

Abstract
The pathogenesis of joint infections is not well understood. In particular, we do not know why these infections respond poorly to antibiotic treatment. Here we show that methicillin-resistant Staphylococcus aureus, a major cause of joint infections, forms exceptionally strong biofilmlike aggregates in human synovial fluid (SF), to an extent significantly exceeding biofilm formation observed in growth medium or serum. Screening a transposon bank identified bacterial fibronectin- and fibrinogen-binding proteins as important for the formation of macroscopic clumps in SF, suggesting an important role of fibrin-containing clots in the formation of bacterial aggregates during joint infection. Pretreatment of SF with plasmin led to a strongly reduced formation of aggregates and increased susceptibility to antibiotics. These results give important insight into the pathogenesis of staphylococcal joint infection and the mechanisms underlying resistance to treatment. Furthermore, they point toward a potential novel approach for treating joint infections.
Keywords: Staphylococcus aureus, MRSA, antibiotic resistance, joint infection, septic arthritis, biofilm
Joint infection (septic arthritis) is a serious condition that if left untreated can lead to rapid, complete destruction of the joint. It may also spread to other sites in the human body and cause life-threatening secondary site infections. When a prosthetic device is present in the joint, the probability of infection increases, with infection rates for total knee or hip arthroplasty reaching 1%–2% [1, 2].
Staphylococcus aureus is the most common cause of septic arthritis in many countries [2]. Together with coagulase-negative staphylococci, it is also the leading pathogen involved in prosthetic joint infection (PJI) [1]. As with other staphylococcal infections, methicillin-resistant S. aureus (MRSA) joint infections are associated with increased risk of treatment failure and cost [3, 4]. In US hospitals, the community-associated (CA) MRSA strain USA300 has been reported to replace the usual hospital-associated (HA) strains in many types of nosocomial infections [5], including PJI [6]. According to data from a 2005 study in an Atlanta, Georgia, hospital, this strain already has emerged as the predominant pathogen involved in PJI in the United States [6].
The pathogenesis of joint infection is poorly understood. It has been speculated that biofilm formation plays a key role [7], a notion that is predominantly based on the isolation of biofilmlike agglomerations from cases of joint infection [8]. Furthermore, the synovial fluid (SF) present in joint cavities has been attributed a function in inhibiting killing of S. aureus by neutrophils [9], while in a somewhat contradicting manner it has also been described as antimicrobial [10]. Finally, microbiological evaluations of joint infections are often complicated by the frequency of false-negative cultures [11]. All these fairly unexplained observations call for a systematic study to investigate the underlying bacterial phenotypes and mechanisms.
The in vitro investigation of bacterial biofilm formation is limited by the chosen growth medium, representing a highly artificial environment with disputable value for the interpretation of biofilm-associated infections [12]. On the other hand, it is difficult to investigate the contribution of biofilm formation to the pathogenesis of joint infections using animal models, because commonly used laboratory animals, such as mice or rats, are too small for that purpose. Notably, despite the frequent use of an S. aureus septic arthritis model in mice [13], biofilm formation has never been linked to septic arthritis based on results from that model. Furthermore, this model is problematic, because bacteria are injected intravenously into the tail and the observed differential degrees of joint infection may be secondary to previous systemic effects. Therefore, in this study we used an ex vivo approach to systematically investigate bacterial behavior in SF during joint infection. We isolated SF from human individuals to monitor bacterial phenotypes in the environment that the pathogen encounters during infection of the joints. We demonstrate extreme biofilm formation and aggregation of S. aureus in SF and show that aggregation in SF dramatically affects resistance to antibiotics. Elucidation of the molecular basis of the observed aggregation pointed to fibrin- and fibronectin-binding proteins, on which ground we tested plasmin to prevent aggregation. Our findings indicate that plasmin treatment efficiently increases the sensitivity of S. aureus to antibiotics in clinical use for joint infections, suggesting a novel pathway for their treatment.
MATERIALS AND METHODS
Ethics Statement
Human SF and samples were collected with permission of the Thomas Jefferson University Institutional Review Board. A waiver for the requirement of informed written consent was obtained, because the samples were deidentified, obtained during routine procedures, and would normally have been discarded.
Materials
The SF was aspirated from human subjects either in the clinic or drained from the joint during total knee arthroplasty in the operating room. Plasmin (Sigma Aldrich), proteinase K (Promega), DNase (Ambion), and dispersin B were used at final concentrations of 150, 100, 200, and 600 µg/mL, respectively. Wheat germ agglutinin staining for polysaccharide intercellular adhesin (PIA) was performed using Alexafluor 488–labeled wheat germ agglutinin (Life Technologies) at a concentration of 0.1 mg/mL, as described elsewhere [14]. Propidium iodide (Sigma) staining of bacterial cells was performed using a 1% propidium iodide solution in sterile phosphate-buffered saline, followed by a 30-minute incubation at 37°C.
Bacterial Strains
The major part of our study was performed with the widely used strain LAC, a representative of the highly clonal CA-MRSA lineage of pulsed-field type USA300 [15], the main source of skin and septic arthritis infections in the United States [6, 16]. Strain LAC is a skin infection isolate and the background strain for the Nebraska transposon bank [17]. We also used strain Xen36, which is a derivative of strain ATCC49525 [strain Wright, a hospital-associated methicillin-susceptible S. aureus (MSSA) isolate from a bacteremia patient] engineered to express luciferase [18]. Finally, we used a collection of several MRSA and MSSA strains to evaluate the aggregation phenotype in SF: MRSA strains SF8300 (another USA300 CA-MRSA strain), MW2 (ATCC BAA-1707, USA400, CA-MRSA), ST72 (a CA-MRSA strain from Korea), COL (1960s HA-MRSA isolate from the United Kingdom), 252 (ATCC BAA-1720, EMRSA-16, USA100, HA-MRSA from the United Kingdom), Newman (ATCC25904, MSSA), Cowan I (ATCC12598, MSSA, a septic arthritis isolate), Wood 46 (ATCC10832, MSSA). The ica-negative control strain used is a derivative of strain SA113, in which the ica operon was deleted [19].
Bacterial Growth Measurement by Colony-Forming Unit Determination or Quantitative Polymerase Chain Reaction
For measurement of bacterial growth, 100 (for quantitative polymerase chain reaction [qPCR]) or 106 colony-forming units (CFUs; for CFU count determination) were added to 1 mL of tryptic soy broth (TSB), SF, or serum, and incubated at 37°C under agitation. Cultures were serially diluted, plated on TSB agar, and CFUs counted. Alternatively, total genomic DNA was isolated and quantified by qPCR of the hla gene, using a TaqMan Master Mix and forward and reverse primers (forward, AAAAAACTGCTAGTTATTAGAACGAAAGG; reverse, GGCCAGGCTAAACCACTTTTG) and a probe with 6-carboxyfluorescein-tetramethylrhodamine dye label (CCTTCTTCGCTATAAACTCTATATTGACCAGCAAT).
Macroscopic Determination of Bacterial Aggregation
Macroscopic clumping of S. aureus was monitored by UV-fluorescent imaging of ethidium bromide–labeled bacteria (10 µg/mL ethidium bromide; 10 minutes in the dark).
Microscopy
Microscopic evaluation was performed by bright-field light microscopy at 20 minutes and 24 hours after the addition of 108 CFUs/mL to 1 mL of TSB, SF, or serum and subsequent incubation at 37°C under static conditions. Alternatively, bacteria were stained with SYTO9 nucleic acid stain, and green fluorescence was imaged using confocal laser scanning microscopy. For this microscopic examination of biofilms, 24-hour biofilms (inoculated with 300 µL containing 108 CFUs/mL) were grown in 8-well borosilicate glass tissue culture wells. Samples were imaged using a Zeiss LSM510 confocal laser scanning microscope (Carl Zeiss). Section images were generated using Imaris (Bitplan) image software.
Scanning Electron Microscopy
For scanning electron microscopy of bacterial aggregates formed in SF, 24-hour aggregates were gently removed with forceps and placed in a 4% formalin solution for 1 hour. The aggregate was then serially dehydrated with ethanol and dried in a carbon dioxide chamber at 4°C for 48 hours, after which the sample was sputter coated with platinum and imaged with a Tabletop Hitachi scanning electron microscope.
Transposon Library Screen
A transposon mutant library of strain USA300 containing approximately 2000 single-gene mutants (Nebraska transposon bank), courtesy of the Network on Antimicrobial Resistance in Staphylococcus aureus, was screened for macroscopic clump formation, as follows: Each strain was grown in a single well of a 96-well plate in 150 L of TSB for 18 hours. Next, 50 µL of SF was added to each well and incubated at 37°C for 1 hour under agitation. Then 10 µL of ethidium bromide (10 µg/mL) was added, and the plate was incubated in the dark for 30 minutes at 37°C. All wells were then monitored for clump formation with UV fluorescent imaging and bright-field microscopy.
Antibiotic Exposure
The susceptibility of S. aureus to antibiotics was assessed by incubating 107 CFUs/mL of strain USA300 for 6 hours in 100 µg/mL cefazolin or 100 µg/mL vancomycin at 37°C, followed by brief proteinase K treatment, dilution, and plating of the bacteria for CFU counts. Alternatively, a luminescent S. aureus strain (Xen36) was used to monitor luminescence as readout for viability. Xen36 was grown under the same conditions and antibiotic concentrations, after which luminescence levels were detected with a luminometer (Victor; PerkinElmer).
Measurement of Aggregate Size Distribution
Aggregate size distribution was measured with a cell counter (Nexcelom T4). To that end, an overnight culture of USA 300 was centrifuged and resuspended to a concentration of approximately 1010 CFUs/mL in sterile phosphate-buffered saline. Next, 1-mL samples of SF or SF pretreated with plasmin were prewarmed to 37°C, and 5 µL of the bacterial suspension and 20 µL of trypan blue was added. The suspension was then pipetted lightly, and incubated for 20 minutes at 37°C under agitation, after which 20 µL of each sample was added to a dual-chamber cell counter and inserted into the Nexcolom T4 cell counter. Parameters were set as follows: minimum cell diameter, 0.1 µm; maximum cell diameter, 100 µm; roundness, 0; contrast enhancement, 0.5; no declustering. The detectable size range was 3–100 µm.
Statistical Analysis
Statistical evaluation was performed using GraphPad Prism software (version 6.02), with t tests used to compare 2 groups and 1-way analysis of variance to compare >2 groups. Multiple comparisons with 1-way analysis of variance were performed using Dunnett or Tukey posttests.
RESULTS
Effect of SF on S. aureus
To analyze growth of S. aureus in SF, we compared growth patterns of the CA-MRSA strain USA300 in SF with those in serum and TSB. The number of CFUs indeed decreased in SF (Figure 1A), as noted in a study reporting antimicrobial activity of SF [10]. However, with qPCR analysis, considerable growth was observed in SF, comparable to that in serum (Figure 1B), indicating that SF does not have antimicrobial activity.
Figure 1.
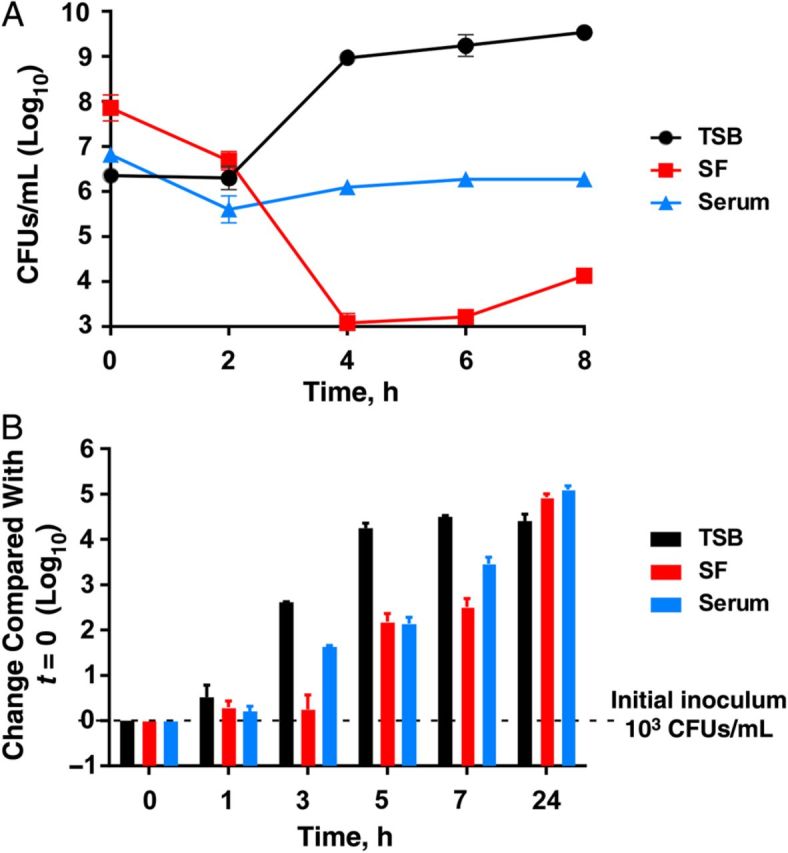
Growth of Staphylococcus aureus in synovial fluid (SF), human serum, and growth medium (tryptic soy broth [TSB]). The methicillin-resistant S. aureus strain USA300 was grown in the indicated fluids and growth was measured according to colony-forming units (CFUs) (A) or quantitative polymerase chain reaction determination (B). The experiment was performed in triplicate, and error bars represent standard deviations.
Formation of S. aureus Aggregates in SF
Differences observed between CFU and qPCR estimation of bacterial growth are usually explained by bacterial aggregation. To determine whether S. aureus forms aggregates in SF, we first used visual and microscopic analysis. When incubated for 1 hour in 24-well tissue culture plates, strain USA300 formed macroscopic clumps in SF (Figure 2A). This formation was more pronounced than in serum, whereas in TSB no clumping was visible. Notably, even SF diluted to 1:10 000 led to aggregation (Figure 2B). The aggregation phenotype was conserved among a collection of MRSA and MSSA strains of different backgrounds, which also contained a septic arthritis isolate (strain Cowan I) (Figure 2C). Interestingly, only one strain, Wood 46, did not show strong aggregation. Because that strain does not adhere to fibrinogen or fibronectin [20, 21], these results are in good accordance with our findings described below, attributing a key contribution of fibrinogen- and fibronectin-binding proteins to aggregation in SF. Light (Figure 2D and 2E) and electron microscopic (Figure 2F) examination verified the extreme clumping behavior of S. aureus USA300 grown in SF.
Figure 2.
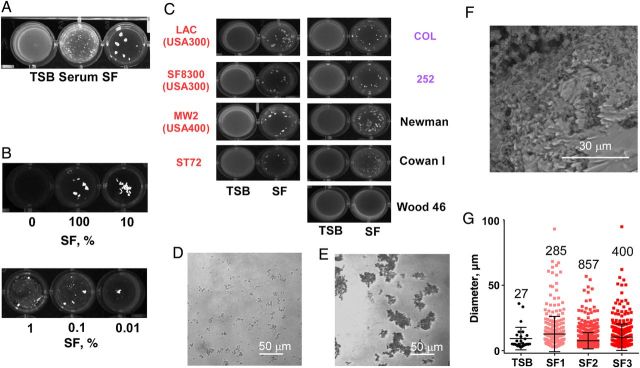
Staphylococcus aureus shows extreme aggregation in synovial fluid (SF). A–E, Washed overnight cultures of S. aureus (A, B, D, E: USA300) were incubated in the indicated fluids for 20 minutes, and aggregation was examined visually (A–C) or with light microscopy (D, E) in tryptic soy broth (TSB) (D) and SF (E). C, Red represents community associated methicillin-resistant S. aureus (MRSA) strains; purple, hospital-associated MRSA strains; black, methicillin-susceptible S. aureus strains. F, Scanning electron microscopy of an USA300 aggregate formed in a 24-hour culture grown in SF. G, Cell counting analysis of aggregates formed after 20-minute incubation in the indicated fluid, with SF obtained from 3 individuals (SF1, SF2, and SF3). Error bars represent standard deviations. Note that macroscopic clumps cannot be analyzed in the cell counter.
To determine S. aureus aggregate formation more exactly, we used a cell counter to analyze aggregates formed in SF obtained from 3 different individuals and TSB. Although the mean aggregate size did not differ significantly between TSB and SF samples, the number of measurable aggregates (ie, those >3 µm in diameter, about the size of a diplococcus), was considerably increased in SF samples (Figure 2G). These results show that SF strongly and rapidly induces S. aureus aggregate formation.
S. aureus Forms Biofilms in SF
During joint infection, aggregation may also occur in a surface-attached, biofilmlike mode on the surface of indwelling medical devices (in PJI) or on the synovial membrane. Growth in SF produced biofilms showing the 3-dimensional structure with towers and channels characteristic of mature biofilms, as shown with confocal laser scanning microscopy. Growth in TSB under the same conditions produced no visible biofilm formation. In serum, biofilm formation was much reduced compared with SF and lacked maturation (Figure 3). These results show that SF promotes both the formation of free-floating biofilmlike aggregates and surface-attached biofilms sensu stricto.
Figure 3.
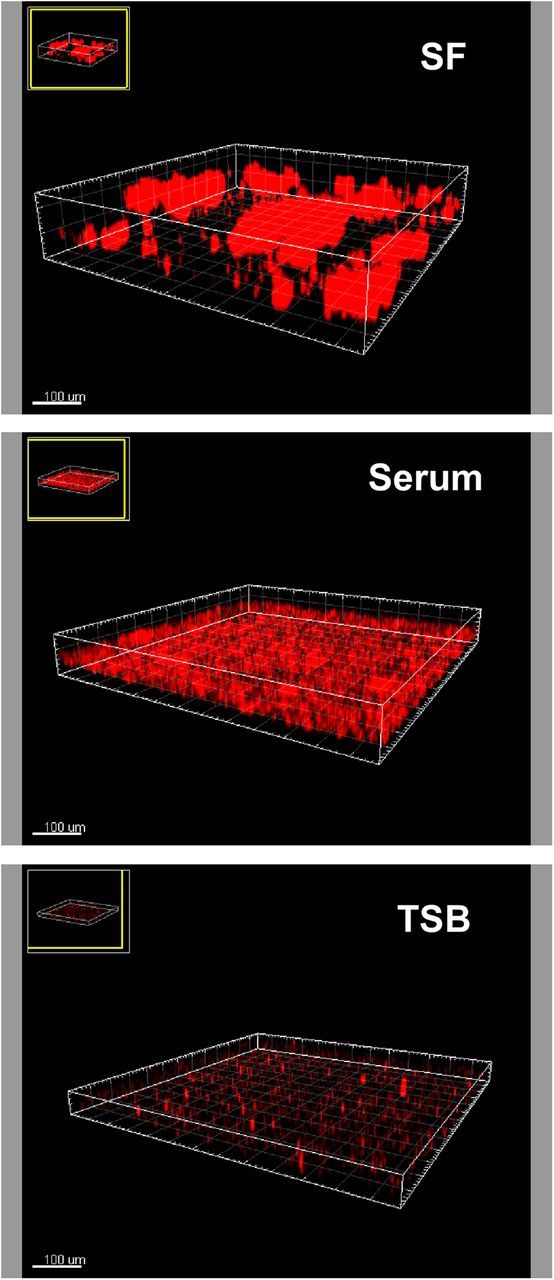
Confocal laser scanning microscopy of biofilms grown in synovial fluid (SF), serum, or growth medium (tryptic soy broth [TSB]). Static 24-hour biofilms were grown in the indicated fluids, stained with propidium iodide, and visualized with confocal laser scanning microscopy. (In the TSB sample, small red clusters represent background staining, not cells.)
Protein-Based SF-Induced Biofilm Formation
The extracellular matrix that defines staphylococcal biofilms is composed of a series of molecules, including most importantly a biofilm exopolysaccharide called polysaccharide intercellular adhesin (PIA), extracellular DNA, and a considerable number of proteins shown to promote intercellular adhesion [22]. The relative importance of these molecules for staphylococcal biofilm formation varies, and can be tested with degradative enzymes. Although wheat germ agglutinin staining of aggregates showed that the matrix contains large amounts of PIA (Figure 4A), S. aureus aggregates persisted after digestion with dispersin B, which degrades PIA [23] (Figure 4B). Similarly, digestion with DNase or proteinase K did not remove macroscopic aggregates entirely (Figure 4B). However, treatment with proteinase K, but not DNase or dispersin B, led to a significant increase in measurable CFUs, indicating partial aggregate disruption (Figure 4C). Together, these results indicate that biofilm formation by the MRSA strain USA300 in SF is multifactorial, with proteins playing the major role.
Figure 4.

Aggregation of USA300 methicillin-resistant Staphylococcus aureus in synovial fluid (SF) is predominantly protein dependent. A, Analysis for presence of polysaccharide intercellular adhesin (PIA) exopolysaccharide in USA300 biofilms using wheat germ agglutinin (WGA) staining. Propidium iodide (PI) staining shows the entire biofilm, and WGA staining shows PIA. In the merge images, PI- and WGA-stained areas appear yellow. Bottom, Biofilms of an ica mutant not expressing PIA do not stain with WGA. B, Macroscopic evaluation of the effect of enzymatic digestion on aggregates of USA300 formed in SF. Dispersin B is an enzyme specifically digesting PIA. C, Estimation of aggregate dispersion based on measurement of increased colony-forming unit (CFU) counts after enzymatic digestion (n = 7). †P < .01; error bars represent standard deviations; Abbreviation: NS, not significant.
Important Role of Surface-Binding Proteins in SF-Induced Biofilms
Because several proteins and other factors have been reported to affect S. aureus biofilm formation [22], it becomes very difficult to pinpoint the molecular basis of aggregation to certain proteins or other molecules by standard methods using directed gene deletion. To circumvent this problem, the Nebraska transposon bank in strain USA300 was created and contains a clone with transposon insertion in every nonessential USA300 gene, providing an excellent means to screen for genes affecting a specific phenotype [17]. In the current study, we used this transposon bank to screen for genes influencing aggregation behavior in SF.
Only 6 clones turned out to have no or almost no visually detectable formation of macroscopic clumps (Figure 5A). Specifically, lack of macroscopic clumps was associated with insertions in the genes coding for fibronectin-binding proteins A and B (FnbA and FnbB), the fibrinogen-binding proteins clumping factor A and B (ClfA and ClfB), and the regulatory proteins RsbU and TRAP. The RsbU protein is part of the SigmaB operon, which has been implicated in the pathogenesis of septic arthritis [24], as have ClfA and ClfB [25, 26]. Extracellular proteolytic activity is strongly increased in rsbU mutants of S. aureus [27], possibly leading to degradation of the FnbA-, FnbB-, ClfA-, and ClfB-binding proteins and thus aggregation. Together, these results indicate an important role of fibrinogen- and fibronectin-binding proteins in the aggregation of S. aureus in SF and strongly point to an involvement of bacterial attachment to fibrin during aggregation in SF. Of note, microscopic examination, and cellometry showed that microscopic aggregates were still present in all mutants, underlining the functional redundancy of the identified factors in the aggregation phenotype (Figure 5A and 5B).
Figure 5.

Factors affecting macroscopic aggregation of USA300 methicillin-resistant Staphylococcus aureus identified by screening of a transposon mutagenesis bank. The Nebraska USA300 transposon mutagenesis bank was screened for absence of aggregation after incubation in synovial fluid. Six clones showed no or strongly reduced aggregation (those with insertions in the fnbA, fnbB, clfA, clfB, rsbU, and trap genes). A, Macroscopic (left) and microscopic (right) evaluation of aggregation in USA300 wild-type and mutant strains. B, Analysis of remaining aggregates by cell counter.
Effect of Plasmin Pretreatment on Susceptibility of S. aureus in SF to Antibiotics
Because our findings suggested that attachment to fibrin plays a role in bacterial aggregation in SF, we next tested whether plasmin treatment would destroy aggregates. Plasmin is a serine protease that destroys fibrin clots by fibrinolysis. Treatment with plasmin had no considerable effect when applied to preformed aggregates, but pretreatment with plasmin was very efficient in preventing aggregation (Figure 6A) and significantly decreased bacterial aggregate sizes (Figure 6B). These results confirm the data from our transposon screen indicating that fibrin plays a key role in bacterial aggregate formation in SF.
Figure 6.

Plasmin treatment strongly increases susceptibility of Staphylococcus aureus aggregates in synovial fluid (SF) to antibiotics. A, Treatment of aggregates with plasmin before and after aggregate formation, macroscopic evaluation. Plasmin pretreatment was performed by incubating solutions before inoculation for 12 hours with 150 µg/mL plasmin; posttreatment was performed after 20-minute clump formation using the same concentration. B, Cellometry analysis. ‡P < .001; error bars represent standard deviations. C, Antibiotic susceptibility was assessed by incubating the methicillin-resistant S. aureus strain USA300 for 6 hours in with cefazolin or vancomycin at 37°C, followed by brief proteinase K treatment and colony-forming unit (CFU) determination. CEF, cefazolin; TSB, tryptic soy broth. D, Alternatively, a luminescent S. aureus strain (Xen36) was used to monitor luminescence as readout under the same conditions. Plasmin pretreatment was performed by incubating solutions before inoculation for 12 hours with 150 µg/mL plasmin. C, D, Assays were performed in triplicate. *P < .05; †P < .01; ‡P < .001; error bars represent standard deviations. Abbreviation: VAN, vancomycin.
To test whether plasmin pretreatment of SF renders S. aureus more susceptible to antibiotic treatment, we measured S. aureus sensitivity to cefazolin, the preferred antibiotic given to patients with joint infections and those undergoing orthopedic surgery [28–30] (Figure 6C). Notably, cefazolin is commonly used at doses that cause serum concentrations to exceed the minimum inhibitory concentration of even many MRSA isolates [31], particularly USA300, a strain with a relatively high sensitivity to β-lactam antibiotics [32]. Therefore, and because USA300 is a major cause of septic arthritis in the United States [6], we also used cefazolin in the antibiotic treatment experiments. In a control experiment, cefazolin completely eradicated bacteria grown in TSB. Cefazolin alone did not significantly reduce CFU counts (after disruption of aggregates) in SF, but pretreatment with plasmin followed by treatment with cefazolin reduced CFU counts by almost 5 logs (approximately 87 000-fold). This was a significant reduction compared with cefazolin treatment alone (P < .01), increasing antibiotic efficacy >2000-fold. In serum, cefazolin caused significant killing of bacteria (P < .01); pretreatment with plasmin did not significantly alter the magnitude of this effect. These findings indicate that plasmin pretreatment significantly increases cefazolin activity against bacteria in SF and again underline the exceptional situation regarding biofilms and antibiotic resistance in SF compared with other body fluids.
To further substantiate these results, we measured bacterial viability using a luminescent MSSA strain, Xen36. Luminescence is dependent on the presence of adenosine triphosphate and thus viability [18]. Notably, in this method bacterial counts are not affected by aggregation behavior. In accordance with the CFU counts, pretreatment of SF with plasmin significantly increased bacterial killing by cefazolin (approximately 6-fold; P < .001) (Figure 6D). Similar results were obtained with vancomycin (approximately 28-fold; P < .001), underlining the general resistance of S. aureus in SF to antibiotics used clinically to treat staphylococcal joint infections and establishing that plasmin treatment before aggregate formation strongly increases antibiotic efficacy.
DISCUSSION
Biofilm formation has been suspected to play a key role during septic arthritis and PJI [2, 7], but to our knowledge this important bacterial virulence determinant has never been directly investigated for its contribution to the pathogenesis of joint infections. Here we show that the synovial environment leads to very strong aggregation of S. aureus, the major pathogen involved with such infections. Our study focused on MRSA, owing to the difficulties that methicillin resistance causes during therapy and the predominance that the MRSA strain USA300 in particular has achieved among PJI in the United States [6]. However, our results show that induction of the aggregation phenotype in SF is conserved among both MRSA and MSSA strains. Only Wood 46, a strain lacking adhesion to fibrinogen and fibronectin, did not show extensive aggregation, which is in accordance with our findings attributing fibrinogen- and fibronectin-binding proteins a key role in the aggregation phenotype. Notably, according to our findings, the conditions that the bacteria encounter during joint infection thus promote the formation of aggregates and biofilms to an extraordinary extent. Furthermore, our results indicate that host-derived fibrin is a key component of these exceptionally large bacterial aggregates. Altogether, our study shows that septic arthritis due to S. aureus is a genuine biofilm-associated infection with exceptionally pronounced protein-dependent aggregate formation (Figure 7).
Figure 7.

Model of Staphylococcus aureus biofilm formation in joint infection. S. aureus biofilms may form in a surface-attached form on the synovial membrane or on indwelling medical devices during prosthetic joint infection or exist as free-floating aggregates in synovial fluid (SF) within the joint cavity. Aggregate formation is dependent on host-derived fibrin, as indicated by preventing aggregate formation by plasmin treatment of SF, and bacterial fibrinogen- and fibronectin-binding proteins (ClfA, ClfB, FnbA, and FnbB). Plasmin treatment increases susceptibility of antibiotics and probably increases efficiency of host defenses by decreasing aggregate sizes for more efficient uptake by phagocytes. Abbreviations: eDNA, extracellular DNA; PIA, polysaccharide intercellular adhesin.
Our findings explain a series of previous observations regarding the pathogenesis and diagnosis of joint infections. First, they explain that cultures of SF will often turn out culture negative despite the assumed presence of an infection [11], because the macroscopic bacterial aggregates may not have been contained in the aspirate. Second, the formation of aggregates in a very short time with sizes significantly exceeding neutrophil dimensions (approximately 12–15 µm) explains why SF has been reported to inhibit killing of S. aureus by neutrophils [9], which cannot ingest such large aggregates. Of note, such a scenario can lead to “frustrated phagocytosis,” in which neutrophils eject lysosomal components, further contributing to inflammation and disease [33]. Third, our results identify biofilm formation as a key mechanism inhibiting the activity of antibiotics in clinical use for staphylococcal joint infection. By inhibiting neutrophil function and antibiotic treatment, biofilm formation may thus contribute to the recalcitrance of staphylococcal joint infections by interfering with both host defenses and therapeutic intervention.
Importantly, our study findings suggest that targeting staphylococcal biofilm formation during joint infection is a promising means for combination therapy together with commonly used antibiotics such as cefazolin. Although we found that already-formed biofilms are very resistant even to harsh enzymatic digestion, proteolytic attack of staphylococcal and/or host proteins that are involved in aggregate formation may prove useful as prophylaxis or in prolonged therapy to prevent further aggregation. We chose to investigate plasmin, because it specifically targets fibrin and because proteases with broader specificity may be too harmful to the host environment. Plasmin normally completely digests fibrin clots [34], but its activity during unresolved staphylococcal joint infection is probably too low to disaggregate the fibrin-containing bacterial biofilms. In our experiments, application of plasmin strongly increased antibiotic efficiency, indicating that it may have value as an additive enhancing antibiotic therapy during joint infection. Notably, plasmin has the advantage of being a human protease, for which there are specific inactivating mechanisms that would prevent harmful side effects, such as degradation of cartilage protein [35, 36]. Although plasmin may contribute to inflammation during joint infection [37], findings obtained in a plasminogen-deficient mouse model of S. aureus–induced septic arthritis indicate a beneficial role of plasmin in that scenario [38], which is in accordance with our results and strengthens our argument for a potential therapeutic use of plasmin.
In conclusion, we here provide evidence indicating that staphylococcal joint infection proceeds with pronounced aggregate and biofilm formation. These findings explain the recalcitrance of joint infections to therapeutic intervention and clearing by host defenses and point to possible new therapeutic strategies based on the enzymatic digestion of biofilms in joint infections.
Notes
Acknowledgments. We thank S. Leppla, PhD, National Institute of Allergy and Infectious Diseases, for permission to use the Nexcolom Cellometer.
Financial support. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH) (ZIA AI000904-13, to M. O.), the National Institute of Child Health and Human Development (grant HD06153 to N. J. H.), the National Institute of Dental and Craniofacial Research (grant DE019901 to N. J. H.), and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant T32 AR 052273 to I. M. S.).
Potential conflicts of interest. J. P. is a board member of 3M and a consultant for Zimmer, Smith & Nephew, ConvaTec, TissueGene, CeramTec, and Medtronic; holds stocks with Hip Innovation Technologies, CD Diagnostics, and PRN; and received research support from the NIH, the Orthopaedic Research and Education Foundation, Stryker Orthopedics, Depuy, Zimmer, Baxter, 3M, Biomemetics, Ceramtec, and Smith & Nephew. N. J. H. received funding from the NIH and Synergy Medical, and her husband holds stocks with Pfizer Pharmaceuticals. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Zimmerli W, Trampuz A, Ochsner PE. Prosthetic-joint infections. N Engl J Med. 2004;351:1645–54. doi: 10.1056/NEJMra040181. [DOI] [PubMed] [Google Scholar]
- 2.Shirtliff ME, Mader JT. Acute septic arthritis. Clin Microbiol Rev. 2002;15:527–44. doi: 10.1128/CMR.15.4.527-544.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lora-Tamayo J, Murillo O, Iribarren JA, et al. A large multicenter study of methicillin-susceptible and methicillin-resistant Staphylococcus aureus prosthetic joint infections managed with implant retention. Clin Infect Dis. 2013;56:182–94. doi: 10.1093/cid/cis746. [DOI] [PubMed] [Google Scholar]
- 4.Salgado CD, Dash S, Cantey JR, Marculescu CE. Higher risk of failure of methicillin-resistant Staphylococcus aureus prosthetic joint infections. Clin Orthop Relat Res. 2007;461:48–53. doi: 10.1097/BLO.0b013e3181123d4e. [DOI] [PubMed] [Google Scholar]
- 5.D'Agata EM, Webb GF, Horn MA, Moellering RC, Jr, Ruan S. Modeling the invasion of community-acquired methicillin-resistant Staphylococcus aureus into hospitals. Clin Infect Dis. 2009;48:274–84. doi: 10.1086/595844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kourbatova EV, Halvosa JS, King MD, Ray SM, White N, Blumberg HM. Emergence of community-associated methicillin-resistant Staphylococcus aureus USA 300 clone as a cause of health care-associated infections among patients with prosthetic joint infections. Am J Infect Control. 2005;33:385–91. doi: 10.1016/j.ajic.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Arnold WV, Shirtliff ME, Stoodley P. Bacterial biofilms and periprosthetic infections. Instr Course Lect. 2014;63:385–91. [PubMed] [Google Scholar]
- 8.Stoodley P, Nistico L, Johnson S, et al. Direct demonstration of viable Staphylococcus aureus biofilms in an infected total joint arthroplasty: a case report. J Bone Joint Surg Am. 2008;90:1751–8. doi: 10.2106/JBJS.G.00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon GL, Miller HG, Borenstein DG. Synovial fluid inhibits killing of Staphylococcus aureus by neutrophils. Infect Immun. 1983;40:1004–10. doi: 10.1128/iai.40.3.1004-1010.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruber BF, Miller BS, Onnen J, Welling RD, Wojtys EM. Antibacterial properties of synovial fluid in the knee. J Knee Surg. 2008;21:180–5. doi: 10.1055/s-0030-1247816. [DOI] [PubMed] [Google Scholar]
- 11.Swan A, Amer H, Dieppe P. The value of synovial fluid assays in the diagnosis of joint disease: a literature survey. Ann Rheum Dis. 2002;61:493–8. doi: 10.1136/ard.61.6.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joo HS, Otto M. Molecular basis of in vivo biofilm formation by bacterial pathogens. Chem Biol. 2012;19:1503–13. doi: 10.1016/j.chembiol.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bremell T, Lange S, Yacoub A, Ryden C, Tarkowski A. Experimental Staphylococcus aureus arthritis in mice. Infect Immun. 1991;59:2615–23. doi: 10.1128/iai.59.8.2615-2623.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jefferson KK, Cerca N. Bacterial-bacterial cell interactions in biofilms: detection of polysaccharide intercellular adhesins by blotting and confocal microscopy. Methods Mol Biol. 2006;341:119–26. doi: 10.1385/1-59745-113-4:119. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy AD, Otto M, Braughton KR, et al. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc Natl Acad Sci U S A. 2008;105:1327–32. doi: 10.1073/pnas.0710217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–68. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fey PD, Endres JL, Yajjala VK, et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 2013;4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francis KP, Joh D, Bellinger-Kawahara C, Hawkinson MJ, Purchio TF, Contag PR. Monitoring bioluminescent Staphylococcus aureus infections in living mice using a novel luxABCDE construct. Infect Immun. 2000;68:3594–600. doi: 10.1128/iai.68.6.3594-3600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun. 1999;67:5427–33. doi: 10.1128/iai.67.10.5427-5433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maxe I, Ryden C, Wadstrom T, Rubin K. Specific attachment of Staphylococcus aureus to immobilized fibronectin. Infect Immun. 1986;54:695–704. doi: 10.1128/iai.54.3.695-704.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yonemasu K, Sasaki T, Ohmae R, Kashiba S. Expression of clumping and fibrinogen-binding activities of Staphylococcus aureus at various growth stages. Microbiol Immunol. 1991;35:405–9. doi: 10.1111/j.1348-0421.1991.tb01571.x. [DOI] [PubMed] [Google Scholar]
- 22.Otto M. Staphylococcal biofilms. Curr Top Microbiol Immunol. 2008;322:207–28. doi: 10.1007/978-3-540-75418-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan JB, Velliyagounder K, Ragunath C, et al. Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J Bacteriol. 2004;186:8213–20. doi: 10.1128/JB.186.24.8213-8220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonsson IM, Arvidson S, Foster S, Tarkowski A. Sigma factor B and RsbU are required for virulence in Staphylococcus aureus-induced arthritis and sepsis. Infect Immun. 2004;72:6106–11. doi: 10.1128/IAI.72.10.6106-6111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Josefsson E, Hartford O, O'Brien L, Patti JM, Foster T. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J Infect Dis. 2001;184:1572–80. doi: 10.1086/324430. [DOI] [PubMed] [Google Scholar]
- 26.Palmqvist N, Foster T, Fitzgerald JR, Josefsson E, Tarkowski A. Fibronectin-binding proteins and fibrinogen-binding clumping factors play distinct roles in staphylococcal arthritis and systemic inflammation. J Infect Dis. 2005;191:791–8. doi: 10.1086/427663. [DOI] [PubMed] [Google Scholar]
- 27.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. sigmaB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325–4. J Bacteriol. 2002;184:5457–67. doi: 10.1128/JB.184.19.5457-5467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeller V, Durand F, Kitzis MD, et al. Continuous cefazolin infusion to treat bone and joint infections: clinical efficacy, feasibility, safety, and serum and bone concentrations. Antimicrob Agents Chemother. 2009;53:883–7. doi: 10.1128/AAC.00389-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marschall J, Lane MA, Beekmann SE, Polgreen PM, Babcock HM. Current management of prosthetic joint infections in adults: results of an Emerging Infections Network survey. Int J Antimicrob Agents. 2013;41:272–7. doi: 10.1016/j.ijantimicag.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dellinger EP, Gross PA, Barrett TL, et al. Quality standard for antimicrobial prophylaxis in surgical procedures. The Infectious Diseases Society of America. Infect Control Hosp Epidemiol. 1994;15:182–8. doi: 10.1086/646887. [DOI] [PubMed] [Google Scholar]
- 31.Yamada K, Matsumoto K, Tokimura F, Okazaki H, Tanaka S. Are bone and serum cefazolin concentrations adequate for antimicrobial prophylaxis? Clin Orthop Relat Res. 2011;469:3486–94. doi: 10.1007/s11999-011-2111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerjee R, Fernandez MG, Enthaler N, Graml C, Greenwood-Quaintance KE, Patel R. Combinations of cefoxitin plus other beta-lactams are synergistic in vitro against community associated methicillin-resistant Staphylococcus aureus. Eur J Clin Microbiol Infect Dis. 2013;32:827–33. doi: 10.1007/s10096-013-1817-9. [DOI] [PubMed] [Google Scholar]
- 33.Barrett AJ. The possible role of neutrophil proteinases in damage to articular cartilage. Agents Actions. 1978;8:11–8. doi: 10.1007/BF01972396. [DOI] [PubMed] [Google Scholar]
- 34.Lack CH. Chondrolysis in arthritis. J Bone Joint Surg Br. 1959;41-B:384–7. doi: 10.1302/0301-620X.41B2.384. [DOI] [PubMed] [Google Scholar]
- 35.Lijnen HR. Pleiotropic functions of plasminogen activator inhibitor-1. J Thromb Haemost. 2005;3:35–45. doi: 10.1111/j.1538-7836.2004.00827.x. [DOI] [PubMed] [Google Scholar]
- 36.Lack CH, Rogers HJ. Action of plasmin on cartilage. Nature. 1958;182:948–9. doi: 10.1038/182948a0. [DOI] [PubMed] [Google Scholar]
- 37.Hsieh YS, Yang SF, Lue KH, Lu KH. Clinical correlation with the PA/plasmin system in septic arthritis of the knee. Clin Orthop Relat Res. 2006;447:172–8. doi: 10.1097/01.blo.0000203473.96549.4e. [DOI] [PubMed] [Google Scholar]
- 38.Guo Y, Li J, Hagstrom E, Ny T. Protective effects of plasmin(ogen) in a mouse model of Staphylococcus aureus-induced arthritis. Arthritis Rheum. 2008;58:764–72. doi: 10.1002/art.23263. [DOI] [PubMed] [Google Scholar]