

Abstract
Organization of existing and emerging toxicological knowledge into adverse outcome pathway (AOP) descriptions can facilitate greater application of mechanistic data, including those derived through high-throughput in vitro, high content omics and imaging, and biomarker approaches, in risk-based decision making. The previously ad hoc process of AOP development is being formalized through development of internationally harmonized guidance and principles. The goal of this article was to outline the information content desired for formal AOP description and some rules of thumb and best practices intended to facilitate reuse and connectivity of elements of an AOP description in a knowledgebase and network context. For example, key events (KEs) are measurements of change in biological state that are indicative of progression of a perturbation toward a specified adverse outcome. Best practices for KE description suggest that each KE should be defined as an independent measurement made at a particular level of biological organization. The concept of “functional equivalence” can help guide both decisions about how many KEs to include in an AOP and the specificity with which they are defined. Likewise, in describing both KEs and evidence that supports a causal linkage or statistical association between them (ie, a key event relationship; KER), best practice is to build from and contribute to existing KE or KER descriptions in the AOP knowledgebase rather than creating redundant descriptions. The best practices proposed address many of the challenges and uncertainties related to AOP development and help promote a consistent and reliable, yet flexible approach.
Keywords: adverse outcome pathway, regulatory toxicology, predictive toxicology, extrapolation, knowledgebase
Interest in so-called twenty-first century approaches to regulatory toxicology that focus on measures of the initial induction or early progression of a biological perturbation known to lead to toxicity, rather than direct measurement of apical outcomes (Krewski et al., 2010), has spurred interest in describing adverse outcome pathways (AOPs). An AOP is a conceptual framework for organizing and describing the biologically plausible and scientifically supported linkage between a molecular initiating event (MIE) that triggers a biological perturbation and occurrence of an adverse outcome (AO) with accepted relevance to regulatory decision making (Ankley et al., 2010). A companion paper (Villeneuve et al. 2014) described a series of core principles that underlie AOP development. These principles focus on AOPs as generalizable, as opposed to chemical specific, motifs of biological response. AOPs are described as a series of measurable changes in biological state (key events; KEs) linked by key event relationships (KERs) that summarize a scientific basis from which to infer a potential change in a “downstream” KE, based on the measured or predicted state of an “upstream” KE. KEs and KERs, each described independently, can be linked in a (generally) non-branching sequence to define an individual AOP, which represents the most basic unit for inferring a potential AO from an MIE and evaluating the relative level of certainty with which inference along that sequence can be made. AOPs can also be linked in a broader network context that characterizes convergence and divergence of KEs among AOPs as a way to systematically consider potential interactions, cumulative impacts of multiple perturbations, and or similarities and differences in response as a function of life stage, sex, taxa, or the site of perturbation. The measurements and scientific support that underlie AOPs can be expected to evolve over time. Therefore, KE and KER descriptions, individual AOPs, AOP networks, and the AOP knowledgebase (AOP-KB) as a whole, can be expected to evolve over time, ideally toward greater levels of predictive certainty and suitability for a broader range of regulatory applications.
In the present article, we consider the ways in which the core principles of AOP development translate into practice. Two illustrative examples of AOP descriptions developed using the AOP-Wiki (www.aopwiki.org, Accessed October 10, 2014) module of the AOP-KB are provided for reference (Supplementary Documents 1 and 2). The information content desired for defining each of the constituents of an AOP is described. Additionally, a set of best practices and “rules of thumb” that address several of the recurring challenges or uncertainties that AOP developers face are provided. Adoption of these practices can help promote a consistent and reliable, yet flexible, approach to AOP development.
CONVENTIONS OF AOP DESCRIPTION
As the ad hoc process of AOP description has been formalized, a number of conventions have developed. Perhaps the most fundamental of these is that individual AOPs are described as a unique, non-branching sequence of KEs generally linking a single MIE to an AO (e.g., Fig. 1). This convention is based on the principle that an individual AOP is a pragmatic unit for the purposes of both development and evaluation (Villeneuve et al. part I). In general, there should be one MIE per AOP. However, a given MIE may link to more than one unique sequence of KEs leading to either the same AO, or a range of AOs (e.g., Fig. 1, MIE 3). Thus, MIEs can be shared by more than one AOP. Likewise, there should be at least one AO per AOP, but individual AOs can link to multiple AOPs ((e.g., Fig. 1, AO1, AO5). More than one AO may be included in an AOP description as long as those AOs still represent an unbranched sequence (e.g., Fig. 1, AO1–AO5; AO2–AO5; AO3–AO6). Any number of KEs, and thus KERs, can be used to link the MIE to the terminal AO, and consistent with the principle of modularity (Villeneuve et al. 2014), individual KEs and KERs can be shared by multiple AOPs (e.g., Fig. 1; KE1, KE2, KE3 shared by AOP1 and AOP2; KE5 and KE6 shared by AOP3 and AOP4). AOPs sharing one or more KEs or KERs can be represented as an AOP network (Fig. 2). This general set of conventions serves as a useful guide for most AOP description. The following sections on best practices describe additional nuances, exceptions, and rules of thumb, as they pertain to each of the main elements of an AOP description.
FIG. 1.
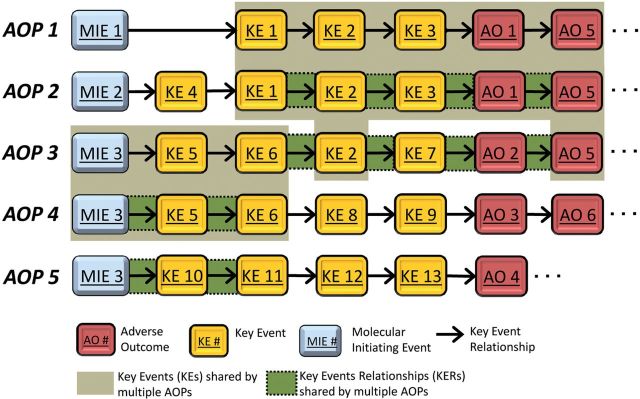
Graphical illustration of the conventions of adverse outcome pathway (AOP) description. Color image is available in the online version of the article.
FIG. 2.
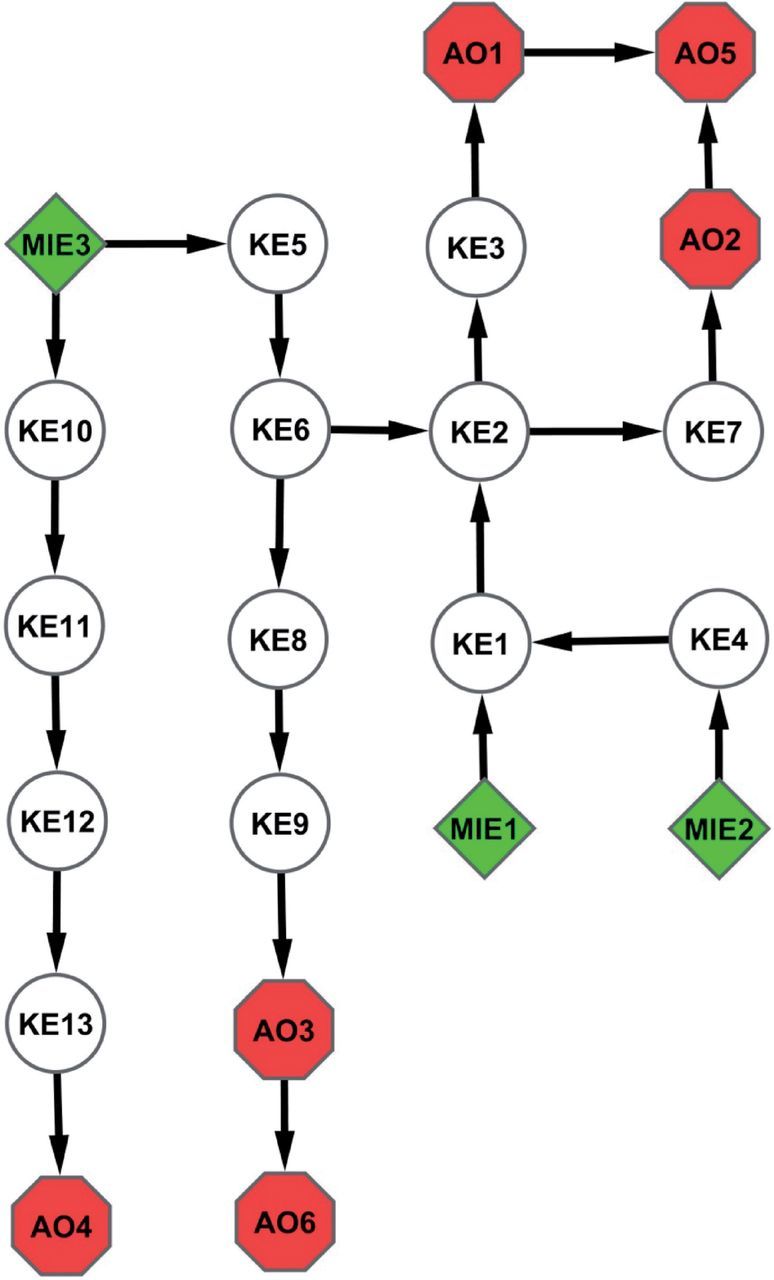
AOP network representation of the five AOPs shown in Figure 1. MIE, molecular initiating event; KE, key event; AO, adverse outcome. Arrows = key event relationships. Color image is available in the online version of the article.
BEST PRACTICES FOR DESCRIBING KEs
KEs are one of the two fundamental modular components that make up an AOP description (see Villeneuve et al. 2014). Each KE represents a measurable change in biological state, which is essential, but not necessarily sufficient for progression from the MIE to an AO (Boobis et al., 2008; OECD, 2013). The information desired as part of a KE description includes the following: (1) a description of the biological state being observed, including definition of the biological context in which it is measured and its general role in the biology; (2) description of the types of methods that can be used to observe or measure the KE; and (3) an indication of the taxonomic applicability of that KE, in terms of taxa in which the KE could or could not be measured.
There are a number of common challenges that AOP developers can encounter in defining the KEs to include in an AOP. One of those is deciding how many KEs to include. Strictly speaking, there is no minimum or maximum number of KEs in an AOP. If the weight of correlative evidence supporting the KER were sufficiently strong one could, conceivably, link from a measure of the MIE directly to the AO, resulting in an AOP with a single KER. However, in terms of best practices, it is preferable to identify at least one KE at each level of biological organization (molecular, cellular, tissue, organ, organ system, and individual).
Each KE included in an AOP should be essential in the sense that if the event does not occur there will be no progression to the further downstream events represented in the AOP. Likewise, if the KE does occur, it implies potential for the perturbation to lead to further biological effect(s) (ie, the next KE in the chain). This convention of including only those KEs deemed essential to the development of the AO has its history in the Mode of Action framework developed for assessing the relevance of animal studies for human health risk assessment by applying Bradford Hill considerations for evaluating causality (Boobis et al., 2006, 2008; Meek et al., 2014). In practice, it provides an effective way to focus AOP description on the biological responses that are best understood and most meaningfully linked to outcomes.
An example of how consideration of essentiality comes into play when developing an AOP is provided in supplementary Document 1. In certain fish species, phenotypic masculinization of females is an observable response considered diagnostic of exposure to androgen agonists (Ankley et al., 2003; Ankley and Gray, 2013; LaLone et al., 2013a; Sone et al., 2005). However, there is not adequate evidence to establish that phenotypic masculinization is part of the causal chain that leads to impaired spawning and reduced cumulative fecundity in females (Supplementary Document 1; Fig. 3A). In the context of an AOP linking androgen receptor agonism to reproductive dysfunction, phenotypic masculinization in females would not be regarded as a KE if it cannot be functionally linked to a downstream KE or AO, even though it has recognized diagnostic value. Instead, the endpoint of phenotypic masculinization was included as a potential indirect biomarker of the MIE (ie, androgen receptor agonism). If, through subsequent work, one was able to demonstrate that such masculinization caused significant impairment in the ability to attract a mate, and therefore ability to reproduce, the endpoint may be a KE in a different AOP. The question of essentiality is one of the critical considerations when trying to decide which measurements/endpoints should be included as KEs in a particular AOP, which to exclude, and which may best be represented as indirect measurements or biomarkers of an essential event.
FIG. 3.

AOP diagrams for example AOPs described in Supplementary Documents 1 (A) and 2 (B) and AOP network representation of the two AOPs (C) GtH, gonadotropins; T, testosterone; E2, 17β-estradiol; VTG, vitellogenin.
An additional consideration concerning whether to include or exclude potential KEs from the AOP description is the relative difficulty of gathering enough evidence to support the KERs that link the KEs. If one of the KEs proposed involves a highly technical and difficult measurement that is only rarely made, such that the KER would typically rely almost exclusively on plausibility, as opposed to empirical evidence, there may be little value in describing it as an independent KE. Instead, the event could be collapsed into the description of biological plausibility of the linkage between two more readily measureable KEs. The exception to this rule of thumb would be when this would lead to the need for a KE that spanned levels of biological organization.
Another common question, which ties closely with the question of how many KEs to include, is how specifically or broadly the KEs should be defined. For example, one could represent the sequence of KEs in an AOP (Figs. 3A and 3B; Supplementary Documents 1 and 2), as either, reduced plasma estradiol—reduced plasma vitellogenin (2 KEs); or as reduced plasma estradiol—reduced ER activation in hepatocytes—reduced hepatic vitellogenin transcription—reduced hepatic vitellogenin protein abundance—reduced plasma vitellogenin (5 KEs). Although the ultimate decision remains at the discretion of the developer, there are several useful rules of thumb. First, KEs should not bridge levels of biological organization. The convention is that KERs are used to span levels of organization whereas individual KEs should not. Second, the description should be sufficiently detailed that the reader knows what to measure. For example, a KE description that simply indicated “gene expression changes” or “altered hormone titers” lacks sufficient detail.
A third rule of thumb is that although a KE description includes defining the biological context in which the measurement is made (ie, in relation to cell, tissue, organ, sex, species, or life stage), it is recommended to keep KEs as broadly applicable as possible while including the necessary level of detail in the KE description. This recommendation follows the principle of the modularity of AOPs and the associated reusability of KEs and KERs (Villeneuve et al. 2014). The guiding principle here in terms of how narrowly to restrict the biological context of a KE is consideration of “functional equivalence.” If the function of the biological event in question is the same regardless of cell type, there is no need to make the KE cell type specific; if the function is the same in either sex, there is no need to make the KE sex specific; if the function is the same regardless of life stage, there is no need to make the KE life-stage specific. It is only at the point that functional equivalence is not maintained that the biological context of the KE should be restricted. Application of this concept of functional equivalence aids the generation of KEs and KE descriptions that can be shared by multiple AOPs.
A final best practices consideration relative to KE description pertains specifically to the development of AOPs within the AOP-KB (eg, using the AOP-Wiki; www.aopwiki.org, Accessed October 10, 2014). To facilitate the collective approach to AOP network development, it is fundamental that the same event representing a KE in more than one AOP is not recreated over and over in the AOP-KB but simply reused by different AOP developers. For example, the two AOPs depicted in Fig. 3 have 6 KEs in common. The same KE descriptions were linked to the respective AOP pages within the AOP-KB, allowing those KEs to be used for both example AOPs (Supplementary Documents 1 and 2) as well as others. Consequently, when linking a KE (including MIEs or AOs) to an AOP under development, the first step should always be to search the AOP-KB to identify whether any of the KEs have already been defined and described. If a KE description is present, but is not suitable for the new AOP under development (eg, perhaps the biological context is too narrowly defined or a different nomenclature is used for a functionally equivalent protein), the developer should consider making contact with the developer(s) of the existing KE to discuss whether it can be modified to accommodate the new AOP without disrupting its original use(s). In general, new KEs should only be created after fully exploring the possibilities to reuse existing ones.
Although these practices and rules of thumb can often be applied in a relatively straightforward manner in cases in which the sequence of KEs is fairly specific (eg, Fig. 3; Supplementary Documents 1 and 2), defining and describing appropriate reusable KEs can be particularly challenging when relatively non-specific toxicological mechanisms are involved in the production of the AO. An example of a nonspecific mechanism is membrane disruption through narcosis, which is commonly accepted to result from accumulation of lipophilic chemicals in membranes disrupting membrane integrity and function (Schultz, 1989). Inflammatory responses, which can occur in a wide range of tissues, are another common biological response to many types of chemical stress. A third example is oxidative stress, which can be defined as a disruption of the cell redox potential and an imbalance between the production and scavenging of reactive oxygen species which damage components of the cell including proteins, lipids, and DNA (Regoli and Giulliani, 2014). Membrane disruption, inflammation, and oxidative stress are quite general in that they can be produced by a wide variety of processes and often occur in addition to a more specific mechanism, with which they can also interact. For example, many pesticides have specific neurotoxic mechanisms of toxicity, but depending on their lipophilicity can additionally disrupt membranes through narcosis; this can be considered as two distinct AOPs. Another example is polycyclic aromatic hydrocarbons causing both narcosis and oxidative stress. Additionally, oxidative stress can be caused in several ways, eg, inhibition of specific ROS scavenging enzymes or a hyperactivity of mitochondrial oxidative phosphorylation. Both oxidative stress and narcosis can lead to cell death because the general function of the cell becomes impaired, and both are considered very important aspects of toxicity. Depending on the cell/tissue type in which they occur, oxidative stress, inflammation, and/or narcosis can be involved in many different types of downstream events, eg, impaired liver or kidney function and impaired cardiovascular function.
A significant challenge in describing AOPs involving such general KEs involves conferring adequate specificity to define progression to a particular toxicological outcome, while at the same time allowing for the generalizability and reuse of a common KE. In a knowledgebase context, it is not optimal to have separate KEs describing oxidative stress in hepatocytes, oxidative stress in peripheral neurons, oxidative stress in lung, oxidative stress in nasal epithelium, etc., if the overall biological phenomenon and the ways one would measure it are largely the same. In such cases, it is important to recognize that specificity for a particular outcome can be conferred through the other KEs that these more general KEs link to. For example, in cases where oxidative stress is linked to impaired liver function, the biological context in which the oxidative stress occurs becomes implicit for the AOP. Likewise, rather than being regarded as an outcome unto itself, narcosis may be represented as a KE describing cellular membrane disruption that is linked to a large number of AOPs, within an AOP network, with differing subsequent KEs depending on the cell, tissue, organ context in which the chemically induced membrane disruption occurred. When applied in this way, these nonspecific KEs can become important building blocks of AOPs and AOP networks.
Best Practices for Describing MIEs
MIEs are regarded as a specialized type of KE. Like other KEs, MIEs represent a measureable change in biological state that is essential, although not necessarily sufficient, to trigger the AO it is connected to via an AOP. However, unlike other KEs, the change in biological state (perturbation) at the MIE is the result of a direct interaction between a chemical and some biomolecule in an organism. The target biomolecule may be specific, as in the case of an enzyme whose inhibition defines the MIE. Alternatively it may represent a group of biomolecules subject to the same reactivity or interaction and leading to a common biological perturbation. For example, in the case of the AOP for skin sensitization, the MIE involves formation of a hapten-protein conjugate (Karlberg et al., 2008). The extent and propensity of adduct formation with different amino acids are predicted from the relative hardness/softness of the electrophile and nucleophile (Schwobel et al., 2011). Likewise, an AOP for liver fibrosis describes the MIE as covalent protein alkylation (www.aopwiki.org, Accessed October 10, 2014). A wide range of proteins is susceptible to reaction, but only a subset of the proteins alkylated contribute to injury (Codreanu et al., 2014). Regardless, of whether it is specific or non-specific, for a given AOP, the MIE is the primary point of interaction between the biology and the specific properties of a chemical. All subsequent KEs linked in that AOP are the result of biological changes emanating from the MIE. As such, the MIE can be viewed as the point of contact between chemical-specific properties that dictate toxicokinetics and absorption, distribution, metabolism, and elimination (ie, determining how much chemical reaches the MIE and for what duration), potency (ie, the strength of interaction at the target), and the toxicodynamic biological responses represented by the AOP. This separation between the chemical-specific properties that dictate the severity and duration of perturbation at the MIE and the downstream toxicodynamics allows AOPs to be described in a manner that is not chemical specific (see Villeneuve et al. 2014).
The special characteristics of the MIE as the point of interaction with chemicals and chemical-specific properties also define the special information requirements of an MIE description. As for any KE, the AOP developer should describe the biological state being observed, how it can be observed or measured and the taxonomic domain for which the measurement would be relevant. However, for the MIE, it is also relevant to describe the chemicals and chemical properties that are known to trigger the MIE (Supplementary Documents 1 and 2). In the AOP-Wiki, this is facilitated by allowing KE pages designated as MIEs to be linked to “chemical initiator” pages that define specific chemicals or groups/categories of chemicals known to trigger the MIE. Relative to the application of AOPs, the MIE is the critical KE for chemical category formation. If the goal of AOP development is to support chemical categorization, the MIE is a critical KE to define.
The fact that the MIE is always defined at the molecular level of biological organization also means that it presents special opportunity in terms of defining the domain of taxonomic relevance. Most notably, consideration of molecular target conservation across taxa (eg, Gunnarsson et al., 2008) is one strategy that can be applied for nearly all MIEs involving chemical interaction with a protein or specific nucleotide (DNA or RNA) sequence. There are a number of current methods for leveraging the large amounts of protein, RNA, and DNA sequence available in publically accessible databases to provide a first-cut estimate of which taxa would be expected to have one or more structurally and functionally similar biomolecule (LaLone et al., 2013b; McRobb et al., 2014). There are recognized limitations to such approaches (Celander et al., 2011; Fent and Sumpter, 2011), but they provide a reasonable, and scientifically based, first approximation where other data concerning taxonomic relevance are lacking.
By convention there should generally be just one MIE per AOP. The notable exception to this would be the case where two or more KEs, each triggered by direct interaction between a chemical and a biomolecule, must occur in order to produce the AO. An example would be certain carcinogenic modes of action which require mutagenic initiation followed by independent promotion resulting in cell proliferation where neither the mutagenic nor the promotion response alone would be sufficient to produce the AO; both must occur (Tanaka et al., 2013). In such a case, more than one MIE would actually be essential and thus more than one would be included. However, multiple MIEs should only be included in an AOP when they all must occur in order for the toxicity to proceed along the AOP.
Metabolic activation is sometimes cited as an example of a molecular interaction with a chemical that is required, along with a second molecular interaction in order for toxicity to proceed along a pathway. However, with regard to the fundamental principle that AOPs are not chemical specific (Villeneuve et al. 2014), we contend that metabolic activation should only rarely be considered an MIE in an AOP. In many cases, defining metabolic activation as the MIE in effect imposes chemical specificity on the AOP. There are likely very few cases where one could not bypass the need for a chemical activation step by directly triggering the pathway using the active metabolite. A notable exception may be situations in which the chemical-biological interaction that triggers an AOP involves metabolites that are so reactive that direct exposure to those metabolites is not possible (ie, they would react with other molecules in the environment before actually getting into an organism). In general, metabolic activation should be fairly rare as an MIE. Nonetheless, it is recognized that like other toxicokinetic factors, the potential for metabolic activation is an important consideration when applying AOPs for chemical-specific predictions. It is further noted that in applying AOPs for a chemical-specific risk assessment, one may need to consider both the AOP(s) triggered by the parent chemical and those triggered by its metabolites, some of which may or may not be in common.
As is the case for KEs in general, MIEs can be linked to more than one AOP. Additionally, KEs that serve as the direct point of interaction with a chemical in one AOP (ie, an MIE) may serve as an “ordinary” KE in a different pathway, where the chemical interaction leading to a perturbation occurs upstream. For example, estrogen receptor activation may be described as the MIE upon perturbation of the nuclear receptor with an exogenous chemical. Alternatively, in another AOP, the estrogen receptor activation would be considered a KE if an endogenous ligand activates the ER in response to upstream KEs. Both roles, of what is effectively a common KE, can be represented in both a knowledgebase and AOP network context.
The specificity with which the MIE is defined follows the same general rule applied to other KEs. That is, define the MIE only as specifically as necessary with regard to consideration of functional equivalence. For example, although the chemical interaction may be with the same target biomolecule (eg, a receptor), agonism versus antagonism is not functionally equivalent. The nature of the interaction would dictate what the next KE in the chain would be. Therefore, one would want to be specific about the nature of the interaction if it impacts function. The same basic consideration could apply to the question of whether chemical interactions with different isoforms of a particular protein should be represented as separate MIEs. If, based on the state of existing knowledge, both isoforms perform the same function they would be represented as a single MIE. This would be the case, even if they had different sensitivity to specific chemicals, but still play the same biological function (eg, bind the same promoter region; trigger the same signaling cascade). In contrast, if it is known that different isoforms perform different functions, a different MIE should be defined for each isoform, as the downstream chain of KEs could be expected to differ.
The consideration of functional equivalence can be relatively straight-forward for MIEs in which the biomolecule targeted by the chemical interaction is specifically defined and dictated by structural recognition (eg, a particular enzyme or receptor). However, in other cases, the biomolecule(s) targeted by the chemical interaction may be fairly non-specific, as is the case with many reactive mechanisms. In such cases, the progression of a downstream biological response may be more a function of the biological context in which the perturbation occurs rather than the specific biomolecules perturbed. If the chemical-biological interaction is non-specific such that a discrete function associated with the biomolecule(s) impacted cannot be ascribed, then the MIE should only be defined as specifically as the measurement associated with it can actually discriminate. In such cases, additional specificity would need to be conferred through the series of KERs and downstream KEs that connected to the more general MIE description. In general, as long as some measureable biological activity can be causally linked to a structural or functional property of a chemical, an MIE should be described and included in the AOP. However, in the event that the causal relationship between a chemical exposure and the earliest defined biological effect in the chain is unknown, it becomes unhelpful or even misleading to identify an MIE. In such cases, this should be portrayed as a known gap or uncertainty in the pathway. Although it is ideal to define an MIE for all AOPs, whether or not it is practical remains dictated by the available knowledge, the cost and difficulty of filling the knowledge gap(s), and the intended application(s) of the AOP.
Best Practices for Describing AOs
AOs are considered a second specialized type of KE. Since the primary intent of AOPs is to help support the use of alternative types of data for risk-based regulatory decision making, it is important that the sequence of biological responses described in an AOP extend to at least one effect that would generally be accepted as adverse from a regulatory perspective (ie, an AO). Although there continues to be debate regarding an objective definition of adversity (Boekelheide and Andersen, 2010; Boekelheide and Campion, 2010), in the context of regulatory application of AOPs, a functional definition would be that an AO could be any KE that corresponds to an established protection goal and/or is structurally and functionally equivalent to an apical outcome measured as part of an accepted regulatory test guideline. AOP development should always proceed until linkage to at least one AO has been achieved. This is why, along with MIEs, AOs were identified as one of the critical anchors for an AOP (Ankley et al., 2010; Hutchinson et al., 2006; OECD, 2013).
However, unlike the MIE, the level of biological organization at which an AO is defined can vary. It is not necessary that AOP descriptions stop at the first KE that meets the functional definition of an AO provided above. A particular AOP may extend beyond the first KE with established regulatory relevance and represent further progression of that AO (Figs. 1 and 3), and it is often desirable to do so if there is adequate supporting information available. For example, in a human health regulatory context, observation of hepatic steatosis may be of regulatory significance, and thus, an AO in an AOP. Nonetheless, AOP development could be extended further, from progression of liver steatosis to steatohepatitis, fibrosis, cirrhosis, and eventually hepatocellular carcinoma leading to premature mortality. As long as the chain of causality proceeded in a sequential fashion it would remain as one AOP, despite the fact that it includes four KEs that could be considered AOs (Fig. 4A). It is also possible, that beyond the initial AO in the chain, the pathway could branch (ie, one could either progress toward malignant tumor formation or to liver failure as a result of cirrhosis; Fig. 4B). In this case, such branches should be developed as separate AOPs, even though they may differ only at the terminal end of the pathway.
FIG. 4.
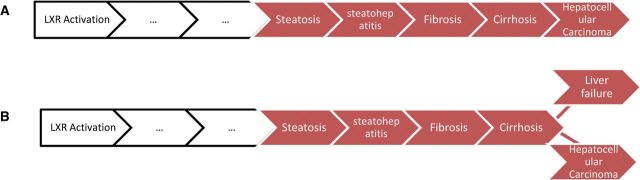
Illustrative example of AOPs containing multiple AOs in a single sequence, versus AOs that branch into multiple AOPs. A, Example of five KEs in a linear sequence which would all be considered AOs in a single AOP. B, Example of branching that would indicate that two AOPs, sharing all but the final AO, should be described. Color image is available in the online version of the article.
In practice, it is desirable to develop AOPs that are as broadly applicable as possible in terms of life stage, sex, taxa, etc. This is one of the major objectives behind the use of functional equivalence as a guide for how narrowly to define KEs. Many AOPs may apply equally well to both human health and ecological risk-based regulatory decision making. However, in the case of ecological risk, with the possible exception of endangered species risk assessments, maintenance of viable populations is typically the protection goal. This is most often assessed in terms of impacts on survival, growth, and reproduction as measured at the individual level and considered demographically significant for predicting potential impacts on populations (Hutchinson et al., 2006). With this in mind, although AOP development intended solely for human health applications may terminate with the linkage to an AO at a relatively low level of biological organization (eg, tissue or organ), it is considered a best practice to extend AOPs to the individual level whenever supporting information allows. Another practical result of the focus of ecological assessments on a relatively small number of outcomes of demographic significance is that, in an AOP network sense, many distinct paths will be expected to converge on a relatively finite number of terminal AOs. This characteristic makes it practical to easily implement a defined ontology and highly generalizable KE description for these nodes.
As AOs are considered specialized cases of KEs, the information content of an AO description is, in most respects, the same as that of any KE. However, with regard to the functional definition presented above, an AO description should also describe the regulatory relevance of the biological response represented by the AO (see Supplementary Documents 1 and 2). For example, one or more specific guideline tests that employ the same measurement as an endpoint could be cited, along with a description of how that endpoint is used in regulatory decision making (http://www.oecd.org/env/ehs/testing/oecdguidelinesforthetestingofchemicals.htm, Accessed October 10, 2014). By adding this type of information, the developer aids application by citing specific examples where the endpoint/effect that the AOP is being used to extrapolate to or predict has been employed for regulatory decision support. Additionally, by defining the regulatory relevance in a specific manner, this information can help establish whether the AO is of both human health and ecological relevance, or would only be considered adverse for one or the other, with the most common case being that an AO considered relevant for human health decision making would not be considered relevant in an ecological risk context.
BEST PRACTICES FOR DESCRIBING KERs
KERs are the second fundamental unit of AOP construction. They serve two key roles. First, they define a directed relationship between any given pair of KEs, establishing one as “upstream” and the other as “downstream.” Second, they provide the scientific foundation for prediction or extrapolation from an upstream cause to a downstream effect. They are intended to facilitate inference, based on prior experience and knowledge. Consequently, they must be supported by biological plausibility and/or empirical evidence, and the weight of evidence and overall level of certainty/uncertainty in the inference or extrapolation from one event to the next should be defined (see Supplementary Documents 1 and 2).
In describing KERs as a reusable module for AOP development, it is useful to assemble three main types of information. First, the developer should define the conditions under which the KER is relevant. This can help provide added specificity, not conveyed in the description of the upstream KE itself, which influences the downstream KE it connects to. For example, if the upstream event involves inhibition of an enzyme activity (a molecular level KE), the KER description may define the cellular or tissue context in which that inhibition occurs. Even though the enzyme has the same molecular function, regardless of tissue, the impact of its inhibition may vary in different tissue contexts. Russom et al. (2014) provide an illustrative example of this for a network of AOPs linking acetylcholinesterase (AChE) inhibition to mortality. Although AChE inhibition is systemic, the sequence of KEs one might observe following exposure to an AChE inhibitor can be tissue-dependent; for example, AChE inhibition leading to bradycardia in heart muscle versus paralysis in skeletal muscle. As with other general KEs mentioned previously (eg, oxidative stress, inflammation, and narcosis/membrane disruption), specificity is conferred through the linkages and associated KER descriptions. Likewise, broad differences in the duration and severity of change in the upstream KE that influence the downstream consequences should be defined here if known. For example, a chronic, low intensity perturbation may elicit different consequences than an acute, high intensity perturbation. By defining this additional specificity as part of the KER, the developer provides adequate specificity to define a discrete biological outcome of this event without sacrificing the ability to link a common KE to its many potential outcomes, in the context of an AOP network. As with KEs, the goal is to provide the level of specificity required to support the connection to the downstream KE, but not to define the context more specifically than is needed to accomplish that. By using KER descriptions in this manner, one can represent the multidimensional context-specific outcomes of a single KE, without needing a separately defined KE for each context. Thus, effective construction of KER descriptions is a critical aspect of modular, collective, development of AOP networks.
The second key type of information included in a KER description is the weight of evidence that supports inference or extrapolation from the state of the upstream KE to the state of the downstream KE. This includes consideration of the biological plausibility of the connection, typically considering the structural or functional relationship between the KEs under “normal” conditions. For example, establishing that a transporter of interest is key for maintaining an ion-gradient across a cell membrane or that a cardiomyocyte is a structural component of heart tissue. It also includes empirical evidence that shows that when there is a change in the upstream KE (ie, a perturbed state), a change in the downstream KE is also likely. In particular, evidence that shows that the change in the downstream event occurs as a result of exposure to concentrations equal to or greater than those that elicited a change in the upstream KE (establishing dose-response concordance), and that the change in the downstream KE follows the change upstream (establishing temporal concordance) (Becker et al.—https://aopkb.org/saop/workshops/somma.html, Accessed October 10, 2014; Meek et al., 2014; OECD, 2013) is desired.
In assembling the weight of evidence, data that are inconsistent with the relationship and/or recognized uncertainties and limitations need to be considered. These data should also be included and described as part of the evidence supporting (or potentially rejecting) a particular KER. In contrast with much of AOP development, which is intended to be non-chemical specific and as general/reusable as possible to promote modularity in the assembly of AOP networks, assembly of the weight of evidence supporting KERs should be specific about the biological contexts and chemical exposure conditions under which the evidence supporting a given KER were collected. In the context of weight of evidence supporting a KER, providing such specificity supports the weight of evidence evaluation and assessment of suitability for use in different regulatory contexts without undermining the modularity/reusability of the KER.
The third relevant type of information is any that helps define the quantitative relationship between the KEs linked via the KER. This may take the form of a regression (ie, x amount of change in upstream KE elicits y amount of change in downstream KE); a threshold (ie, when upstream KE reaches a certain threshold, a defined change in the downstream KE is elicited); computational models that capture complex relationships such as feedback loops, time-lags, etc. between KEs; or others. This quantitative information contributes to the overall weight of evidence supporting a KER. As for qualitative evidence, it is desirable to capture specific information regarding the biological context and exposure conditions under which the quantitative relationships were defined. The ultimate goal is to establish quantitative relationships that are generally valid across the entire applicability domain of the two KEs. However, until that generalizability can be established, providing details about the context in which quantitative relationships were defined helps insure transparency in how they are applied. This is also a section of the KER description in which the developer may want to capture information concerning modulating factors (eg, genetic variations, disease states, and nutritional or environmental factors) known to significantly influence the quantitative relationship between the two KEs linked via the KER. It is recommended that information on modulating factors be supported with citation of appropriate evidence. It should not be speculative in nature. However, well-supported information on known modulating factors can significantly enhance the accurate application of quantitative understanding of a KER and thus should be captured when known and well defined.
With regard to specific AOP development, typically the most challenging aspect of KER description is assembling the weight of evidence. In many cases, the biological plausibility between KEs may be so well established that it is virtually dogma making it difficult to select and provide appropriate supporting evidence, due to the sheer volume of information supporting the plausible linkage. In such cases, it is not necessary to provide a complete account of the primary scientific literature concerning the relationship. Rather, scientifically credible syntheses such as presented by text-books or review articles can be offered as supporting information.
However, the more challenging, and generally more frequent case is that in which supporting evidence for a KER is limited. Empirical evidence demonstrating that a change in a specific upstream KE is associated with a change in the downstream KE can be difficult to come by. Even rarer is empirical evidence that demonstrates dose-response and/or temporal concordance of two responses or demonstrates essentiality by showing that if the upstream event is prevented, the downstream will not occur. In many cases, the reason such data are lacking may be technical. For example, the measurement at one KE may be very routine and easy to make, while that at the next KE may be difficult, requiring expensive equipment and/or specialized skills or reagents. Therefore, both measures may only rarely be reported in a given study. Another is a case where the two KEs are displaced in time by an uncertain or unpredictable interval. Although both measurements could be made, one may not reliably observe changes in both KEs without doing time-course experiments that are both costly and challenging to implement. Consequently, one of the important purposes of defining the desired information content for KER descriptions is that an awareness of the information requirements and the types of data considered in the weight of evidence evaluation and how those data are weighted (Becker et al.—https://aopkb.org/saop/workshops/somma.html, Accessed October 10, 2014; Meek et al., 2014; OECD, 2013) can aid the design of experiments better suited for supporting AOP development.
Literature searches, data-mining approaches, targeted experimentation, and even modeling can all be viable tools for generating weight of evidence to support a KER. However, all can be time-consuming, technically challenging, and/or costly. Therefore, there are significant advantages to be gained through a collective and coordinated approach to KER development and description facilitated through a publically accessible knowledgebase. The major advantage is that if a KER of interest has already been described in the AOP-KB, one can use the biological plausibility and weight of evidence information already assembled to help support a new AOP. Likewise, since development of a KER in the AOP-KB provides the potential for other developers to view the developing weight of evidence for a KER and contribute to it, the AOP-KB provides an opportunity to crowd-source the scientific challenge of KER description and draw upon expertise, data, and approaches that may not be available to any single developer.
However, KER development in a collaborative knowledgebase context is not without some challenges. There may be cases where the specific nature and purpose of one’s AOP development may differ from that of the individual that developed the original KER, such that the weight of evidence provided may not address one’s specific needs. For example, the AOP-Wiki contains a KER for aromatase inhibition leading to decreased 17β-estradiol synthesis in ovarian granulosa cells (Supplementary Document 2). The original developer was primarily interested in linking aromatase inhibition to reproductive impairment in fish. However, a new developer may be interested in linking aromatase inhibition to impaired uterine development in rodents. The existing KER makes a strong case for the relevance of the relationship in fish, but lacks specific evidence for the relationship in rodents. In such a case, linking to the existing KER, as is, may not be ideal for establishing the predictive value of the rodent AOP.
In concordance with the principle of modularity and reusability of KEs and KERs and the collective development of AOP networks (Villeneuve et al. 2014), it is not desirable to simply create a new KER customized for the needs of a particular AOP. This does not mean that new KERs should not be added to the AOP-KB as science evolves and novel relationships are introduced (see principle of AOPs as living documents; Villeneuve et al. 2014). However, best practices would dictate that the AOP developer try to use and build upon existing KER descriptions where possible. This typically means adding to an existing KER description in a way that accommodates, and strengthens all the AOPs linked to it, where possible, creating new KERs only when necessary, and avoiding creation of redundant KERs in the knowledgebase.
CONCLUSIONS
Development of formal AOP descriptions is a new process for most scientists and regulators in the field. Construction of a knowledgebase of AOP descriptions (AOP-KB) has great potential as an information source to complement the mechanistically oriented high throughput, high content, and in silico data being generated using new technologies. However, unless AOPs are developed in a shared environment, according to a common set of guiding principles, conventions, and best practices, it will be difficult for scientists, regulators, and decision makers to access and utilize this knowledge in an efficient and meaningful manner. The conventions and best practices described above reflect the products of deliberation, debate, and experience among a pioneering community of AOP developers. Although these conventions and practices will continue to evolve over time, the information provided here should serve as a useful introduction to the emerging practice of AOP description. Together with continued development of approaches for evaluating the weight of evidence supporting an AOP (Becker et al.—https://aopkb.org/saop/workshops/somma.html, Accessed October 10, 2014), their acceptance for various regulatory applications (Perkins et al.—https://aopkb.org/saop/workshops/somma.html, Accessed October 10, 2014), and utility for guiding integrated approaches to testing and assessment (Tollefsen et al. 2014), they represent the state of the science with regard to AOP development and its role in the twenty-first century toxicology.
SUPPLEMENTARY DATA
Examples of two AOP descriptions prepared in the AOP-Wiki (www.aopwiki.org) module of the AOP-KB are provided (Supplementary Documents 1 and 2). Documents are provided in HTML format and can be opened in most web-browsers. Each document represents a static capture of an AOP page and all linked KE and KER pages from the AOP-Wiki. Links within the document are active and will direct users to the archived version of AOP-Wiki content as it appeared at the time of document preparation (July 2014); AOP-Wiki login id and password required. Instructions for obtaining an AOP-Wiki login and password are provided at www.aopwiki.org.
FUNDING
The American Chemistry Council, BioDetection Systems, European Centre for Ecotoxicology and Toxicology of Chemicals, Environment Canada, European Commission Directorate General Joint Research Centre, Human Toxicology Project Consortium, International Life Sciences Institute—Health and Environmental Science Institute, The Research Council of Norway (Grant No. 221455), Society for Environmental Toxicology and Chemistry, US Army Engineer Research and Development Center, and the US Environmental Protection Agency contributed support for the workshop.
Supplementary Material
ACKNOWLEDGMENTS
The content of this article is the result of an international expert workshop on Advancing Adverse Outcome Pathways (AOPs) for Integrated Toxicology and Regulatory Applications (https://aopkb.org/saop/workshops/somma.html). The authors acknowledge the workshop organizing committee, and all workshop participants for both plenary and informal discussions that influenced the content presented here. Special thanks to Michael Hornung for reviewing an earlier draft of this manuscript and Stephen Edwards for assistance in preparing supplementary materials. The views expressed are those of the authors, and do not necessarily represent the views of the organizations the authors are affiliated with or the sponsors. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
REFERENCES
- Ankley G. T., Bennett R. S., Erickson R. J., Hoff D. J., Hornung M. W., Johnson R. D., Mount D. R., Nichols J. W., Russom C. L., Schmieder P. K., et al. (2010). Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 29, 730–741. [DOI] [PubMed] [Google Scholar]
- Ankley G. T., Jensen K. M., Makynen E. A., Kahl M. D., Korte J. J., Hornung M. W., Henry T. R., Denny J. S., Leino R. L., Wilson V. S., et al. (2003). Effects of the androgenic growth promoter 17-beta-trenbolone on fecundity and reproductive endocrinology of the fathead minnow. Environ. Toxicol. Chem. 22, 1350–1360. [PubMed] [Google Scholar]
- Ankley G.T., Gray L.E. (2013). Cross-species conservation of endocrine pathways: a critical analysis of tier 1 fish and rat screening assays with 12 model chemicals. Environ. Toxicol. Chem. 32, 1084–1087. [DOI] [PubMed] [Google Scholar]
- Boekelheide K., Andersen M. E. (2010). A mechanistic redefinition of adverse effects—a key step in the toxicity testing paradigm shift. Altex 27, 243–252. [DOI] [PubMed] [Google Scholar]
- Boekelheide K., Campion S. N. (2010). Toxicity testing in the 21st century: using the new toxicity testing paradigm to create a taxonomy of adverse effects. Toxicol. Sci. 114, 20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boobis A. R., Cohen S. M., Dellarco V., McGregor D., Meek M. E., Vickers C., Willcocks D., Farland W. (2006). IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit. Rev. Toxicol. 36, 781–792. [DOI] [PubMed] [Google Scholar]
- Boobis A. R., Doe J. E., Heinrich-Hirsch B., Meek M. E., Munn S., Ruchirawat M., Schlatter J., Seed J., Vickers C. (2008). IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit. Rev. Toxicol. 38, 87–96. [DOI] [PubMed] [Google Scholar]
- Celander M. C., Goldstone J. V., Denslow N. D., Iguchi T., Kille P., Meyerhoff R. D., Smith B. A., Hutchinson T. H., Wheeler J. R. (2011). Species extrapolation for the 21st century. Environ. Toxicol. Chem. 30, 52–63. [DOI] [PubMed] [Google Scholar]
- Codreanu S. G., Ullery J. C., Zhu J., Tallman K. A., Beavers W. A., Porter N. A., Marnett L. J., Zhang B., Liebler D. C. (2014). Alkylation damage by electrophiles targets functional protein systems. Mol. Cell Proteomics 13, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fent K., Sumpter J. P. (2011). Progress and promises in toxicogenomics in aquatic toxicology: is technical innovation driving scientific innovation? Aquat. Toxicol. 105(Suppl.), 25–39. [DOI] [PubMed] [Google Scholar]
- Gunnarsson L., Jauhiainen A., Kristiansson E., Nerman O., Larsson D. G. (2008). Evolutionary conservation of human drug targets in organisms used for environmental risk assessments. Environ. Sci. Technol. 42, 5807–5813. [DOI] [PubMed] [Google Scholar]
- Hutchinson T. H., Ankley G. T., Segner H., Tyler C. R. (2006). Screening and testing for endocrine disruption in fish-biomarkers as “signposts,” not “traffic lights,” in risk assessment. Environ. Health Perspect. 114(Suppl. 1), 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlberg A. T., Bergstrom M. A., Borje A., Luthman K., Nilsson J. L. (2008). Allergic contact dermatitis—formation, structural requirements, and reactivity of skin sensitizers. Chem. Res. Toxicol. 21, 53–69. [DOI] [PubMed] [Google Scholar]
- Krewski D., Acosta D., Jr, Andersen M., Anderson H., Bailar J. C., 3rd, Boekelheide K., Brent R., Charnley G., Cheung V. G., Green S., Jr, et al. (2010). Toxicity testing in the 21st century: a vision and a strategy. J Toxicol. Environ. Health B Crit. Rev. 13, 51–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLone C. A., Villeneuve D. L., Burgoon L. D., Russom C. L., Helgen H. W., Berninger J. P., Tietge J. E., Severson M. N., Cavallin J. E., Ankley G. T. (2013b). Molecular target sequence similarity as a basis for species extrapolation to assess the ecological risk of chemicals with known modes of action. Aquat. Toxicol. 144–145, 141–154. [DOI] [PubMed] [Google Scholar]
- LaLone C. A., Villeneuve D. L., Cavallin J. E., Kahl M. D., Durhan E. J., Makynen E. A., Jensen K. M., Stevens K. E., Severson M. N., Blanksma C. A., et al. (2013a). Cross-species sensitivity to a novel androgen receptor agonist of potential environmental concern, spironolactone. Environ. Toxicol. Chem. 32, 2528–2541. [DOI] [PubMed] [Google Scholar]
- McRobb F. M., Sahagun V., Kufareva I., Abagyan R. (2014). In silico analysis of the conservation of human toxicity and endocrine disruption targets in aquatic species. Environ. Sci. Technol. 48, 1964–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek M. E., Boobis A., Cote I., Dellarco V., Fotakis G., Munn S., Seed J., Vickers C. (2014). New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. J. Appl. Toxicol. 34, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD. (2013). Guidance document on developing and assessing adverse outcome pathways. In Series on Testing and Assessment, No. 184, Vol. ENV/JM/MONO(20136, p. 45. Organisation for Economic Cooperation and Devleopment, Environment Directorate Paris, France. [Google Scholar]
- Regoli F., Giuliani M. E. (2014). Oxidative pathways of chemical toxicity and oxidative stress biomarkers in marine organisms. Mar. Environ. Res. 93, 106–117. [DOI] [PubMed] [Google Scholar]
- Russom C. L., Lalone C. A., Villeneuve D. L., Ankley G. T. (2014). Development of an adverse outcome pathway for acetylcholinesterase inhibition leading to acute mortality. Environ. Toxicol. Chem. 33, 2157–2169. [DOI] [PubMed] [Google Scholar]
- Schultz T. W. (1989). Non-polar narcosis: a review of the mechanism of action for baseline aquatic toxicity. In (Cowgill U. M., Williams L. R., Eds.), American Society for Testing and Materials, Philadelphia, PA. [Google Scholar]
- Schwobel J. A., Koleva Y. K., Enoch S. J., Bajot F., Hewitt M., Madden J. C., Roberts D. W., Schultz T. W., Cronin M. T. (2011). Measurement and estimation of electrophilic reactivity for predictive toxicology. Chem. Rev. 111, 2562–2596. [DOI] [PubMed] [Google Scholar]
- Sone K., Hinago M., Itamoto M., Katsu Y., Watanabe H., Urushitani H., Tooi O., Guillette L. J., Jr, Iguchi T. (2005). Effects of an androgenic growth promoter 17beta-trenbolone on masculinization of Mosquitofish (Gambusia affinis affinis). Gen. Comp. Endocrinol. 143, 151–160. [DOI] [PubMed] [Google Scholar]
- Tanaka T., Shimizu M., Kochi T., Moriwaki H. (2013). Chemical-induced carcinogenesis. J. Exp. Clin. Med. 5, 203–209. [Google Scholar]
- Tollefsen K. E., Scholz S., Cronin M. T., Edwards S. W., de Knecht J., Crofton K., Garcia-Reyero N., Hartung T., Worth A., Patlewicz G. (2014). Applying adverse outcome pathways (AOPs) to support integrated approaches to testing and assessment (IATA). Reg. Toxicol. Pharmacol. (in press). [DOI] [PubMed] [Google Scholar]
- Villeneuve D. L., Crump D., Garcia-Reyero N., Hecker M., Hutchinson T. H., LaLone C. A., Landesmann B., Lettieri T., Munn S., Nepelska M., et al. (2014). Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol. Sci. (this issue). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.