

Abstract
Context:
Kidney dysfunction induces insulin resistance, but it is unknown if β cell function is affected.
Objective:
To investigate insulin release (β cell function) and glucose tolerance following a standardized oral glucose tolerance test (OGTT) across kidney function strata.
Setting and Design:
Community-based cohort study from the Uppsala Longitudinal Study of Adult Men (ULSAM).
Participants and Main Outcome Measure:
Included were 1015 nondiabetic Swedish men aged 70–71 years. All participants underwent OGTT and euglycaemic hyperinsulinaemic clamp (HEGC) tests, allowing determination of insulin sensitivity, β cell function, and glucose tolerance. Kidney function was estimated by cystatin C-algorithms. Mixed models were used to identify determinants of insulin secretion after the hyperglycemic load.
Results:
As many as 466 (46%) of participants presented moderate-advanced kidney disease. Insulin sensitivity (by HEGC) decreased across decreasing kidney function quartiles. After the OGTT challenge, however, β cell function indices (area under the curve for insulin release, the estimated first phase insulin release, and the insulinogenic index) were incrementally higher. Neither the oral disposition index nor the 2-h postload glucose tolerance differed across the kidney function strata. Mixed models showed that dynamic insulin release during the OGTT was inversely associated with kidney function, despite the correction for each individual's insulin sensitivity or its risk factors.
Conclusions:
In older men, β cell function after a hyperglycemic load appropriately compensated the loss in insulin sensitivity that accompanies kidney dysfunction. As a result, the net balance between insulin sensitivity and β cell function was preserved.
Insulin resistance (IR) is common among individuals with chronic kidney disease (CKD) (1), and it worsens as kidney function declines (2, 3). IR in CKD is multifactorial, and whereas modifiable factors such as obesity, sedentary lifestyle, and inappropriate diet (4) play an important role, it is attributable also to metabolic alterations associated with the loss of kidney function per se, such as electrolyte disturbances, metabolic acidosis, accumulation of various metabolites, and hypovitaminosis D (5–7). The primary molecular mechanism of IR in CKD is believed to be a postreceptor signaling defect in skeletal muscle (1, 8).
In the setting of IR, whereas glucose uptake by muscle and fat cells is reduced, the net release of glucose from hepatic cells is increased due to the reduced glycogen synthesis and storage, leading altogether to hyperglycemia. To maintain a normal glucose concentration, the pancreatic β cell increases insulin secretion to stimulate glucose uptake in target cells. In some individuals, β cell compensation can fail, leading to impaired glucose tolerance and thereby constituting an additional cause of IR. It is plausible (9), but recently contested (10), that kidney dysfunction leads to β cell dysfunction resulting in impaired glucose tolerance. We have previously shown in a population of community-dwelling older men with similar age that cystatin C-estimated glomerular filtration rate (eGFR) is independently associated with insulin sensitivity (IS) as assessed by the gold-standard hyperinsulinaemic euglycaemic clamp technique (3, 11, 12). Using the same cohort, we aim to investigate potential links between impaired renal function, glucose tolerance, and β cell function following a standardized oral glucose tolerance test (OGTT).
Study Design and Methods
Participants
Individuals in the study were those included in the third examination of the Uppsala Longitudinal Study of Adult Men (ULSAM: www.pubcare.uu.se/ULSAM/). ULSAM invited 2322 men (age 50–51 years) born between 1920 and 1924 living in Uppsala County, Sweden to a thorough investigation of risk factors. These individuals were invited to attend re-examination when they became 60 years and then again at 70 years (third examination cycle). At the third examination, 1221 men of the original cohort attended and they all were 70–71 years old. For the present study, men who were diagnosed with overt diabetes mellitus (DM; n = 178), lacked serum cystatin-C measurements (n = 27) or did not undergo OGTT (n = 1) were excluded, leaving 1015 individuals available for analysis. DM was defined as one or more of the following: fasting plasma glucose ≥7.0 mmol/L, 2-h postload glucose levels ≥11.1 mmol/L, and use of oral-hypoglycemic agents or insulin. The study was approved by the Ethics Committee of the Faculty of Medicine at Uppsala University and informed consent was obtained from all subjects.
Oral glucose tolerance test
Participants completed a standard OGTT by ingesting a solution containing 75 g of dextrose after having refrained from food or any liquids except water for 10 h. Venous blood samples were collected at time points 0, 30, 60, 90, and 120 min for measurement of plasma glucose and plasma insulin. Fasting glucose and insulin were measured by standard methods. The intra-class correlation (ICC) of the OGTT test following repeated measurements in a subsample of 17 individuals was 0.57, and the coefficient of variation (CV) was 0.50 (13).
Insulin resistance, β cell function, and glucose tolerance by OGTT
Insulin resistance was assessed by fasting insulin concentration and the Matsuda index (14). To estimate the β cell function, individual phases of insulin release across the different time periods in OGTT test were quantified with the areas under the insulin curve (AUCins) as calculated with the Simpsons rule (modified and improved from the trapezoidal rule) (15). The AUC insulin-time curves considered the intervals 0–30 min and 90–120 min. Logarithmic transformation was used during the decline phase of insulin response (from 60 min to the end of OGTT). The AUCins for the first 30 min in the course of OGTT has been shown to be superior to HOMA calculated indices in the evaluation of insulin secretion by pancreatic β cells (25, 26). The effect of insulin on glucose control was quantified by the ratio of AUCins and AUCgluc. The insulinogenic index at 30 min was also used to assess β-cell function with the following formula: [(insulin 30 min − insulin 0 min) / (glucose 30 min − glucose 0 min)]. The estimated insulin release during the first phase of the OGTT test was calculated (16) as 1283 + 1.829 × I30 − 138.7 × G30 + 3.772 × I0; where I30 is insulin (pmol/L) at 30 min and G30 is glucose (mmol/L) at 30 min, and I0 is insulin at 0 min. Glucose tolerance was defined by the 2 h glucose concentration after the OGTT test. To evaluate the composite β cell function, we calculated the oral disposition index (DIo) as insulinogenic index X 1/fasting insulin from the OGTT test (17).
Insulin resistance by hyperinsulinaemic euglycaemic clamp
A hyperinsulinaemic euglycaemic clamp (HEGC) was performed as detailed by DeFronzo et al (18) with slight modifications (19). Briefly, two intravenous (IV) infusion lines were placed, one into an antecubital vein and the other into a hand or wrist vein. After 10-min priming infusion, insulin (Actrapid Human) was held constant at a concentration of 660 pmol/L for 120 min. Plasma glucose was measured every 5 min to be clamped at the euglycemic level (5.1 mmol/L) by infusion of variable amounts of 20% dextrose solution. The glucose metabolic clearance rate (MCR) was then calculated as the measure of IS. The coefficient of variation of clamp test was 12% on repeated clamp investigations within 30 days in the same individual (20).
Study covariates
Education, smoking status, and physical activity were assessed by a self-reporting record at the study visit. Previous history of cardiovascular disease (CVD) was defined as any cardiovascular event registered in the Swedish Hospital Discharge Registry according to the International Classification of Disease-8 (ICD-8) codes 390–458 or ICD-9 codes 390–459. Hypertension was defined as systolic blood pressure (BP) ≥140 mmHg, diastolic BP ≥90 mmHg, and/or use of antihypertensive medication. Serum cystatin C was measured by latex-enhanced reagent (Dade Behring) with Behring BN ProSpec analyzer (Dade Behring) (21). The estimated GFR (eGFR) was calculated from serum cystatin C by the following formula: Y = 77.24X−1.2623, which yields values closely correlated with those of iohexol clearance (22). Interleukin-6 (IL-6) was analyzed by an ELISA kit (IL-6 HS, R&D Systems).
Statistical methods
Data are presented as means ± SD or median and interquartile range (IQR). Comparisons between quartiles of eGFR distribution were performed by P for trend analysis, using analysis of variance (ANOVA), followed by post hoc testing with Scheffe's test for categorical variables and Jonckeheere-Terpstra test for continuous variables. Multivariable linear regressions models were used to analyze the association of HEGC-assessed IS, eGFR, and BMI to predict OGTT-assessed beta cell function. Finally, linear mixed model (LMM) regressions analyzed the factors associated time-dependent insulin secretion during the OGTT challenge. A random intercept growth model was used to define the random part and included the patient's study number. In the primary model (direct), we studied the association between eGFR and time-dependent insulin secretion directly controlling for each individual's clamp-derived MCR. In the secondary model (indirect), we studied the association between eGFR and time-dependent insulin secretion controlling for the known risk factors of insulin resistance, which were BMI, cardiovascular disease, hypertension, smoking, physical activity, education, and IL-6. eGFR, clamp-derived MCR, BMI, and IL-6 were used as continuous variables in the LMM. A log-likelihood ratio (LR) test was used to determine whether the model improved the fit. A nonlinear mixed model was used to graphically represent with a quadratic fit the insulin and glucose concentration curves during the OGTT across eGFR quartiles. P values <0.05 were set as criterion for statistical significance. All statistical analyses were performed using STATA 12.0 software (StataCorp LP.).
Results
In this homogeneous population of elderly men, eGFR presented a normal distribution, with a median value of 61.3 (Interquartile range: 52.8–72.8) mL/min/1.73 m2. Based on the KDIGO/KDOQI classification (23), as many as 356 individuals would be considered to have CKD stage 3a, 99 CKD stage 3b, and 11 CKD stage 4. The demographic and clinical characteristics of the study population stratified by quartiles of eGFR are presented in Table 1. Across decreasing eGFR strata, subjects presented incrementally increasing IL-6 concentration and a trend towards increasing BMI. Subjects with decreased eGFR who were more likely to have CVD history and hypertension and to be smokers, tended to be less physically active and had higher BMI values. IS, as assessed by either HEGC or the OGTT-derived Matsuda index, significantly decreased across decreasing eGFR quartiles, but fasting insulin concentration did not differ between the groups.
Table 1.
General Characteristics of the Study Individuals Categorized by Quartiles of eGFR Distribution
| Parameters | Decreasing Quartiles of eGFR |
P for Trend | |||
|---|---|---|---|---|---|
| Q1 (n = 254) | Q2 (n = 254) | Q3 (n = 254) | Q4 (n = 253) | ||
| eGFR, mL/min/1.73 m2 | 76.3 (72.5–82.1) | 64.7 (62.3–66.9) | 57.1 (54.9–58.9) | 46.7 (41.3–49.6) | <.001 |
| CKD, % | 0 | 0 | 85% | 100% | <.001 |
| BMI, kg/m2 | 25.9 ± 3.1 | 25.8 ± 3.3 | 25.8 ± 3.2 | 26.4 ± 3.3 | .07 |
| IL-6, ng/L | 2.9 (1.9–4.7) | 3.2 (2.2–5.7) | 3.6 (2.1–6.4) | 4.1 (2.4–7.6) | <.001 |
| Cardiovascular disease, % | 20 | 20 | 28 | 31 | <.001 |
| Hypertension, % | 25 | 22 | 30 | 40 | <.001 |
| Current smoking, % | 13 | 20 | 23 | 25 | <.001 |
| Sedentary/moderate physical activity, % | 34 | 37 | 37 | 42 | .08 |
| University education, % | 26 | 30 | 28 | 23 | .27 |
| Insulin resistance assessment | |||||
| Clamp-derived MCR, mg min−1 kg−1 | 6.25 ± 2.21 | 6.09 ± 2.22 | 6.07 ± 1.99 | 5.30 ± 2.00 | .006 |
| Fasting glucose at OGTT, mmol/L | 5.29 (4.8–5.7) | 5.39 (5–5.7) | 5.3 (5–5.6) | 5.3 (4.9–5.7) | .90 |
| Fasting insulin at OGTT, pmol/L | 10.9 (8.2–14.1) | 10.9 (8.2–14.1) | 10.9 (8.2–14.1) | 10.9 (8.2–14.1) | .37 |
| OGTT-derived Matsuda Index | 36.5 (23.3–56.4) | 32.2 (22.2–50.5) | 34.5 (24.6–48.3) | 32.8 (21.7–45.8) | .01 |
Values are represented as mean ± standard deviation, median (interquartile range), or percentage.
Abbreviations: BMI, body mass index; CKD, presence of chronic kidney disease defined as eGFR < 60 mL/min/1.73 m2; IL-6, interleukin-6.
Insulin secretion after the OGTT challenge is presented in Table 2 according to quartiles of eGFR distribution. Both within and across decreasing eGFR quartiles, individuals showed gradually higher insulin levels (AUC) at all time-intervals (Figure 1, panel A). The insulinogenic index after 30 min was significantly increased, and the estimated insulin secretion during the first and second phase from the OGTT increased with decreasing eGFR quartiles. The effect of insulin on glucose control decreased (ie, AUCins/AUCgluc increased) within each eGFR quartile, and across decreasing eGFR categories, but at the end of the OGTT challenge there was no difference across the eGFR strata. Likewise, the 2-h postload glucose concentration (Figure 1, panel B) and the proportion of individuals with impaired glucose tolerance did not differ across the eGFR groups. The composite index of β cell function, the DIo which takes into consideration β cell function corrected by IS, increased with reducing eGFR quartiles. Multivariable regression models (Table 3) showed that the association between eGFR and β cell function adaptation prevailed even after taking into consideration IS and BMI.
Table 2.
Surrogates of β Cell Function and Glucose Tolerance During an OGTT Stratified by Quartiles of eGFR Distribution
| Parameters | Decreasing Quartiles of eGFR |
P for Trend | |||
|---|---|---|---|---|---|
| Q1 (n = 254) | Q2 (n = 254) | Q3 (n = 254) | Q4 (n = 253) | ||
| β cell function assessment | |||||
| AUCins0–30, μ × 30 min/L | 855 (621–1172) | 921 (657–1348) | 923 (663–1333) | 1002 (650–1464) | .004 |
| AUCins90–120, μ × 30 min/L | 3075 (2789–3450) | 3155 (2877–3519) | 3246 (2882–3667) | 3325 (2926–3652) | <.001 |
| Estimated first phase insulin release, pM | 976 (752–1214) | 978 (772–1398) | 1015 (781–1468) | 1098 (751–1494) | <.001 |
| Insulinogenic index30, μ/L/min | 3.8 (2.5–6.0) | 4.2 (2.7–7.0) | 4.5 (3.0–7.2) | 4.7 (3.0–7.7) | .008 |
| Glucose tolerance | |||||
| AUCins0–30/AUCglu0–30 | 4.0 (3.0–5.4) | 4.2 (3.2–6.3) | 4.4 (3.2–6.4) | 4.6 (3.2–6.8) | <.001 |
| AUCins90–120/AUCglu90–120 | 14.2 (11.7–17.1) | 14.5 (12.1–17.7) | 14.7 (12.4–18.0) | 14.4 (12.2–17.6) | .24 |
| 2-h glucose concentration, mmol/L | 6.88 ± 1.91 | 6.85 ± 1.81 | 6.91 ± 1.78 | 7.10 ± 1.81 | .12 |
| Impaired glucose tolerance, % | 32 | 30 | 28 | 35 | .34 |
| Composite β cell function | |||||
| Oral disposition index, DI0 | 0.98 (0.60–1.62) | 1.13 (0.64–1.76) | 1.23 (0.78–1.90) | 1.16 (0.69–1.82) | .01 |
Values are presented as median (interquartile range).
Abbreviations: AUC, area under the curve; AUCins0–30, area under curve of insulin for 0 min to 30 min; AUCins0–30/AUCglu0–30, area under curve of insulin for 0 min to 30 min/area under curve for glucose from 0 min to 30 min; AUCins90–120, area under curve of insulin for 90 min to 120 min; AUCins90–120/AUCglu90–120, area under curve of insulin for 90 min to 120 min/area under curve for glucose from 90 min to 120 min; glu, glucose; ins, insulin; Insulinogenic index30 = Insulin 30 (pmol/L) − Insulin 0 (pmol/L)/Glucose 30 (mg/dL) − Glucose 0 (mg/dL); Oral disposition index, DIo = insulinogenic index × 1/fasting insulin.
Impaired glucose tolerance was defined as 2-h glucose between 7.8–11 mmol/L.
Figure 1.
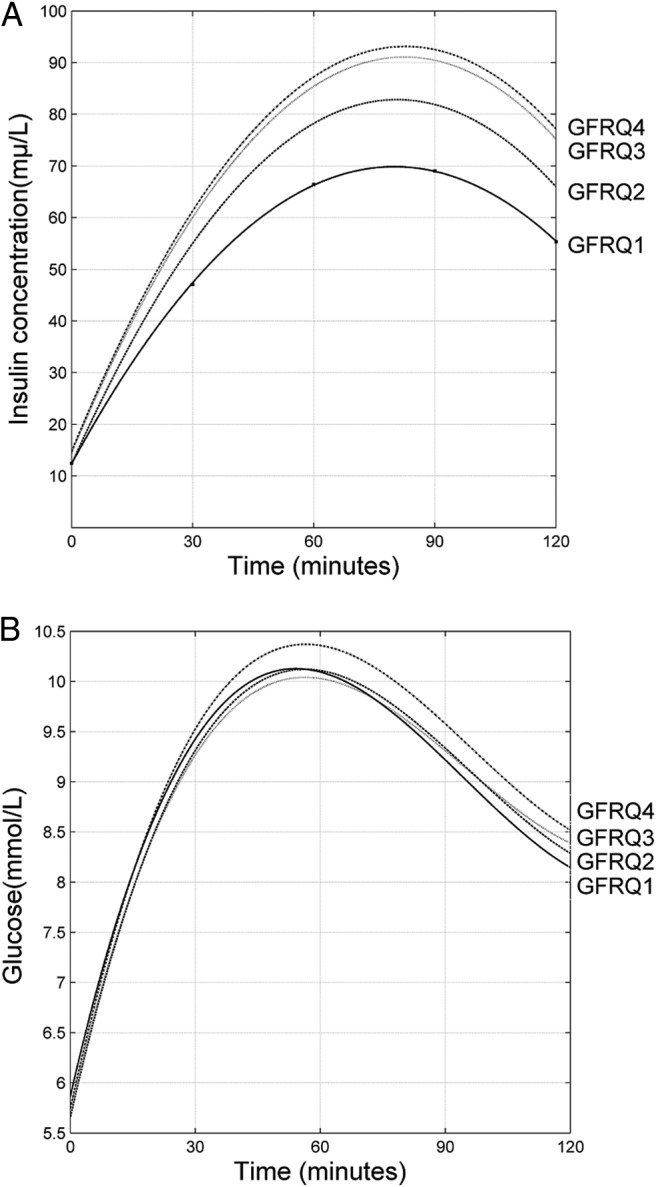
Quadratic trends of fitted insulin concentration (Panel A), and fitted glucose concentration (Panel B) by nonlinear mixed-models after an OGTT and stratified by quartiles of eGFR.
Table 3.
Multivariate Linear Regressions Predicting for β Cell Function and Glucose Tolerance During an OGTT With Clamp-Derived Glucose Clearance Rate (MCR), Estimated Glomerular Filtration Rate and Body Mass Index as Independent Variables
| Parameters | MCR, mg min−1 kg−1 (insulin sensitivity by clamp) |
eGFR, mL/min/1.73 m2 |
BMI (kg/m2) |
|||
|---|---|---|---|---|---|---|
| Standard B | P | Standard B | P | Standard B | P | |
| β cell function | ||||||
| AUCins0–30, μ × 30 min/L | −0.41 | <.001 | −0.06 | .02 | .12 | .001 |
| Estimated first phase insulin release, pM | −0.33 | <.001 | −0.08 | .01 | .14 | <.001 |
| Insulinogenic index30, μ/L/min | −0.31 | <.001 | −0.08 | .01 | .06 | .18 |
| Glucose tolerance | ||||||
| 2-h glucose concentration | −0.52 | <.001 | −0.01 | .69 | −0.07 | .04 |
Abbreviations: AUCins0–30, area under curve of insulin for 0 min to 30 min; Insulinogenic index30 = Insulin 30 (pmol/L) − Insulin 0 (pmol/L)/Glucose 30 (mg/dL) − Glucose 0 (mg/dL).
The multivariate determinants of insulin secretion during an OGTT were assessed by a linear mixed regression model (LMM, Table 4) modeling eGFR as a continuous variable. In the primary model, it is shown that dynamic insulin release increases in proportion to each individual's eGFR, even when directly controlling for their clamp-assessed IS. In the secondary model, controlling instead for IR-traditional and nontraditional risk factors, similar associations were observed.
Table 4.
Maximum Likelihood Estimates for Insulin Release During an Oral Glucose Tolerate Test
| Parameters | Primary Model (direct) |
Secondary Model (indirect) |
||||
|---|---|---|---|---|---|---|
| Estimate | se | P Value | Estimate | se | P Value | |
| Fixed part | ||||||
| Constant | −142.4 | 4.8 | <.001 | −186.3 | 4.72 | <.001 |
| Time (every 30 mina) | 77.6 | 2.27 | <.001 | 233.9 | 2.10 | <.001 |
| Time2 | −10.6 | 0.33 | <.001 | −46.5 | 0.33 | <.001 |
| Clamp-derived MCR, mg min−1 kg−1 | −8.40 | 0.40 | <.001 | |||
| eGFR, per mL/min/1.73 m2 increase | −0.13 | 0.05 | .02 | −0.14 | 0.03 | <.001 |
| BMI, per kg/m2 increase | 0.90 | 0.12 | <.001 | |||
| Hypertension, presence | −0.33 | 0.85 | .70 | |||
| Smoking, presence | −1.11 | 0.96 | .25 | |||
| Sedentary/moderate physical activity | −2.50 | 0.60 | <.001 | |||
| University education | 0.80 | 0.34 | .02 | |||
| Cardiovascular disease, presence | 2.76 | 0.87 | <.001 | |||
| IL-6, per ng/L increase | −0.16 | 0.05 | <.001 | |||
| Random part | ||||||
| Residual | 1380.6 | 31.8 | <.001 | 184.3 | 6.55 | <.001 |
| Patient No. variance | 0.0002 | −0.0002 | <.001 | 0.0001 | 0.0001 | <.001 |
Every 30 min (time 0, 30, 60, 90, 120 min) set as equidistant categories 1, 2, 3, 4, 5.
Discussion
Our results in a large population of community-dwelling older men indicate that β cell function after a hyperglycemic load appropriately compensates for the loss in insulin sensitivity that accompanies kidney dysfunction. As a result, the net balance between insulin sensitivity and β cell function was preserved.
Advanced CKD is characterized by resistance to peripheral actions of insulin and is accompanied by both hyperinsulinemia and hyperglycemia. In this cohort, and more extensively reported in our previous analyses (3, 11, 12), a lower kidney function is associated with worse IS as assessed by the gold-standard HEGC. This was also the case for the OGTT-derived Matsuda index, which may be a more valid assessment of basal IS compared to indices derived from fasting insulin levels (12). In the setting of dynamic states, such as those instigated by OGTT and HEGC, glucose is primarily disposed of in skeletal muscle, the main body determinant of peripheral insulin resistance (24). In the fasting state, however, IS is determined by the ability of insulin to regulate hepatic glucose production, thereby reflecting primarily hepatic insulin resistance. Pham et al (10) previously reported that higher fasting insulin concentration was associated with lower eGFR. Contrary to that study, we fail to observe any association between these two variables. Although we have no clear explanation to this divergence, we believe it is possible that the population homogeneity of our cohort renders a patient selection with too narrow eGFR range to adequately address this issue.
The impairment of β cell function is thought to be one of the main reasons for diabetes incidence and progressive deterioration in glucose control despite treatment in type 2 diabetes (25). The loss of the initial insulin response (first 30 min) to glucose loading is considered the first detectable abnormality of β cell function (26), and plays an important role in subsequently inhibiting endogenous glucose production (27). In our study, we used three different dynamic β cell function estimations that evaluate the insulin response during the first 30 min of the OGTT. Again, dynamic β cell function assessments (26, 28) are preferred to those derived from fasting glucose or insulin (29), as the latter are more dependent on IR (28).
While early studies indicated that glucose intolerance is present in CKD patients (9, 30), our study demonstrates a compensatory increase of β cell function in the face of the progressive loss of IS inherent to kidney dysfunction. Such observation confirms and expands the large study of Pham et al (10) including older US adults, where β cell function was appropriately augmented with lower creatinine-based eGFR. A strength of our study over that of Pham et al (10) is that we are able to consider the concomitant assessment of both β cell function and IS from different gold-standard methods, allowing us to better separate these inter-related processes. This is shown by both the direct inclusion of HEGC-derived IS into the linear mixed model of insulin release (Table 4) as well as by the lack of differences in oral disposition index across eGFR strata (Table 2), representing the overall composite beta cell function. Given that most prandial glucose is disposed of in the muscle, low muscle mass may reduce an individual's ability to dispose of a fixed-dose glucose load. Therefore, an additional advantage of our study over the previous report is that we estimated kidney function not from serum creatinine, but from cystatin C measurements, which are less influenced by muscle mass. Finally, and at the other side of the eGFR spectrum, our data would be in accordance with Niemczyk et al (31) who observed increased β cell function in dialysis patients as compared to healthy subjects.
Because glucose uptake by extrahepatic tissues is reduced in patients with advanced CKD (32), this may partly explain the disproportionate rise of plasma insulin during the first OGTT phase in our study. In addition, impaired clearance could also affect. Elucidating the relative contribution of hypersecretion vs clearance would, however, imply infusion studies, such as for instance radioactive iodine-labeled insulin (33) or titrated semisynthetic biologically active insulin (34). Nevertheless, these measurements are not feasible to perform in large cohorts such as this one. The fact that our population was composed of Caucasian nondiabetic older men of identical age is also a strength towards the study of unbiased associations. However, because of the applied strict inclusion criteria, the narrow eGFR range earlier alluded, and the relatively healthy profile of included individuals, the extrapolation of our data to other patient groups should be done with caution.
As a clinical implication that can be derived from our study, therapeutic and preventive strategies aiming at improving glucose tolerance in CKD may benefit more if they are focused on improving IS rather than β cell function. In this sense, because reduced whole body IS is mainly linked to the inability of skeletal muscle to take up excess glucose from the circulation (1, 35), attempts to increase muscle mass or muscle strength, or both, by means of improved nutrition, increased physical activity or other related approaches would seem appropriate (36–38). Furthermore, our results suggest that a separate measurement of β cell function may not be an indispensable component needed for the clinical assessment of glucose intolerance in CKD patients. In conclusion, our results indicate that β cell function adequately compensates for the loss of IS that occurs during the earlier stages of CKD. As a final cautionary remark, whether β cell function is preserved also in individuals with more advanced stages of CKD (ie, CKD stages 4 and 5) cannot be derived from this analysis and needs verification in future studies.
Acknowledgments
We are indebted to all participants in the study and are grateful to the ULSAM group.
This work was supported by Grants from the Swedish Research Council. T.A.I. is supported by K24 DK 62849 from the National Institute of Diabetes and Digestive and Kidney Diseases and by Veterans Administration Merit Award No. 1I01CX000414. Baxter Novum is the result of a grant from Baxter Healthcare Corporation to Karolinska Institutet.
Author contributions: T.J. designed and carried out the analyses, as well as drafted the manuscript. J.J.C. conceived the study, participated in study design/data interpretation, and co-wrote the manuscript draft. H.X. and T.A.I. provided support on data analysis and interpretation. The other authors revised and contributed to develop the manuscript to its final form. U.R., P.S., T.C., T.E.L., J.A., and B.L. provided economic support, as well as access to and management of the database. J.J.C. stands as guarantor for this work.
Disclosure Summary: Bengt Lindholm is employed by Baxter Healthcare Corporation. T.E.L. is an employee of Astellas. The other authors have nothing to disclose.
Footnotes
- CKD
- chronic kidney disease
- eGFR
- estimated glomerular filtration rate
- HEGC
- hyperinsulinaemic euglycaemic clamp
- IR
- insulin resistance
- IS
- insulin sensitivity
- LMM
- linear mixed model
- MCR
- metabolic clearance rate
- OGTT
- oral glucose tolerance test
- ULSAM
- Uppsala Longitudinal Study of Adult Men.
References
- 1. DeFronzo RA, Alvestrand A, Smith D, Hendler R, Hendler E, Wahren J. Insulin resistance in uremia. J Clin Invest. 1981;67:563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eidemak I, Feldt-Rasmussen B, Kanstrup IL, Nielsen SL, Schmitz O, Strandgaard S. Insulin resistance and hyperinsulinaemia in mild to moderate progressive chronic renal failure and its association with aerobic work capacity. Diabetologia. 1995;38:565–572. [DOI] [PubMed] [Google Scholar]
- 3. Nerpin E, Risérus U, Ingelsson E, et al. Insulin sensitivity measured with euglycemic clamp is independently associated with glomerular filtration rate in a community-based cohort. Diabetes Care. 2008;31:1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trirogoff ML, Shintani A, Himmelfarb J, Ikizler TA. Body mass index and fat mass are the primary correlates of insulin resistance in nondiabetic stage 3–4 chronic kidney disease patients. Am J Clin Nutr. 2007;86:1642–1648. [DOI] [PubMed] [Google Scholar]
- 5. Wilcken DE, Sim AS, Wang J, Wang XL. Asymmetric dimethylarginine (ADMA) in vascular, renal and hepatic disease and the regulatory role of L-arginine on its metabolism. Mol Genet Metab. 2007;91:309–317; discussion 308. [DOI] [PubMed] [Google Scholar]
- 6. Mak RH. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int. 1998;54:603–607. [DOI] [PubMed] [Google Scholar]
- 7. Nazarian S, St Peter JV, Boston RC, Jones SA, Mariash CN. Vitamin D3 supplementation improves insulin sensitivity in subjects with impaired fasting glucose. Transl Res. 2011;158:276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedman JE, Dohm GL, Elton CW, et al. Muscle insulin resistance in uremic humans: glucose transport, glucose transporters, and insulin receptors. Am J Physiol. 1991;261:E87–E94. [DOI] [PubMed] [Google Scholar]
- 9. DeFronzo RA, Alvestrand A. Glucose intolerance in uremia: site and mechanism. Am J Clin Nutr. 1980;33:1438–1445. [DOI] [PubMed] [Google Scholar]
- 10. Pham H, Robinson-Cohen C, Biggs ML, et al. Chronic kidney disease, insulin resistance, and incident diabetes in older adults. Clin J Am Soc Nephrol. 2012;7:588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu H, Huang X, Arnlöv J, et al. Clinical correlates of insulin sensitivity and its association with mortality among men with CKD stages 3 and 4. Clin J Am Soc Nephrol. 2014;9:690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jia T, Huang X, Qureshi AR, et al. Validation of insulin sensitivity surrogate indices and prediction of clinical outcomes in individuals with and without impaired renal function. Kidney Int. 2014;86:383–391. [DOI] [PubMed] [Google Scholar]
- 13. Berglund L, Berne C, Svärdsudd K, Garmo H, Zethelius B. Early insulin response and insulin sensitivity are equally important as predictors of glucose tolerance after correction for measurement errors. Diabetes Res Clin Pract. 2009;86:219–224. [DOI] [PubMed] [Google Scholar]
- 14. Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. [DOI] [PubMed] [Google Scholar]
- 15. Molodianovitch K, Faraggi D, Reiser B. Comparing the areas under two correlated ROC curves: Parametric and non-parametric approaches. Biometrical J. 2006;48:745–757. [DOI] [PubMed] [Google Scholar]
- 16. Stumvoll M, Mitrakou A, Pimenta W, et al. Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care. 2000;23:295–301. [DOI] [PubMed] [Google Scholar]
- 17. Utzschneider KM, Prigeon RL, Faulenbach MV, et al. Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care. 2009;32:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. [DOI] [PubMed] [Google Scholar]
- 19. Zethelius B, Lithell H, Hales CN, Berne C. Insulin sensitivity, proinsulin and insulin as predictors of coronary heart disease. A population-based 10-year, follow-up study in 70-year old men using the euglycaemic insulin clamp. Diabetologia. 2005;48:862–867. [DOI] [PubMed] [Google Scholar]
- 20. Brown JL. Efficacy of combined interferon and ribavirin for treatment of hepatitis C. Lancet. 1998;351:78–79. [DOI] [PubMed] [Google Scholar]
- 21. Stevens LA, Coresh J, Schmid CH, et al. Estimating GFR using serum cystatin C alone and in combination with serum creatinine: a pooled analysis of 3,418 individuals with CKD. Am J Kidney Dis. 2008;51:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Larsson A, Malm J, Grubb A, Hansson LO. Calculation of glomerular filtration rate expressed in mL/min from plasma cystatin C values in mg/L. Scand J Clin Lab Invest. 2004;64:25–30. [DOI] [PubMed] [Google Scholar]
- 23. Levey AS, Coresh J, Balk E, et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137–147. [DOI] [PubMed] [Google Scholar]
- 24. Fliser D, Pacini G, Engelleiter R, et al. Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int. 1998;53:1343–1347. [DOI] [PubMed] [Google Scholar]
- 25. Turner R, Cull C, Holman R. United Kingdom Prospective Diabetes Study 17: a 9-year update of a randomized, controlled trial on the effect of improved metabolic control on complications in non-insulin-dependent diabetes mellitus. Ann Int Med. 1996;124:136–145. [DOI] [PubMed] [Google Scholar]
- 26. Bingley PJ. Interactions of age, islet cell antibodies, insulin autoantibodies, and first-phase insulin response in predicting risk of progression to IDDM in ICA+ relatives: the ICARUS data set. Islet Cell Antibody Register Users Study. Diabetes. 1996;45:1720–1728. [DOI] [PubMed] [Google Scholar]
- 27. DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non-insulin-dependent diabetes mellitus: Contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism. 1989;38:387–395. [DOI] [PubMed] [Google Scholar]
- 28. Ahrén B, Pacini G. Importance of quantifying insulin secretion in relation to insulin sensitivity to accurately assess beta cell function in clinical studies. Eur J Endocrinol. 2004;150:97–104. [DOI] [PubMed] [Google Scholar]
- 29. Pacini G, Mari A. Methods for clinical assessment of insulin sensitivity and β-cell function. Best practice, research. Clin Endocrinol, Metab. 2003;17:305–322. [DOI] [PubMed] [Google Scholar]
- 30. Allegra V, Mengozzi G, Martimbianco L, Vasile A. Glucose-induced insulin secretion in uremia: effects of aminophylline infusion and glucose loads. Kidney Int. 1990;38:1146–1150. [DOI] [PubMed] [Google Scholar]
- 31. Niemczyk S, Szamotulska K, Giers K, et al. Homeostatic model assessment indices in evaluation of insulin resistance and secretion in hemodialysis patients. Med Sci Monit. 2013;19:592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alvestrand A, Mujagic M, Wajngot A, Efendic S. Glucose intolerance in uremic patients: the relative contributions of impaired beta-cell function and insulin resistance. Clinical Nephrology. 1989;31:175–183. [PubMed] [Google Scholar]
- 33. Reiner L, Keston AS, Green M. The Absorption and Distribution of Insulin Labelled with Radioactive Iodine. Science. 1942;96:362–363. [DOI] [PubMed] [Google Scholar]
- 34. Halban PA, Offord RE. The preparation of a semisynthetic tritiated insulin with a specific radioactivity of up to 20 Curies per millimole. The Biochemical journal. 1975;151:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siew ED, Pupim LB, Majchrzak KM, Shintani A, Flakoll PJ, Ikizler TA. Insulin resistance is associated with skeletal muscle protein breakdown in non-diabetic chronic hemodialysis patients. Kidney Int. 2007;71:146–152. [DOI] [PubMed] [Google Scholar]
- 36. Marinik EL, Kelleher S, Savla J, Winett RA, Davy BM. The Resist Diabetes trial: rationale, design, and methods of a hybrid efficacy/effectiveness intervention trial for resistance training maintenance to improve glucose homeostasis in older prediabetic adults. Contemporary Clinical Trials. 2014;37:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Donges CE, Duffield R, Guelfi KJ, Smith GC, Adams DR, Edge JA. Comparative effects of single-mode vs. duration-matched concurrent exercise training on body composition, low-grade inflammation, and glucose regulation in sedentary, overweight, middle-aged men. Appl Physiol Nutr Metab. 2013;38:779–788. [DOI] [PubMed] [Google Scholar]
- 38. Ryan AS, Li G, Blumenthal JB, Ortmeyer HK. Aerobic exercise + weight loss decreases skeletal muscle myostatin expression and improves insulin sensitivity in older adults. Obesity. 2013;21:1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]