 48 (15.5%) | 43 (13.87%) | 38 (12.3%) | 36 (11.7%) | 47 (15.2%) |
| Creatinine, mg/dl | 0.9 (0.2) | 0.9 (0.2) | 0.9 (0.2) | 0.9 (0.2) | 0.9 (0.2) |
| eGFR, mL/min/1.73 m2 | 82.2 (12.8) | 80.9 (12.8) | 81.3 (12.3) | 81.1 (12.8) | 80.8 (12.6) |
| Serum sodium, Eq/L | 146.7 (3.3) | 146.9 (3.5) | 147.2 (3.2) | 147.7 (3.5) | 146.6 (3.5) |
| Serum potassium, mEq/L | 4.35 (0.34) | 4.31 (0.36) | 4.32 (0.41) | 4.27 (0.34) | 4.18 (0.39) |
| Serum calcium, mg/dl | 9.6 (0.4) | 9.6 (0.4) | 9.7 (0.4) | 9.6 (0.4) | 9.6 (0.4) |
| Urine calcium-to-creatinine ratio, mg/g | 0.102 (0.068) | 0.104 (0.075) | 0.099 (0.079) | 0.098 (0.059) | 0.098 (0.082) |
| Serum phosphate, mg/dl | 3.6 (0.5) | 3.7 (0.5) | 3.6 (0.5) | 3.7 (0.5) | 3.7 (0.5) |
| Urine phosphate-to-creatinine ratio, mg/g | 0.435 (0.169) | 0.467 (0.181) | 0.461 (0.193) | 0.460 (0.171) | 0.470 (0.179) |
| Urine sodium-to-creatinine ratio, mg/g | 1.21 (0.62) | 1.24 (0.78) | 1.09 (0.63) | 1.07 (0.63) | 0.95 (0.64) |
| Urine potassium-to-creatinine ratio, mg/g | 0.52 (0.23) | 0.58 (0.27) | 0.56 (0.28) | 0.57 (0.27) | 0.53 (0.24) |
| Annualized 25(OH)D, ng/ml | 24.2 (9.9) | 26.3 (14.1) | 25.0 (10.3) | 27.7 (9.3) | 26.8 (10.3) |
| PTH, pg/ml | 43.2 (18.4) | 43.3 (16.2) | 43.5 (15.6) | 43.0 (17.1) | 46.2 (18.0) |
| Aldosterone, ng/dl | 6.3 (1.5); 6.6 [5.3, 7.6] | 10.1 (0.9); 10 [9.4, 10.9] | 13.2 (1.0); 13.0 [12.3, 14.0] | 17.2 (1.4); 17.2 [16.0, 18.3] | 27.3 (8.8); 24.4 [21.3, 29.8] |
| PRA, ng/ml · h | 1.2 (3.2); 0.4 [0.2, 0.8] | 1.0 (1.8); 0.5 [0.3, 0.8] | 0.9 (1.6); 0.5 [0.3, 0.9] | 1.6 (3.8); 0.7 [0.3, 1.3] | 2.5 (4.8); 1.0 [0.5, 1.9] |
| Aldosterone-to renin ratio | 31.8 (96.7); 15.7 [6.7, 27.8] | 41.4 (141.9); 21.1 [12.1, 33.9] | 58.5 (157.9); 25.2 [14.6, 47.6] | 74.8 (250.9); 23.1 [13.8, 52.6] | 51.5 (95.7); 25.2 [13.2, 55.4] |
Demographic and biochemical values are shown by quintile of aldosterone.
Values shown are Mean (sd), except for aldosterone, PRA, and aldosterone-to-renin ratio, which are represented as Mean (sd) and Median [interquartile range].
RAAS inhibitor use and PTH
We first compared mean PTH levels of normotensive vs untreated hypertensive individuals to determine the magnitude of the known association between hypertension and PTH. PTH was significantly higher among untreated hypertensive participants than normotensives (Figure 1, left panel). Among those participants taking at least one or more antihypertensive medications (n = 1888), 47.2% (891) were taking a medication of the RAAS inhibitor class, whereas the remainder were taking exclusively non-RAAS inhibitor antihypertensive medications. As shown in Figure 1 (right panel), RAAS inhibitor use was associated with a significantly lower mean PTH than use of non-RAAS inhibitor antihypertensives (45.0 ± 19.1 vs 47.1 ± 19.4; P = .016). This result remained significant (P = .011) in an adjusted sensitivity analysis whereby the population was limited to those participants on exactly one antihypertensive drug (n = 1164), suggesting that the difference in PTH was not due to a difference in the severity of the underlying hypertension or due to interactions with combination drug therapy. Further, among these individuals taking only a single antihypertensive medication, RAAS inhibitors associated with lower PTH when compared with untreated hypertensives, and calcium-channel blockers associated with higher PTH (Supplemental Figure 3). Although β-blockers and diuretics seemed to have lower PTH values when compared with untreated hypertension, this finding was not statistically significant.
Figure 1.
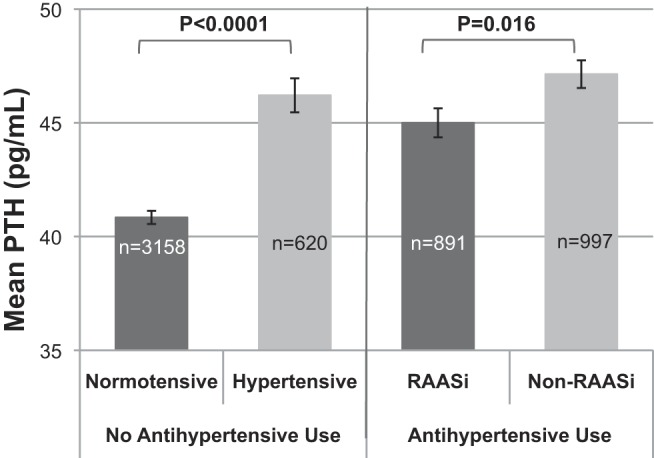
Mean PTH by hypertension status and RAAS inhibitor medication use. Error bars represent SE of means.
In unadjusted linear regression models, the use of any RAAS inhibitor compared with the use of any other antihypertensive medication class independently associated with lower PTH and this association remained significant following multivariable adjustment (Table 2). Further adjustments to the model to include a broader set of potential confounders (diabetes status, smoking history, level of education, physical activity level, waist-to-hip ratio, study site, serum calcium, serum phosphate, urine calcium-to-creatinine ratio, urine phosphate-to-creatinine ratio) did not appreciably change the effect (Table 2). Lastly, addition of other individual antihypertensive medication classes to our models did not change the relation between RAAS inhibitors and PTH (ß= −2.16 pg/ml of PTH per use of RAAS inhibitor, P = .02).
Table 2.
The Association Between RAAS Inhibitor Use and PTH Level
| ß (95% CI) | P Value | |
|---|---|---|
| Model | ||
| Univariate | −2.138 (−3.880–0.396) | .016 |
| Multivariablea | −2.327 (−3.988–0.667) | .006 |
| Extended multivariableb | −2.003 (−3.639–0.368) | .017 |
Abbreviation: CI, confidence interval.
Units for ß are pg/ml of PTH per use of a RAAS inhibitor medication.
Multivariable modeling included adjustment for: age, race, sex, body-mass index (BMI), eGFR, and 25(OH)D.
Extended multivariable modeling included adjustment for: age, race, sex, BMI, eGFR, 25(OH)D, diabetes status, smoking history, level of education, physical activity level, waist-to-hip ratio, study site, serum calcium, serum phosphate, urine calcium-to-creatinine ratio, and urine phosphate-to-creatinine ratio.
Aldosterone-PTH relationship
We observed a significant positive relationship between aldosterone and PTH in univariate analyses that remained significant following multivariable adjustment (Table 3). Further adjustments to the model to include a broader set of potential confounders (diabetes status, smoking history, level of education, physical activity level, waist-to-hip ratio, study site, serum calcium, serum phosphate, urine calcium-to-creatinine ratio, urine phosphate-to-creatinine ratio) did not appreciably change the effect (Table 3). We conducted categorical analyses to assess the mean PTH levels per quintile of aldosterone. Adjusted mean PTH levels were 42.2, 43.1, 43.1, 44.2, and 46.7 pg/ml for aldosterone quintiles 1–5, respectively (adjusted Ptrend = .005). We repeated our analyses using stricter criteria for primary hyperparathyroidism (serum calcium >10.2 mg/dl and PTH ≥30 pg/ml; n = 56 additional participants excluded); however, this stricter definition of primary hyperparathyroidism did not change the results of either the continuous or categorical analyses.
Table 3.
Continuous Association Between Aldosterone and PTH
| ß (95% CI) | P Value | |
|---|---|---|
| Model | ||
| Univariate | 0.140 (0.037–2.43) | .008 |
| Multivariablea | 0.193 (0.097–0.288) | <.0001 |
| Extended multivariableb | 0.176 (0.084–0.269) | .0002 |
Units for ß are pg/ml of PTH per 1 ng/dl of aldosterone.
*Multivariable modeling included adjustment for age, race, sex, BMI, eGFR, and 25(OH)D.
Extended multivariable modeling included adjustment for age, race, sex, body-mass index (BMI), eGFR, and 25(OH)D, diabetes status, smoking history, level of education, physical activity level, waist-to-hip ratio, study site, serum calcium, serum phosphate, urine calcium-to-creatinine ratio, urine phosphate-to-creatinine ratio.
Exploratory analyses: potential clinical implications of the RAAS-PTH relationship
Having observed a significant positive relationship between aldosterone and PTH in this large, general population cohort, we next performed an exploratory analysis to delve further into the mechanisms that might underlie this association. We categorized the participants into three nonoverlapping groups based on a phenotype most similar to primary hyperaldosteronism (ie, low PRA [<0.6 ng/ml · h] and high aldosterone [>15 ng/dl]), to secondary hyperaldosteronism (ie, high PRA [>2 ng/ml · h] and high aldosterone [>15 ng/dl]), or to a “normal” RAAS (neither high nor low PRA [1–2 ng/ml · h] or aldosterone [<12 ng/dl]). As shown in Table 4, those with a secondary HA-like phenotype exhibited the highest serum aldosterone levels (P < .0001); however, their PTH levels did not differ from those with a “normal” phenotype (P = .32). In contrast, those with a primary HA-like phenotype exhibited the highest PTH levels despite not having the highest aldosterone levels (P < .0001) (Table 4).
Table 4.
PTH by Phenotypes of Hyperaldosteronism
| Phenotypes | “Primary HA-Like” (ALDO >15 ng/dl, PRA <0.6 ng/ml · h) | “Normal” (ALDO <12 ng/dl, PRA 1–2 ng/ml · h) | “Secondary HA-Like” (ALDO >15 ng/dl, PRA >2 ng/ml · h) | P Value (Primary vs Normal) | P Value (Secondary vs Normal) | P Value (Primary Versus Secondary) |
|---|---|---|---|---|---|---|
| n | 298 | 66 | 124 | — | — | — |
| Aldosterone, ng/dl | 21.2 (6.7) | 8.3 (2.6) | 26.6 (12.0) | <.0001 | <.0001 | <.0001 |
| PRA, ng/ml · h | 0.3 (0.2) | 1.4 (0.2) | 7.5 (7.7) | <.0001 | <.0001 | <.0001 |
| ARR | 146.0 (283.9) | 6.3 (2.3) | 6.4 (4.4) | <.0001 | .85 | <.0001 |
| Serum calcium, mg/dl | 9.6 (0.4) | 9.7 (0.3) | 9.6 (0.4) | .39 | .71 | .30 |
| Serum phosphate, mg/dl | 3.7 (0.5) | 3.6 (0.5) | 3.6 (0.5) | .24 | .74 | .30 |
| PTH, pg/ml | 50.2 (21.1) | 40.3 (16.3) | 43.0 (18.1) | <.0001 | .32 | <.001 |
Abbreviation: ALDO, aldosterone; ARR, aldosterone-to-renin ratio.
P values indicate statistical significance between indicated columns. Values represent means with standard deviation.
Given that the aldosterone-PTH relationship is proposed to be positive and bidirectional (14–16), in a second exploratory analysis we investigated the interrelationships between aldosterone, PTH, and an available cardiovascular outcome: the prevalence of hypertension. Hypertension was more prevalent in higher quintiles of aldosterone (Q1, 43.7% vs Q5, 58.2%; P < .0001) and PTH (Q1, 35.9% vs Q5, 59.6%; P < .0001). When each hormone was categorized as either high (>75th percentile) or low (<25th percentile), low aldosterone levels with low PTH levels associated with the lowest prevalence of hypertension, whereas high aldosterone levels with high PTH levels associated with the highest prevalence of hypertension (Ptrend <.0001) (Figure 2). The interaction between aldosterone and PTH with hypertension prevalence was significant after multivariable modeling (P = .02).
Figure 2.
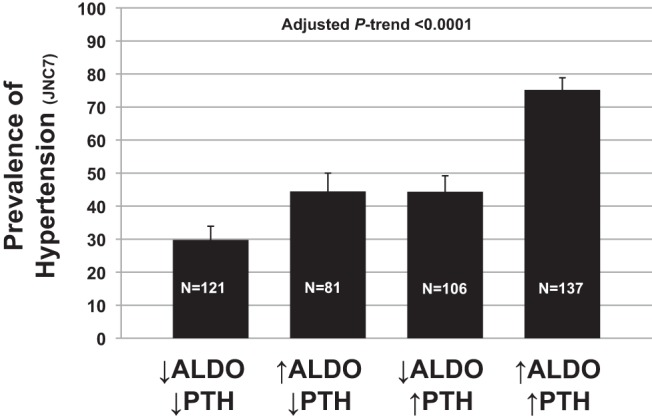
The prevalence of hypertension by categories (high or low) of aldosterone and PTH levels (high, >75th percentile; low, <25th percentile).
Discussion
The proposed positive and bidirectional relationship between aldosterone and PTH implicates a potential vicious cycle of aldosterone-PTH stimulation contributing to human cardiovascular and skeletal disease (14–16). We observed associations of higher serum aldosterone concentration with higher serum PTH concentration and the use of RAAS inhibitors with lower PTH concentration. These findings support prior small observational and interventional studies that suggest that high aldosterone activity stimulates PTH (14–16, 24) and extend the findings of a recent population-based study linking a higher aldosterone-to-renin ratio with higher PTH (39). The results suggest that pharmacologic inhibition of the RAAS may lower PTH and therefore may impart pleiotropic effects on human cardiovascular and skeletal health (2, 40–42).
In this cross-sectional study, we observed an approximately 4.5 pg/ml higher PTH level in the highest compared with lowest aldosterone quintile, and an approximately −2.0 pg/ml lower PTH level associated with RAAS inhibitor use when compared with non-RAAS inhibitor medication use. Although at first glance the magnitudes of these associations seem small, they must be interpreted in the context of reflecting a mean estimate of a large general population. Given that the findings of this study extend prior mechanistic and physiologic observations, we suspect that investigation of a concentrated population with bona fide pathophysiology (hyperparathyroidism or hyperaldosteronism) may have resulted in much stronger associations with greater magnitudes of differences. Indeed, the clinical relevance and future implications of our findings may be most significant with respect to the treatment and management of patients with hyperparathyroidism or hyperaldosteronism, where a vicious cycle of aldosterone-PTH positive feedback may escalate cardiovascular and skeletal disease risk.
To better understand the implications of the magnitudes of the effect estimates we observed, we explored whether a higher PTH depended solely on the magnitude of the aldosterone level or whether the mechanism of aldosterone elevation was also important. We observed that individuals with a primary hyperaldosteronism-like phenotype (suppressed PRA and high aldosterone) had the highest PTH levels, whereas those with a secondary hyperaldosteornism-like phenotype (high PRA and aldosterone levels) displayed PTH values no different from those without hyperaldosteronism even though they had the highest aldosterone levels. We speculate that the primary hyperaldosteronism-like phenotype may represent a chronic state of autonomous and sustained aldosterone hypersecretion, regardless of absolute aldosterone levels or blood pressure (BP) at the time of the MESA examination. In contrast, the secondary hyperaldosteronism phenotype may represent a transient or subacute state of low circulating blood volume and renal perfusion. In this scenario, the former would be expected to associate with higher PTH levels than the latter, reflecting the greater effect of direct and chronic mineralocorticoid receptor activation by sustained aldosterone exposure. This observation may further support prior reports describing primary hyperaldosteronism resulting in primary hyperparathyroidism (and vice versa) (17, 19, 43).
Previous observational studies have found higher PTH levels in states of primary aldosteronism that were lower following treatment with adrenalectomy or mineralocorticoid receptor blockade (18, 19), and Rossi et al (44) have even shown that PTH levels can be used to distinguish between unilateral aldosterone-producing adenoma from bilateral adrenal hyperplasia, possibly due to differences in severity of the underlying aldosterone excess. Beyond small studies in primary aldosteronism, larger cohort studies in Caucasian populations have extended this relationship by observing that higher aldosterone and higher PTH coassociate and that PTH levels are highest in those with aldosterone-to-renin ratios greater than the 90th percentile (39, 45). We here build on these finding by demonstrating that higher aldosterone associates with a higher mean adjusted PTH in a large multiethnic cohort, that this relationship seems to be most pronounced in those with a primary hyperaldosteronism-like phenotype, and that the use of RAAS inhibitors may represent a pharmacologic method to counteract this physiologic relationship.
Prior evidence for the effect of medications that target the RAAS on PTH is more limited but has suggested an effect. Our own controlled intervention studies in 14 and 27 subjects demonstrated acute PTH lowering within 2 hours of a dose of captopril, and chronic PTH lowering after 6 weeks of spironolactone therapy (24). In an observational study of 1076 patients with end-stage renal disease, Koiwa et al (46) found an association between RAAS inhibitor use and lower PTH levels. We now have shown that use of antihypertensive medications that inhibit the RAAS associates with lower PTH levels compared with medications that reduce BP through a different primary mechanism, not only in renal failure but also under physiologic conditions of normal renal function, independent of calcium, phosphate, and vitamin D.
It is important to realize that in endocrine systems where feedback mechanisms are complex, the absolute circulating level of any hormone may not be sufficient to evaluate biologic and clinical outcomes; the setting of a new homeostatic equilibrium may not be detected by circulating levels alone. For example, although ACE inhibitors acutely lower angiotensin II and aldosterone concentrations, chronic ACE inhibition can result in a re-equilibration and normalization of aldosterone levels (47–49), yet the clinical benefits associated with chronic ACE inhibition (BP and cardiovascular event reduction) remain highly apparent (50). Therefore, the associations we observed between aldosterone levels, RAAS inhibitors, and PTH levels may only partially reflect the true endocrine dynamic that exists. Future interventional or longitudinal prospective studies that include outcomes associated with PTH excess are needed to resolve whether chronic use of RAAS inhibitors is clinically significant.
Though there is now mounting evidence for a relationship between the RAAS and PTH from small and large studies in both pathophysiologic and physiologic states, the mechanisms of such a relationship remains unknown. Both indirect and direct pathways have been proposed. In primary aldosteronism, urinary calcium wasting due to chronic aldosterone exposure has been proposed as a stimulus for the observed elevation in PTH (51). In their study of 10 patients with primary aldosteronism and 182 essential hypertensive controls, Pilz et al (18) found lower serum calcium levels and a trend toward higher urinary calcium excretion in those with primary aldosteronism, leading them to conclude a secondary hyperparathyroidism in primary aldosteronism. In the current study, the positive association between aldosterone and PTH was independent of the urine calcium-to-creatinine ratio, although small changes may not have been discernable and thus this type of indirect relationship cannot be excluded. However, we and others have detected both the mineralocorticoid receptor (24, 43) and the angiotensin type 1 receptor (24) in human parathyroid tissue, suggesting a plausible direct mechanism of RAAS action on the parathyroid.
Additional studies are needed to further characterize the effect of aldosterone and mineralocorticoid receptor activation on PTH levels and to evaluate the use of RAAS inhibitors to target the cardiovascular and skeletal outcomes of intertwined aldosterone and PTH excess. The on-going Renin-Angiotensin-Aldosterone System and PARathyroid Control Study (RAAS-PARC) (NCT01691781) and the Effects of Eplerenone in Patients with Primary HyperPAraTHyroidism EPATH (ISRCTN33941607) interventional studies will shed further light on this topic by evaluating the effect of ACE inhibitors and mineralocorticoid receptor antagonists, respectively, on PTH levels in subjects with primary hyperparathyroidism.
Strengths and limitations
This study has the benefit of a large, diverse population that has been carefully characterized within the MESA cohort. Furthermore, the finding of an independent positive relationship between aldosterone and PTH is logically concordant with the result of the RAAS inhibitor analysis, such that both higher levels of aldosterone predict higher levels of PTH and down-regulated activity of the RAAS predicts lower PTH levels. However, many limitations must be acknowledged. First, whereas baseline characteristics and PTH levels were measured at visit 1, assessment of the RAAS was performed at a subsequent follow-up visit; therefore, there is a risk of some misclassification bias. Furthermore, as with any cross-sectional study, we can only speculate on causal pathways which might underlie our finding of an association between the RAAS and PTH in this general population cohort. Because the RAAS is highly dynamic depending on sodium intake and volume status, it is also impossible to determine the time course of the RAAS-PTH relationship. We did not have measures of ionized calcium or vitamin D supplementation to further refine our adjustment for known predictors of PTH. Finally, unlike ACEi and ARB medications, mineralocorticoid receptor antagonist use could not be distinguished from use of other potassium-sparing diurectics in the MESA examinations and was therefore categorized as non-RAAS inhibitor medication use. However, the total number of participants taking these medications was very small (n = 158) and their inclusion in the non-RAAS inhibitor category would have biased our results toward the null.
In summary, we have shown for the first time an independent association between higher aldosterone levels and higher PTH and between RAAS inhibitor medication use and lower PTH in a large, diverse, community-based cohort. This affirms a RAAS-PTH relationship that has been increasingly described in pathologic states including primary aldosteronism and end stage kidney disease. The use of medications that target the RAAS may represent a method to mitigate cardiovascular and skeletal disease mediated by aldosterone and PTH.
Acknowledgments
The authors wish to thank the investigators, staff, and participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Nos. R01 HL096875, N01 HC95159, N01 HC95166, N01 HC95169, R01 HL071739, R01 HL072403, and K23 HL11177 (A.V.). Research was also supported by a Brigham and Women's Hospital Biomedical Research Institute Grant (A.V.), a William Randolph Hearst Young Investigator Award from the Brigham and Women's Hospital Department of Medicine (A.V.), and a Harvard Medical School Research Fellowship (J.M.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- 25(OH)D
- 25-hydroxyvitamin D
- ACE
- angiotensin converting enzyme
- BMI
- body mass index
- BP
- blood pressure
- CKD-EPI
- Chronic Kidney Disease Epidemiology Collaboration equation
- CV
- coefficient of variation
- eGFR
- estimated glomerular filtration rate
- HA
- hyperaldosteronism
- MESA
- Multi-Ethnic Study of Atherosclerosis
- PRA
- plasma renin activity
- RAAS
- renin-angiotensin-aldosterone system.
References
- 1. Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98(1):121–128. [DOI] [PubMed] [Google Scholar]
- 2. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 3. Tomaschitz A, Pilz S, Ritz E, Meinitzer A, Boehm BO, März W. Plasma aldosterone levels are associated with increased cardiovascular mortality: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Eur Heart J. 2010;31:1237–1247. [DOI] [PubMed] [Google Scholar]
- 4. Carbone LD, Cross JD, Raza SH, et al. Fracture risk in men with congestive heart failure risk reduction with spironolactone. J Am Coll Cardiol. 2008;52:135–138. [DOI] [PubMed] [Google Scholar]
- 5. Salcuni AS, Palmieri S, Carnevale V, et al. Bone involvement in aldosteronism. J Bone Miner Res. 2012;27:2217–2222. [DOI] [PubMed] [Google Scholar]
- 6. van Ballegooijen AJ, Kestenbaum B, Sachs MC, et al. Association of 25-hydroxyvitamin D and parathyroid hormone with incident hypertension: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol. 2014;63(12):1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Ballegooijen AJ, Visser M, Cotch MF, et al. Serum vitamin D and parathyroid hormone in relation to cardiac structure and function: The ICELAND-MI substudy of AGES-Reykjavik. J Clin Endocrinol Metab. 2013;98(6):2544–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Ballegooijen AJ, Visser M, Kestenbaum B, et al. Relation of vitamin D and parathyroid hormone to cardiac biomarkers and to left ventricular mass (from the Cardiovascular Health Study). Am J Cardiol. 2013;111:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pilz S, Tomaschitz A, Drechsler C, et al. Parathyroid hormone level is associated with mortality and cardiovascular events in patients undergoing coronary angiography. Eur Heart J. 2010;31(13):1591–1598. [DOI] [PubMed] [Google Scholar]
- 10. van Ballegooijen AJ, Reinders I, Visser M, et al. Serum parathyroid hormone in relation to all-cause and cardiovascular mortality: The Hoorn study. J Clin Endocrinol Metab. 2013;98:E638–E645. [DOI] [PubMed] [Google Scholar]
- 11. Silverberg SJ, Shane E, de la Cruz L, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res. 1989;4(3):283–291. [DOI] [PubMed] [Google Scholar]
- 12. Rubin MR, Bilezikian JP, McMahon DJ, et al. The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab. 2008;93(9):3462–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rossi GP. Hyperparathyroidism, arterial hypertension and aortic stiffness: A possible bidirectional link between the adrenal cortex and the parathyroid glands that causes vascular damage? Hypertens Res. 2011;34(3):286–288. [DOI] [PubMed] [Google Scholar]
- 14. Brown JM, Vaidya A. Interactions between adrenal-regulatory and calcium-regulatory hormones in human health. Curr Opin Endocrinol Diabetes Obes. 2014;21(3):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomaschitz A, Ritz E, Pieske B, et al. Aldosterone and parathyroid hormone interactions as mediators of metabolic and cardiovascular disease. Metabolism. 2014;63(1):20–31. [DOI] [PubMed] [Google Scholar]
- 16. Tomaschitz A, Pilz S. Interplay between sodium and calcium regulatory hormones: A clinically relevant research field. Hypertension. 2014;63(2):212–214. [DOI] [PubMed] [Google Scholar]
- 17. Chau K, Holmes D, Melck A, Chan-Yan C. Secondary hypertension due to concomitant aldosterone-producing adenoma and parathyroid adenoma. Am J Hypertens. 2014;hpu102. [DOI] [PubMed] [Google Scholar]
- 18. Pilz S, Kienreich K, Drechsler C, et al. Hyperparathyroidism in patients with primary aldosteronism: Cross-sectional and interventional data from the GECOH study. J Clin Endocrinol Metab. 2012;97:E75–E79. [DOI] [PubMed] [Google Scholar]
- 19. Maniero C, Fassina A, Seccia TM, et al. Mild hyperparathyroidism: A novel surgically correctable feature of primary aldosteronism. J Hypertens. 2012;30:390–395. [DOI] [PubMed] [Google Scholar]
- 20. Ceccoli L, Ronconi V, Giovannini L, et al. Bone health and aldosterone excess. Osteoporos Int. 2013;24(11):2801–2807. [DOI] [PubMed] [Google Scholar]
- 21. Petramala L, Zinnamosca L, Settevendemmie A, et al. Bone and mineral metabolism in patients with primary aldosteronism. Int J Endocrinol. 2014;2014:836529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brunaud L, Germain A, Zarnegar R, et al. Serum aldosterone is correlated positively to parathyroid hormone (PTH) levels in patients with primary hyperparathyroidism. Surgery. 2009;146(6):1035–1041. [DOI] [PubMed] [Google Scholar]
- 23. Grant FD, Mandel SJ, Brown EM, Williams GH, Seely EW. Interrelationships between the renin-angiotensin-aldosterone and calcium homeostatic systems. J Clin Endocrinol Metab. 1992;75:988–992. [DOI] [PubMed] [Google Scholar]
- 24. Brown JM, Williams JS, Luther JM, et al. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension. 2014;63(2):273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rifkin DE, Khaki AR, Jenny NS, et al. Association of renin and aldosterone with ethnicity and blood pressure: The multi-ethnic study of atherosclerosis. Am J Hypertens. 2014;27(6):801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bild DE, Bluemke DA, Burke GL, et al. Multi-ethnic study of atherosclerosis: Objectives and design. Am J Epidemiol. 2002;156(9):871–881. [DOI] [PubMed] [Google Scholar]
- 27. Eastell R, Arnold A, Brandi ML, et al. Diagnosis of asymptomatic primary hyperparathyroidism: Proceedings of the third international workshop. J Clin Endocrinol Metab. 2009;94(2):340–350. [DOI] [PubMed] [Google Scholar]
- 28. Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int. 2007;71(1):31–38. [DOI] [PubMed] [Google Scholar]
- 29. Robinson-Cohen C, Hoofnagle AN, Ix JH, Sachs MC, et al. Racial differences in the association of serum 25-hydroxyvitamin D concentration with coronary heart disease events. JAMA. 2013;310(2):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33 Suppl 1:S62–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sachs MC, Shoben A, Levin GP, et al. Estimating mean annual 25-hydroxyvitamin D concentrations from single measurements: The Multi-Ethnic Study of Atherosclerosis. Am J Clin Nutr. 2013;97(6):1243–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bosworth C, Sachs MC, Duprez D, et al. Parathyroid hormone and arterial dysfunction in the multi-ethnic study of atherosclerosis. Clin Endocrinol (Oxf). 2013;79(3):429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cavalier E, Delanaye P, Vranken L, et al. Interpretation of serum PTH concentrations with different kits in dialysis patients according to the KDIGO guidelines: Importance of the reference (normal) values. Nephrol Dial Transplant. 2012;27(5):1950–1956. [DOI] [PubMed] [Google Scholar]
- 35. Snijder MB, Lips P, Seidell JC, et al. Vitamin D status and parathyroid hormone levels in relation to blood pressure: A population-based study in older men and women. J Intern Med. 2007;261(6):558–565. [DOI] [PubMed] [Google Scholar]
- 36. Mateus-Hamdan L, Beauchet O, Bouvard B, Legrand E, Fantino B, Annweiler C. High parathyroid hormone, but not low vitamin D concentrations, expose elderly inpatients to hypertension. Geriatr Gerontol Int. 2013;13(3):783–791. [DOI] [PubMed] [Google Scholar]
- 37. Saleh F, Jorde R, Svartberg J, Sundsfjord J. The relationship between blood pressure and serum parathyroid hormone with special reference to urinary calcium excretion: The Tromsø study. J Endocrinol Invest. 2006;29(3):214–220. [DOI] [PubMed] [Google Scholar]
- 38. Zhao G, Ford ES, Li C, Kris-Etherton PM, Etherton TD, Balluz LS. Independent associations of serum concentrations of 25-hydroxyvitamin D and parathyroid hormone with blood pressure among US adults. J Hypertens. 2010;28(9):1821–1828. [DOI] [PubMed] [Google Scholar]
- 39. Fischer E, Hannemann A, Rettig R, et al. A high aldosterone-to-renin ratio is associated with high serum parathyroid hormone concentrations in the general population. J Clin Endocrinol Metab. 2013;jc20133214. [DOI] [PubMed] [Google Scholar]
- 40. Pitt B, Reichek N, Willenbrock R, et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: The 4E-left ventricular hypertrophy study. Circulation. 2003;108:1831–1838. [DOI] [PubMed] [Google Scholar]
- 41. Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. [DOI] [PubMed] [Google Scholar]
- 42. Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. [DOI] [PubMed] [Google Scholar]
- 43. Maniero C, Fassina A, Guzzardo V, et al. Primary hyperparathyroidism with concurrent primary aldosteronism. Hypertension. 2011;58:341–346. [DOI] [PubMed] [Google Scholar]
- 44. Rossi GP, Ragazzo F, Seccia TM, et al. Hyperparathyroidism can be useful in the identification of primary aldosteronism due to aldosterone-producing adenoma. Hypertension. 2012;60:431–436. [DOI] [PubMed] [Google Scholar]
- 45. Pilz S, Tomaschitz A, März W, Cavalier E, Ritz E. Aldosterone and parathyroid hormone: A complex and clinically relevant relationship. Calcif Tissue Int. 2010;87(4):373–374. [DOI] [PubMed] [Google Scholar]
- 46. Koiwa F, Komukai D, Hirose M, et al. Influence of renin-angiotensin system on serum parathyroid hormone levels in uremic patients. Clin Exp Nephrol. 2012;16:130–135. [DOI] [PubMed] [Google Scholar]
- 47. Staessen J, Lijnen P, Fagard R, Verschueren LJ, Amery A. Rise in plasma concentration of aldosterone during long-term angiotensin II suppression. J Endocrinol. 1981;91(3):457–465. [DOI] [PubMed] [Google Scholar]
- 48. MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart. 1999;82(1):57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3(9):486–492. [DOI] [PubMed] [Google Scholar]
- 50. Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145–153. [DOI] [PubMed] [Google Scholar]
- 51. Rossi E, Sani C, Perazzoli F, Casoli MC, Negro A, Dotti C. Alterations of calcium metabolism and of parathyroid function in primary aldosteronism, and their reversal by spironolactone or by surgical removal of aldosterone-producing adenomas. Am J Hypertens. 1995;8:884–893. [DOI] [PubMed] [Google Scholar]