

Abstract
Serum uric acid is commonly elevated in subjects with chronic kidney disease (CKD), but was historically viewed as an issue of limited interest. Recently, uric acid has been resurrected as a potential contributory risk factor in the development and progression of CKD. Most studies documented that an elevated serum uric acid level independently predicts the development of CKD. Raising the uric acid level in rats can induce glomerular hypertension and renal disease as noted by the development of arteriolosclerosis, glomerular injury and tubulointerstitial fibrosis. Pilot studies suggest that lowering plasma uric acid concentrations may slow the progression of renal disease in subjects with CKD. While further clinical trials are necessary, uric acid is emerging as a potentially modifiable risk factor for CKD.
Gout was considered a cause of CKD in the mid-nineteenth century [1], and, prior to the availability of therapies to lower the uric acid level, the development of end-stage renal disease was common in gouty patients. In their large series of gouty subjects Talbott and Terplan found that nearly 100% had variable degrees of CKD at autopsy (arteriolosclerosis, glomerulosclerosis and interstitial fibrosis) [2]. Additional studies showed that during life impaired renal function occurred in half of these subjects [3]. As many of these subjects had urate crystals in their tubules and interstitium, especially in the outer renal medulla, the disease became known as gouty nephropathy. The identity of this condition fell in question as the presence of these crystals may occur in subjects without renal disease; furthermore, the focal location of the crystals could not explain the diffuse renal scarring present. In addition, many subjects with gout also had coexistent conditions such as hypertension and vascular disease, leading some experts to suggest that the renal injury in gout was secondary to these latter conditions rather than to uric acid per se [4]. Indeed, gout was removed from the textbooks as a cause of CKD, and the common association of hyperuricemia with CKD was solely attributed to the retention of serum uric acid that is known to occur as the glomerular filtration rate falls.
Renewed interest in uric acid as a cause of CKD occurred when it was realized that invalid assumptions had been made in the arguments to dismiss uric acid as a risk factor for CKD [5]. The greatest assumption was that the mechanism by which uric acid would cause kidney disease would be via the precipitation as crystals in the kidney, similar to the way it causes gout. However, when laboratory animals with CKD were made hyperuricemic, the renal disease progressed rapidly despite an absence of crystals in the kidney [6]. Since this seminal study, there has been a renewed interest in the potential role uric acid may have in both acute and CKD. We briefly review some of the major advances that have occurred in this field in the last 15 years.
Keywords: uric acid, gout, allopurinol, hyperuricemia, chronic kidney disease
EXPERIMENTAL STUDIES SUPPORTING : A ROLE FOR URIC ACID IN CKD
Studying the role of uric acid in chronic kidney disease (CKD) is very difficult since uric acid is excreted primarily by the kidney, and hence a decrease in the glomerular filtration rate (GFR) is inevitably accompanied by a rise in the serum uric acid level. As such, studies in experimental animals in which serum uric acid can be modulated are critical to understanding if there is a role for uric acid in the causation or progression of kidney disease. In this regard, humans and great apes metabolize uric acid differently from most other mammals. All animals generate uric acid normally from the turnover of ATP and nucleic acids, and uric acid can also be generated de novo from amino acid precursors. However, in most animals uric acid levels are relatively low as there is an enzyme in the liver (uricase) that degrades uric acid to 5-hydroxyisourate, and eventually to allantoin. However, ancestors to the great apes and humans lost the uricase enzyme ∼15 million years ago due to a mutation. As a consequence, all humans are ‘uricase knockouts’ and have higher serum uric acid levels that can be altered more easily by diet than that in other mammals.
Nevertheless, in laboratory animals it is possible to modulate the uric acid level by raising it (with a uricase inhibitor such as oxonic acid) or by lowering it (such as with xanthine oxidase inhibitors or uricosuric agents). Using this approach, we found that raising the uric acid level could induce oxidative stress and endothelial dysfunction, resulting in the development of both systemic and glomerular hypertension in association with elevated renal vascular resistance and reduced renal blood flow [7–9]. In normal rats there was activation of the renin–angiotensin system (RAS), with the development of vascular disease of the afferent arteriolar system (arteriolosclerosis) and glomerular hypertrophy, and over time mild interstitial disease and glomerulosclerosis [10–11]. Hyperuricemia was also able to induce an epithelial-to-mesenchymal transition, with direct effects on the tubular cell population [12]. As mentioned, the effects of hyperuricemia were particularly impressive in animals with pre-existing renal disease, where it accelerated glomerular hypertension and the vascular lesions, resulting in worsening proteinuria and renal failure associated with worsening glomerulosclerosis and tubulointerstitial disease [6].
Additional studies showed that uric acid might have a role in other experimental models of kidney disease. For example, we found that diabetic mice also developed elevated serum uric acid and that lowering the uric acid level could improve the kidney disease [13]. Cyclosporine is also known to raise the uric acid level, and we found that the renal disease induced by cyclosporine could be worsened by raising the uric acid level and lessened by lowering it [14, 15].
The specific mechanism(s) by which uric acid may be causing these effects has been studied primarily in cell culture systems (Figure 1). One of the more striking findings is that uric acid, while being a potent antioxidant in the extracellular environment, is a pro-oxidant inside the cell where it can induce stimulation of NADPH oxidases with the induction of mitochondrial dysfunction [16]. Uric acid can also induce endothelial dysfunction via a variety of mechanisms [16] and can also stimulate the release of alarmins (such as high mobility group box-1 protein) from endothelial cells that activate Toll-like receptor pathways [17]. Uric acid also stimulates vascular smooth muscle cell proliferation with the production of chemotactic factors and oxidants and the activation of the RAS [18, 19]. Uric acid can also induce phenotypic alterations and chemokines in tubular cells and can induce intrarenal inflammation following its infusion in mice [12, 20].
FIGURE 1:
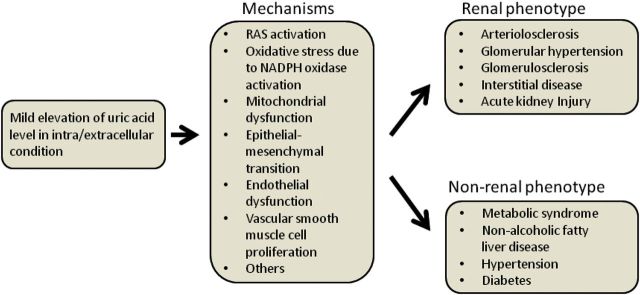
Mechanism by which uric acid contributes to the development of renal and non-renal diseases. RAS, renin–angiotensin system.
Further experimental studies outside the scope of this manuscript have found a key role for uric acid not only in CKD, but also in acute kidney injury, metabolic syndrome, non-alcoholic fatty liver disease (NAFLD) and hypertension. This has led to emerging clinical evidence that uric acid may have a role in systemic hypertension, diabetes and NAFLD. The role of uric acid in these conditions is discussed elsewhere [21, 22].
EPIDEMIOLOGICAL STUDIES LINKING : URIC ACID WITH CKD
As mentioned, serum uric acid is eliminated principally by the kidneys, and while there is a compensatory increased removal by the gut in the setting of renal insufficiency, this is not completely effective, and serum uric acid increases as the GFR falls, with approximately half of the subjects becoming hyperuricemic by the time dialysis is initiated [23]. This makes it very difficult to assess the role of uric acid in the progression of renal disease in subjects with CKD based on epidemiological studies. In addition, the experimental studies suggest that uric acid may cause kidney disease primarily by causing systemic and glomerular hypertension, but in renal disease this mechanism may become less relevant as systemic hypertension commonly develops as a consequence of sodium and water retention. As such, it is not surprising that, in subjects with established CKD, serum uric acid has often [24, 25] not been found to predict progression. Nevertheless, some studies have found an elevated uric acid level to predict progression in subjects with established CKD, especially in diabetes and IgA nephropathy [26, 27].
In contrast, in subjects with normal renal function, an elevated serum uric acid has almost uniformly been found to independently predict the development of CKD, including end-stage renal disease (Table 1). An elevated serum uric acid has also been found to independently predict the development of CKD in subjects with IgA nephropathy [28–31] and of graft failure in transplant patients with chronic allograft nephropathy [32].
Table 1:
An elevated serum uric acid predicts the development of CKD
| Location | Population | Follow-up (years) | Type | Indep? | Author (year) |
|---|---|---|---|---|---|
| Japan | 6403 adults | 2 | CKD | Yes | Iseki (2001) |
| Japan | 48 177 adults | 10 | ESRD | Women | Iseki (2004) |
| Thailand | 3499 adults | 12 | CKD | Yes | Domrongkitchaiporn (2005) |
| USA | 5808 adults | 5 | CKD | No | Chonchol (2007) |
| Austria | 21 457 adults | 7 | CKD | Yes | Obermayr (2008) |
| USA | 13 338 adults | 8.5 | CKD | Yes | Weiner (2008) |
| Austria | 17 375 adults | 7 | CKD | Yes | Obermayr (2008) |
| USA | 177 500 adults | 25 | ESRD | Yes | Hsu (2009) |
| USA | 355 type 1 diabetesa | 6 | CKD | Yes | Ficociello (2010) |
| Italy | 900 adults | 5 | CKD | Yes | Bellomo (2010) |
| Japan | 7078 adults | 5 | CKD | Yes | Sonoda (2011) |
| Taiwan | 94 422 adults | 3.5 | CKD | Men | Wang (2011) |
| Israel | 2449 adults | 26 | ESRD | Yes | Ben-Dov (2011) |
| Japan | 14 399 adults | 5 | CKD | Yes | Yamada (2011) |
| USA | 488 renal transplants | 1 | Graft Loss | Yes | Haririan (2011) |
| China | 1410 adults | 4 | CKD | Yes | Zhang (2012) |
| Korea | 14 939 adults | 10.2 | CKD | Men | Mok (2012) |
| Italy | 1449 type 2 diabetics | 5 | CKD | Yes | Zoppini (2012) |
aSubjects with abnormal albumin excretion.
One of the most interesting relationships of uric acid is with diabetic nephropathy. In diabetes, serum uric acid is often low because of the effects of glycosuria in increasing urate excretion. Indeed, the serum uric acid level tends to be low in subjects with poor glucose control [33]. Since good diabetes control is strongly associated with renoprotection, one might predict that elevated uric acid levels would be associated with better renal outcomes. However, in subjects with type 1 diabetes, an elevated serum uric acid, even when within the normal range, is a strong predictor for the development of both incipient (microalbuminuric) and established (albuminuric) diabetic nephropathy, and also predicts the development of CKD [34–36]; uric acid also predicts the development and progression of CKD in subjects with type 2 diabetes [26, 37].
Thus, an elevated uric acid is strongly associated with the development of CKD, but not always with the progression of CKD. In addition, an elevated serum uric acid level has been associated with both the presence of intrarenal arteriolar lesions [31, 38] and with an increased risk for cardiovascular mortality in subjects with CKD [24, 39, 40], consistent with the vascular effects observed in laboratory animals with hyperuricemia [41]. However, once subjects develop end-stage renal disease, there is a reverse J-curve, in which both high and lower uric acid levels convey increased cardiovascular risk and mortality [23, 42]. This latter finding is not unlike the ‘reverse epidemiology’ that has been noted for other cardiovascular risk factors, such as hypertension and obesity.
MECHANISMS FOR HYPERURICEMIA
The observation that hyperuricemia frequently precedes the development of CKD suggests that factors other than renal insufficiency are likely involved in the pathogenesis of the elevation in uric acid. Studies suggest that a variety of mechanisms may be operative.
One of the most common risk factors for CKD is obesity and metabolic syndrome, which is strongly associated with hyperuricemia likely as a consequence of insulin resistance and the effects of insulin to reduce urinary urate excretion [43]. Hypertension is also commonly associated with renal vasoconstriction which also leads to uric acid retention [44]. However, more recent studies suggest that the rise in serum uric acid also precedes these conditions and hence may not represent the underlying cause of hyperuricemia [21]. Furthermore, one study found uric acid to be minimally elevated in secondary hypertension [45], a condition in which renal vasoconstriction is also present.
Diets high in meats, sugar (fructose) and beer are well-known risk factors for hyperuricemia [46]. Fructose from added sugars is an important candidate since intake in animals and humans is strongly associated with the development of metabolic syndrome [47]. Some but not all studies also suggest that fructose from added sugars increases the risk for hypertension and kidney disease [48–50]. The administration of fructose to rats also reproduces most of the renal hemodynamic effects of experimental hyperuricemia and can also cause renal disease that can be blocked by lowering uric acid levels [51, 52].
Low-level intoxication of lead and cadmium can also raise serum uric acid levels, likely by blocking the renal excretion of uric acid. Chronic low levels of lead have also been strongly associated with the development of CKD [53]. The renal pathology in chronic lead intoxication is associated with the development of microvascular disease, glomerulosclerosis and interstitial fibrosis similar to what is observed in subjects with gout [54]. Furthermore, the administration of lead to animals with CKD is associated with the development of hyperuricemia and an acceleration of the renal disease [55]. In these animals, the administration of allopurinol could reduce the systemic hypertension, but renoprotection was unable to be assessed due to the toxicity from treatment as a consequence of the deposition of allopurinol and xanthine crystals [55].
Epigenetic factors are also possible. For example, low birth weight is associated with elevated serum uric acid in the neonate and mother; and the elevation in the uric acid level persists throughout childhood and adolescence and is associated with endothelial dysfunction and the development of hypertension [56–58]. The mechanism for the hyperuricemia is unknown but may result from genetic and familial factors [59].
Genetic factors are also likely involved. Familial juvenile hyperuricemic nephropathy (also known as medullary cystic kidney disease type 2) results from a mutation in uromodulin and is associated with progressive renal disease with the prominent development of glomerulosclerosis and tubulointerstitial fibrosis. While some studies have not observed any benefit from lowering the uric acid level on renal outcomes [60], other studies suggest that an early initiation of allopurinol treatment can prevent the progression of renal disease in this condition [61]. Uromodulin polymorphisms have recently been identified by genome-wide screening to be associated with the development of CKD in humans [62]. Interestingly, polymorphisms in uromodulin have also been recently shown to be associated with hyperuricemia [63].
Genome-wide association studies have also found association between polymorphisms in urate transport and the risk for gout. One recent study reported a urate-gene–diuretic interaction in the ARIC study, in which the risk of gout in hypertensive individuals was greater in those subjects who had polymorphisms in renal uric acid transporters that associate with higher uric acid levels [64]. The role of genetics in determining the level of serum uric acid level and its impact on the estimated glomerular filtration rate (eGFR) are illustrated by a recent study in offspring of centenarian Ashkenazi Jews [65].
INTERVENTION STUDIES IN CKD AND : DIABETIC NEPHROPATHY
To date, only pilot studies have evaluated the benefit of lowering uric acid in CKD. Siu et al. [66] reported that allopurinol therapy slowed renal disease progression in hyperuricemic subjects with modest (stage 3) CKD at 1 year compared with randomized controls. Similarly, a small prospective controlled study was conducted by Goicoechea et al. [67] in 113 CKD patients. Subjects with an eGFR <60 mL/min were randomly assigned to allopurinol 100 mg/day or the continuation of usual therapy. A decrease of the serum uric acid level from 7.8 ± 2.1 to 6.0 ± 1.2 mg/dL was not associated with a significant change of the eGFR (40.8 ± 11.2 to 42.2 ± 13.2 mL/min/1.73 m2), whereas the eGFR fell in the control group (from 39.5 ± 12.4 to 35.9 ± 12.3 mL/min); allopurinol treatment was also associated with less cardiovascular events (7 in the allopurinol versus 15 in the control group). In a short 4-month study, Momeni et al. [68] reported that allopurinol therapy could reduce proteinuria in subjects with diabetic nephropathy but without an effect on creatinine.
In a meta-analysis, based on 11 papers with a total of only 753 participants, Wang et al. [69] reported that uric acid lowering is associated with significant lowering of the serum creatinine concentration and an increase of the eGFR. Similarly, in the J-HEALTH study [70], which included 7629 subjects, a change in the eGFR was (negatively) correlated with a change in the serum uric acid level and associated with less cardiovascular events.
There is some evidence that lowering the uric acid level may act in part by blocking the RAS. For one thing, uric acid stimulates the production of angiotensin II in vascular cells and also increases renin expression in laboratory animals [10, 19, 71]. Serum uric acid is also associated with elevated serum renin in humans [72], and Perlstein et al. [73] found evidence that hyperuricemia was associated with activation of the intrarenal RAS in humans. In a study by Shi et al. [30], subjects with IgA nephropathy were randomized to receive allopurinol therapy or no treatment for 1 year. None of these subjects were receiving angiotensin converting enzyme (ACE) inhibitors. In the first month, there was a fall in the GFR consistent with a hemodynamic effect similar to that observed with the initiation of ACE inhibitors, followed by a leveling of the slope in the change of GFR over the subsequent year. In this study, no benefit of allopurinol therapy on the GFR was observed, which may have reflected the fact that both groups had mild disease and so renal progression was not observed in either group. However, allopurinol therapy was associated with a significant reduction in blood pressure medications [30]. Finally, Talaat and El-Sheikh [74] performed an interesting study in which they withdrew allopurinol in subjects with CKD. In this study there was a marked worsening in blood pressure, proteinuria and GFR in those subjects who were not receiving ACE inhibitors [74].
While these studies suggest that lowering the uric acid level may simply be another way to block the RAS, there is also some evidence that such a lowering may have other benefits in addition to RAS blockade [75]. Indeed, one unique aspect of the angiotensin receptor blocker, losartan is that it also lowers the serum uric acid level. There are now a few reports that the lowering of uric acid by losartan may provide additional benefits for slowing renal disease [76] and reducing cardiovascular events [77]. For example, in a post hoc analysis of the RENAAL study, Miao et al. [76] found that renal events, i.e. doubling of serum creatinine or end-stage renal disease, were less frequent in individuals in whom—as a result of the known uricosuric effect of losartan—serum uric acid concentrations were lowered by >0.5 mg/dL compared with patients with lesser lowering or increase (9.5 versus 14.3 events per 100 patient/years). The risk of renal events was reduced by 6% for every 0.5 mg/dL decrement in serum uric acid.
SELECTING TREATMENT
Hyperuricemia is strongly associated with CKD, but we still need large clinical trials before we should embrace the lowering of uric acid therapy in management. Treatment of uric acid is not always benign. For example, allopurinol therapy can be associated with fatal Stevens-Johnson syndrome, and, while screening for HLA-B68 may allow the elimination of subjects at highest risk for this condition [78], this procedure is rarely done. Allopurinol may also accumulate in subjects with a low eGFR.
The new xanthine oxidase inhibitor, febuxostat does not appear to be associated with Stevens-Johnson-syndrome to date, and its dosage does not need to be modified in CKD. It may also be more effective at lowering the uric acid level in the setting of CKD [79]. For example, one randomized double-blind trial [80] comprised 1072 patients with gout, serum uric acid >8 mg/dL and either normal serum creatinine or serum creatinine 1.5–2 mg/dL. Patients were randomized to febuxostat (up to 240 mg/day), allopurinol (100–300 mg/day) or placebo. The serum uric acid target was <6 mg/dL. A higher percentage of subjects with impaired renal function achieved a primary end point with febuxostat compared with allopurinol, but diarrhea and dizziness were more frequent in the febuxostat group. In another study, 2269 subjects, 65% of whom had CKD with an eGFR >30 mL/min/1.73 m2, were randomized to febuxostat 40 mg/day, febuxostat 80 mg/day or allopurinol 200 mg/day [81]. The treatment target of <6 mg/dL was reached in 45% of the patients on febuxostat 40 mg/day, 67% on febuxostat 80 mg/day and 42% of patients on allopurinol 200 mg/day. Febuxostat 80 mg/day was superior to the other arms (P < 0.001). In patients with CKD, the target of <6 mg/dL uric acid was more frequently (72%) reached with febuxostat 80 mg/day (P < 0.01). There was no difference in side effects between the groups. While these studies suggest that febuxostat may be safe in CKD, one caution is that all xanthinase oxidase inhibitors can increase urinary xanthine levels, which can be nephrotoxic. As such, if xanthine oxidase inhibitors are administered to subjects with CKD, a low dose should be initiated and the dosage increased slowly over 4–8 weeks.
CONCLUSIONS
In addition to the need for large clinical trials, more studies are required to better understand the biology of uric acid. Does uric acid have the primary role in causing kidney disease, or is it the activation of xanthine oxidase which also produces oxidants in addition to uric acid? Does lowering uric acid provide any additional benefit over ACE inhibitors in subjects with CKD? Would it be more effective to alter diet, or chelate lead, as opposed to reducing uric acid itself in these subjects? Clearly there are more questions than answers. However, we are better off than we were 20 years ago when uric acid was a dead subject [82]. Given the relatively ineffective current treatments for CKD, a new therapy would be greatly beneficial.
CONFLICT OF INTEREST STATEMENT
R.J.J. and T.N. have patent applications related to lowering uric acid as a means to prevent or treat diabetic nephropathy, insulin resistance and features of metabolic syndrome and also have stock in Revascor, a new company interested in lowering uric acid as a treatment for hypertension and metabolic syndrome. R.J.J. is also on the Scientific Board of Amway, has grants with the NIH, State of Colorado, Amway, Cardero, Danone and Questcor, and stock with Cardero and NCD Therapeutics. RJJ is also author of The Sugar Fix (Rodale, 2008) and The Fat Switch (Mercola.com, 2012) that discusses the role of fructose and uric acid in the epidemic of obesity and diabetes. All other authors have no disclosures. The results presented in this paper have not been published previously in whole or in part, and have not been influenced by the COI.
REFERENCES
- 1.Johnson G. On the Diseases of the Kidney. London: John W Parker and Son; 1852. [Google Scholar]
- 2.Talbott JH, Terplan KL. The kidney in gout. Medicine (Baltimore) 1960;39:405–467. [PubMed] [Google Scholar]
- 3.Barlow KA, Beilin LJ. Renal disease in primary gout. Q J Med. 1968;37:79–96. [PubMed] [Google Scholar]
- 4.Yu TF, Berger L. Impaired renal function gout: its association with hypertensive vascular disease and intrinsic renal disease. Am J Med. 1982;72:95–100. doi: 10.1016/0002-9343(82)90593-9. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RJ, Kivlighn SD, Kim YG, et al. Reappraisal of the pathogenesis and consequences of hyperuricemia in hypertension, cardiovascular disease, and renal disease. Am J Kidney Dis. 1999;33:225–234. doi: 10.1016/s0272-6386(99)70295-7. [DOI] [PubMed] [Google Scholar]
- 6.Kang DH, Nakagawa T, Feng L, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002;13:2888–2897. doi: 10.1097/01.asn.0000034910.58454.fd. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez-Lozada LG, Soto V, Tapia E, et al. Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. Am J Physiol. 2008;295:F1134–F1141. doi: 10.1152/ajprenal.00104.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-Lozada LG, Tapia E, Lopez-Molina R, et al. Effects of acute and chronic L-arginine treatment in experimental hyperuricemia. Am J Physiol. 2007;292:F1238–F1244. doi: 10.1152/ajprenal.00164.2006. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez-Lozada LG, Tapia E, Santamaria J, et al. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int. 2005;67:237–247. doi: 10.1111/j.1523-1755.2005.00074.x. [DOI] [PubMed] [Google Scholar]
- 10.Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38:1101–1106. doi: 10.1161/hy1101.092839. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa T, Mazzali M, Kang DH, et al. Hyperuricemia causes glomerular hypertrophy in the rat. Am J Nephrol. 2003;23:2–7. doi: 10.1159/000066303. [DOI] [PubMed] [Google Scholar]
- 12.Ryu E-S, Kim MJ, Shin H-S, et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol. 2013;304:F471–80. doi: 10.1152/ajprenal.00560.2012. [DOI] [PubMed] [Google Scholar]
- 13.Kosugi T, Nakayama T, Heinig M, et al. Effect of lowering uric acid on renal disease in the type 2 diabetic db/db mice. Am J Physiol Renal Physiol. 2009;297:F481–F488. doi: 10.1152/ajprenal.00092.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazzali M, Kim YG, Suga S, et al. Hyperuricemia exacerbates chronic cyclosporine nephropathy. Transplantation. 2001;71:900–905. doi: 10.1097/00007890-200104150-00014. [DOI] [PubMed] [Google Scholar]
- 15.Mazali FC, Johnson RJ, Mazzali M. Use of uric acid-lowering agents limits experimental cyclosporine nephropathy. Nephron Exp Nephrol. 2012;120:e12–e19. doi: 10.1159/000330274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia M, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol. 2012;121:e71–e78. doi: 10.1159/000345509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabadi MM, Kuo MC, Ghaly T, et al. Interaction between uric acid and HMGB1 translocation and release from endothelial cells. Am J Physiol. 2012;302:F730–F741. doi: 10.1152/ajprenal.00520.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang DH, Park SK, Lee IK, et al. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol. 2005;16:3553–3562. doi: 10.1681/ASN.2005050572. [DOI] [PubMed] [Google Scholar]
- 19.Corry DB, Eslami P, Yamamoto K, et al. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J Hypertens. 2008;26:269–275. doi: 10.1097/HJH.0b013e3282f240bf. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y, Fang L, Jiang L, et al. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway. PLoS ONE. 2012;7:e39738. doi: 10.1371/journal.pone.0039738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson RJ, Perez-Pozo SE, Sautin YY, et al. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocr Rev. 2009;30:96–116. doi: 10.1210/er.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–1821. doi: 10.1056/NEJMra0800885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suliman ME, Johnson RJ, Garcia-Lopez E, et al. J-shaped mortality relationship for uric acid in CKD. Am J Kidney Dis. 2006;48:761–771. doi: 10.1053/j.ajkd.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Madero M, Sarnak MJ, Wang X, et al. Uric acid and long-term outcomes in CKD. Am J Kidney Dis. 2009;53:796–803. doi: 10.1053/j.ajkd.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sturm G, Kollerits B, Neyer U, et al. Uric acid as a risk factor for progression of non-diabetic chronic kidney disease? The Mild to Moderate Kidney Disease (MMKD) Study. Exp Gerontol. 2008;43:347–352. doi: 10.1016/j.exger.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Altemtam N, Russell J, El Nahas M. A study of the natural history of diabetic kidney disease (DKD) Nephrol Dial Transplant. 2012;27:1847–1854. doi: 10.1093/ndt/gfr561. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y, Chen W, Jalal D, et al. Clinical outcome of hyperuricemia in IgA nephropathy: a retrospective cohort study and randomized controlled trial. Kidney Blood Press Res. 2012;35:153–160. doi: 10.1159/000331453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohno I, Hosoya T, Gomi H, et al. Serum uric acid and renal prognosis in patients with IgA nephropathy. Nephron. 2001;87:333–339. doi: 10.1159/000045939. [DOI] [PubMed] [Google Scholar]
- 29.Syrjanen J, Mustonen J, Pasternack A. Hypertriglyceridaemia and hyperuricaemia are risk factors for progression of IgA nephropathy. Nephrol Dial Transplant. 2000;15:34–42. doi: 10.1093/ndt/15.1.34. [DOI] [PubMed] [Google Scholar]
- 30.Shi Y, Chen W, Jalal D, et al. Clinical outcome of hyperuricemia in IgA nephropathy: a Retrospective Cohort Study and Randomized Controlled Trial. Kidney Blood Press Res. 2011;35:153–160. doi: 10.1159/000331453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J, Chen X, Xie Y, et al. Characteristics and risk factors of intrarenal arterial lesions in patients with IgA nephropathy. Nephrol Dial Transplant. 2005;20:719–727. doi: 10.1093/ndt/gfh716. [DOI] [PubMed] [Google Scholar]
- 32.Akalin E, Ganeshan SV, Winston J, et al. Hyperuricemia is associated with the development of the composite outcomes of new cardiovascular events and chronic allograft nephropathy. Transplantation. 2008;86:652–658. doi: 10.1097/TP.0b013e3181814f5b. [DOI] [PubMed] [Google Scholar]
- 33.Bo S, Cavallo-Perin P, Gentile L, et al. Hypouricemia and hyperuricemia in type 2 diabetes: two different phenotypes. Eur J Clin Invest. 2001;31:318–321. doi: 10.1046/j.1365-2362.2001.00812.x. [DOI] [PubMed] [Google Scholar]
- 34.Hovind P, Rossing P, Tarnow L, et al. Serum uric acid as a predictor for development of diabetic nephropathy in type 1 diabetes: an inception cohort study. Diabetes. 2009;58:1668–1671. doi: 10.2337/db09-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jalal DI, Rivard CJ, Johnson RJ, et al. Serum uric acid levels predict the development of albuminuria over 6 years in patients with type 1 diabetes: findings from the Coronary Artery Calcification in Type 1 Diabetes study. Nephrol Dial Transplant. 2010;25:1865–1869. doi: 10.1093/ndt/gfp740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ficociello LH, Rosolowsky ET, Niewczas MA, et al. High-normal serum uric acid increases risk of early progressive renal function loss in type 1 diabetes: results of a 6-year follow-up. Diabetes Care. 2010;33:1337–1343. doi: 10.2337/dc10-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zoppini G, Targher G, Chonchol M, et al. Serum uric acid levels and incident chronic kidney disease in patients with type 2 diabetes and preserved kidney function. Diabetes care. 2012;35:99–104. doi: 10.2337/dc11-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohagura K, Kochi M, Miyagi T, et al. An association between uric acid levels and renal arteriolopathy in chronic kidney disease: a biopsy-based study. Hypertens Res. 2013;36:43–49. doi: 10.1038/hr.2012.135. [DOI] [PubMed] [Google Scholar]
- 39.Kanbay M, Yilmaz MI, Sonmez A, et al. Serum uric acid independently predicts cardiovascular events in advanced nephropathy. Am J Nephrol. 2012;36:324–331. doi: 10.1159/000342390. [DOI] [PubMed] [Google Scholar]
- 40.Ito H, Abe M, Mifune M, et al. Hyperuricemia is independently associated with coronary heart disease and renal dysfunction in patients with type 2 diabetes mellitus. PLoS ONE. 2011;6:e27817. doi: 10.1371/journal.pone.0027817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazzali M, Kanellis J, Han L, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol. 2002;282:F991–F997. doi: 10.1152/ajprenal.00283.2001. [DOI] [PubMed] [Google Scholar]
- 42.Lee SM, Lee AL, Winters TJ, et al. Low serum uric acid level is a risk factor for death in incident hemodialysis patients. Am J Nephrol. 2009;29:79–85. doi: 10.1159/000151292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinones GA, Natali A, Baldi S, et al. Effect of insulin on uric acid excretion in humans. The Am J Physiol. 1995;268:E1–E5. doi: 10.1152/ajpendo.1995.268.1.E1. [DOI] [PubMed] [Google Scholar]
- 44.Messerli FH, Frohlich ED, Dreslinski GR, et al. Serum uric acid in essential hypertension: an indicator of renal vascular involvement. Ann Int Med. 1980;93:817–821. doi: 10.7326/0003-4819-93-6-817. [DOI] [PubMed] [Google Scholar]
- 45.Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension. 2003;42:247–252. doi: 10.1161/01.HYP.0000085858.66548.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi HK, Atkinson K, Karlson EW, et al. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350:1093–1103. doi: 10.1056/NEJMoa035700. [DOI] [PubMed] [Google Scholar]
- 47.Malik VS, Popkin BM, Bray GA, et al. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care. 2010;33:2477–2483. doi: 10.2337/dc10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin WT, Huang HL, Huang MC, et al. Effects on uric acid, body mass index and blood pressure in adolescents of consuming beverages sweetened with high-fructose corn syrup. Int J Obes (Lond) 2012 doi: 10.1038/ijo.2012.121. doi:10.1038/ijo.2012.121. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 49.Nguyen S, Choi HK, Lustig RH, et al. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J Pediatr. 2009;154:807–813. doi: 10.1016/j.jpeds.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shoham DA, Durazo-Arvizu R, Kramer H, et al. Sugary soda consumption and albuminuria: results from the National Health and Nutrition Examination Survey, 1999–2004. PLoS ONE. 2008;3:e3431. doi: 10.1371/journal.pone.0003431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanchez-Lozada LG, Tapia E, Bautista-Garcia P, et al. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am J Physiol. 2008;294:F710–F718. doi: 10.1152/ajprenal.00454.2007. [DOI] [PubMed] [Google Scholar]
- 52.Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol. 2006;290:F625–F631. doi: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 53.Lin JL, Ho HH, Yu CC. Chelation therapy for patients with elevated body lead burden and progressive renal insufficiency. A randomized, controlled trial. Ann Int Med. 1999;130:7–13. doi: 10.7326/0003-4819-130-1-199901050-00003. [DOI] [PubMed] [Google Scholar]
- 54.Inglis JA, Henderson DA, Emmerson BT. The pathology and pathogenesis of chronic lead nephropathy occurring in Queensland. J Pathol. 1978;124:65–76. doi: 10.1002/path.1711240202. [DOI] [PubMed] [Google Scholar]
- 55.Roncal C, Mu W, Reungjui S, et al. Lead, at low levels, accelerates arteriolopathy and tubulointerstitial injury in chronic kidney disease. Am J Physiol. 2007;293:F1391–F1396. doi: 10.1152/ajprenal.00216.2007. [DOI] [PubMed] [Google Scholar]
- 56.Chang FM, Chow SN, Huang HC, et al. The placental transfer and concentration difference in maternal and neonatal serum uric acid at parturition: comparison of normal pregnancies and gestosis. Biol Res Pregnancy Perinatol. 1987;8:35–39. [PubMed] [Google Scholar]
- 57.Franco MC, Christofalo DM, Sawaya AL, et al. Effects of low birth weight in 8- to 13-year-old children: implications in endothelial function and uric acid levels. Hypertension. 2006;48:45–50. doi: 10.1161/01.HYP.0000223446.49596.3a. [DOI] [PubMed] [Google Scholar]
- 58.Park B, Park E, Cho SJ, et al. The association between fetal and postnatal growth status and serum levels of uric acid in children at 3 years of age. Am J Hypertens. 2009;22:403–408. doi: 10.1038/ajh.2009.12. [DOI] [PubMed] [Google Scholar]
- 59.Feig DI, Nakagawa T, Karumanchi SA, et al. Hypothesis: Uric acid, nephron number, and the pathogenesis of essential hypertension. Kidney Int. 2004;66:281–287. doi: 10.1111/j.1523-1755.2004.00729.x. [DOI] [PubMed] [Google Scholar]
- 60.Schaffer P, Gombos E, Meichelbeck K, et al. Childhood course of renal insufficiency in a family with a uromodulin gene mutation. Pediatr Nephrol. 2010;25:1355–1360. doi: 10.1007/s00467-009-1436-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fairbanks LD, Cameron JS, Venkat-Raman G, et al. Early treatment with allopurinol in familial juvenile hyerpuricaemic nephropathy (FJHN) ameliorates the long-term progression of renal disease. QJM. 2002;95:597–607. doi: 10.1093/qjmed/95.9.597. [DOI] [PubMed] [Google Scholar]
- 62.Kottgen A, Glazer NL, Dehghan A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nature Genetics. 2009;41:712–717. doi: 10.1038/ng.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han J, Liu Y, Rao F, et al. Common genetic variants of the human UMOD gene are functional on transcription and predict plasma uric acid in two distinct populations. Kidney Int. 2013 doi: 10.1038/ki.2012.449. in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McAdams-Demarco MA, Maynard JW, et al. A urate gene-by-diuretic interaction and gout risk in participants with hypertension: results from the ARIC study. Ann Rheum Dis. doi: 10.1136/annrheumdis-2011-201186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lai JY, Atzmon G, Melamed ML, et al. Family history of exceptional longevity is associated with lower serum uric acid levels in Ashkenazi Jews. J Am Geriatr Soc. 2012;60:745–750. doi: 10.1111/j.1532-5415.2012.03902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Siu YP, Leung KT, Tong MK, et al. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51–59. doi: 10.1053/j.ajkd.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 67.Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5:1388–1393. doi: 10.2215/CJN.01580210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Momeni A, Shahidi S, Seirafian S, et al. Effect of allopurinol in decreasing proteinuria in type 2 diabetic patients. Iran J Kidney Dis. 2010;4:128–132. [PubMed] [Google Scholar]
- 69.Wang H, Wei Y, Kong X, et al. Effects of urate-lowering therapy in hyperuricemia on slowing the progression of renal function: a meta-analysis. J Ren Nutr. 2013 doi: 10.1053/j.jrn.2012.08.005. (in press) [DOI] [PubMed] [Google Scholar]
- 70.Ito S, Naritomi H, Ogihara T, et al. Impact of serum uric acid on renal function and cardiovascular events in hypertensive patients treated with losartan. Hypertens Res. 2012;35:867–873. doi: 10.1038/hr.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu MA, Sanchez-Lozada LG, Johnson RJ, et al. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens. 2010;28:1234–1242. [PubMed] [Google Scholar]
- 72.Saito I, Saruta T, Kondo K, et al. Serum uric acid and the renin-angiotensin system in hypertension. J Am Geriatr Soc. 1978;26:241–247. doi: 10.1111/j.1532-5415.1978.tb02396.x. [DOI] [PubMed] [Google Scholar]
- 73.Perlstein TS, Gumieniak O, Hopkins PN, et al. Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney Int. 2004;66:1465–1470. doi: 10.1111/j.1523-1755.2004.00909.x. [DOI] [PubMed] [Google Scholar]
- 74.Talaat KM, El-Sheikh AR. The effect of mild hyperuricemia on urinary transforming growth factor beta and the progression of chronic kidney disease. Am J Nephrol. 2007;27:435–440. doi: 10.1159/000105142. [DOI] [PubMed] [Google Scholar]
- 75.Roncal CA, Reungjui S, Sanchez-Lozada LG, et al. Combination of captopril and allopurinol retards fructose-induced metabolic syndrome. Am J Nephrol. 2009;30:399–404. doi: 10.1159/000235731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miao Y, Ottenbros SA, Laverman GD, et al. Effect of a reduction in uric acid on renal outcomes during losartan treatment: a post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the Angiotensin II Antagonist Losartan Trial. Hypertension. 2011;58:2–7. doi: 10.1161/HYPERTENSIONAHA.111.171488. [DOI] [PubMed] [Google Scholar]
- 77.Smink PA, Bakker SJ, Laverman GD, et al. An initial reduction in serum uric acid during angiotensin receptor blocker treatment is associated with cardiovascular protection: a post hoc analysis of the RENAAL and IDNT trials. J Hypertens. 2012;30:1022–1028. doi: 10.1097/HJH.0b013e32835200f9. [DOI] [PubMed] [Google Scholar]
- 78.Jung JW, Song WJ, Kim YS, et al. HLA-B58 can help the clinical decision on starting allopurinol in patients with chronic renal insufficiency. Nephrol Dial Transplant. 2011;26:3567–3572. doi: 10.1093/ndt/gfr060. [DOI] [PubMed] [Google Scholar]
- 79.Naoyuki K, Shin F, Toshikazu H, et al. An allopurinol-controlled, multicenter, randomized, open-label, parallel between-group, comparative study of febuxostat (TMX-67), a non-purine-selective inhibitor of xanthine oxidase, in patients with hyperuricemia including those with gout in Japan: phase 2 exploratory clinical study. J Clin Rheumatol. 2011;17:S44–S49. doi: 10.1097/RHU.0b013e31821d352f. [DOI] [PubMed] [Google Scholar]
- 80.Schumacher HR, Jr., Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59:1540–1548. doi: 10.1002/art.24209. [DOI] [PubMed] [Google Scholar]
- 81.Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 2010;12:R63. doi: 10.1186/ar2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beck LH. Requiem for gouty nephropathy. Kidney Int. 1986;30:280–287. doi: 10.1038/ki.1986.179. [DOI] [PubMed] [Google Scholar]