

Abstract
BACKGROUND
A disintegrin and metalloprotease 17 (ADAM17) is a membrane-spanning metalloprotease overexpressed in various cardiovascular diseases such as hypertension and atherosclerosis. However, little is known regarding the regulation of ADAM17 expression in the cardiovascular system. Here, we test our hypothesis that angiotensin II induces ADAM17 expression in the vasculature.
METHODS
Cultured vascular smooth muscle cells were stimulated with 100nM angiotensin II. Mice were infused with 1 μg/kg/minute angiotensin II for 2 weeks. ADAM17 expression was evaluated by a promoter–reporter construct, quantitative polymerase chain reaction, immunoblotting, and immunohistochemistry.
RESULTS
In vascular smooth muscle cells, angiotensin II increased ADAM17 protein expression, mRNA, and promoter activity. We determined that the angiotensin II response involves hypoxia inducible factor 1α and a hypoxia responsive element. In angiotensin II–infused mice, marked induction of ADAM17 and hypoxia inducible factor 1α was seen in vasculatures in heart and kidney, as well as in aortae, by immunohistochemistry.
CONCLUSIONS
Angiotensin II induces ADAM17 expression in the vasculatures through a hypoxia inducible factor 1α–dependent transcriptional upregulation, potentially contributing to end-organ damage in the cardiovascular system.
Keywords: blood pressure, epidermal growth factor receptor, hypertension, renin–angiotensin system, signal transduction, tumor necrosis factor α converting enzyme, vascular biology.
A disintegrin and metalloprotease 17 (ADAM17), also called tissue necrosis factor α (TNFα)–converting enzyme, is a membrane-spanning metalloprotease implicated in the regulatory shedding of cell surface proteins, which include TNFα and the ligands for epidermal growth factor (EGF) receptors.1 It has been documented that trans-activation of EGF receptor plays critical roles for many cellular functions mediated through multiple G protein–coupled receptors.1 We have demonstrated that heparin-binding EGF-like growth factor (HB-EGF) shedding through ADAM17 activation is responsible for the EGF receptor transactivation and subsequent hypertrophy induced by angiotensin II (Ang II) in cultured vascular smooth muscle cells (VSMCs).2
Interestingly, enhanced ADAM17 expression has recently been demonstrated in animal models for and humans with various types of cardiovascular diseases, including hypertension,3 atherosclerosis,4 and abdominal aortic aneurysm.5 We have shown that ADAM17 expression was enhanced in neointima of rat carotid arteries upon angioplasty. Gene transfer of dominant-negative ADAM17 prevented the neointima formation.6 Moreover, human ADAM17 single nucleotide polymorphisms linked to higher risks for cardiovascular mortality have been reported.7 Although these studies suggest that a treatment to reduce ADAM17 activity or its expression may be beneficial to prevent cardiovascular diseases, little is known regarding the regulatory mechanism of ADAM17 expression in the cardiovascular system. In this study, we have tested our hypothesis that Ang II is a key positive regulator of ADAM17 expression in the vasculature and elucidated the molecular mechanism by which Ang II induces ADAM17 in VSMCs.
METHODS
Reagents
Ang II was purchased from Sigma-Aldrich (St. Louis, MO) and Bachem (Torrance, CA). A selective AT1 receptor antagonist, RNH6270, was a gift from Sankyo Pharmaceutical (Tokyo, Japan). A hypoxia inducible factor 1α (HIF-1α) inhibitor, CAS 934593-90-5, was purchased from Santa Cruz Biotechnology (Dallas, TX). Antibodies for immunoblotting and immunohistochemistry against ADAM17 were purchased from Santa Cruz Biotechnology (sc-13973) and Abcam (No. 39163; Cambridge, MA), respectively. Antibodies for immunoblotting and immunohistochemistry against HIF-1α were purchased from Novus (NB100-123 and NB100-134, respectively; Littleton, CO). Antibodies for immunoblotting and immunohistochemistry against Tyr1068-phosphorylated EGF receptor were purchased from Invitrogen (44788G; Grand Island, NY) and Cell Signaling (No. 2236; Danvers, MA), respectively. Antibodies against total EGF receptor and GAPDH were purchased from Santa Cruz Biotechnology (sc-03) and Millipore (MRB374; Billerica, MA), respectively.
Cell culture
VSMCs were prepared from thoracic aorta of male Sprague-Dawley rats by the explant method as described previously.8 VSMCs were subcultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum, penicillin, and streptomycin. Cells from passages 3–10 at 80%–90% confluence were made quiescent by incubation with serum-free medium for 2–3 days. To avoid any potential phenotypic alteration, VSMCs were renewed every 2–3 months, and VSMCs from frozen stock were never used. The results were confirmed in at least 2 distinct cell preparations.
Immunoblotting
Immunoblotting was performed as previously described.8 Quiescent VSMCs grown on 6-well plates were stimulated for specified durations. The reaction was terminated by the replacement of medium with 100 μl of 1× sodium dodecyl sulfate sample buffer. Forty microliters of the cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrophoretically transferred to a nitrocellulose membrane. The membranes were then exposed to primary antibodies overnight at 4 °C. After incubation with the peroxidase linked secondary antibody for 1 hour at room temperature, immunoreactive proteins were visualized by a chemiluminescence reaction kit.
Promoter assay
pTACE-Luc vectors encoding murine ADAM17 promoter (−2,340 to −1bp), its 5’ deletion constructs, or hypoxia responsive element (HRE) mutants located at H4 site were constructed as reported previously.9 A luciferase reporter gene driven by 3 repeats of the HRE retrieved from the erythropoietin gene, HRE3-Luc, was generously provided by Dr. Steven McKnight (University of Texas). pSEAP2-control vector (Clontech, Mountain View, CA) was used to normalize transfection efficiency. VSMC (2×106 cells) were transfected by Amaxa Nucleofector program U-25 in basic SMC NucleofectorT solution (Lonza, Basel, Switzerland) containing 5 μg pTACE-Luc or HRE3-Luc vector and 1 μg pSEAP2-control vector. Twenty-four hours after the transfection, cells were incubated with serum-free Dulbecco’s modified Eagle medium for 24 hours. The cells were then stimulated with 100nM Ang ll for 24 hours. Cellular luciferase activity was measured by Luciferase Reporter Gene Assay kit (Roche, Indianapolis, IN), and secreted alkaline phosphatase activity in the medium was measured as described.8
HB-EGF shedding assay
Ang II-mediated HB-EGF shedding was quantified in VSMCs using an alkaline-phosphatase–tagged HB-EGF encoding adenovirus (HB-EGF-ALP) as previously described.8 After serum starvation, VSMCs were infected with adenoviral vectors encoding HB-EGF-ALP. Two days after infection, cells were incubated with or without 100nM Ang II for 24 hours. After the medium change, secreted HB-EGF was measured for 1 hour using a colorimetric alkaline phosphatase activity assay as previously described.8
Quantitative real-time polymerase chain reaction
Total RNA was extracted from VSMCs using TRIzol reagent (Invitrogen). cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo, Waltham, MA). Quantitative real-time polymerase chain reaction (qPCR) was performed with SYBR Green qPCR Master Mix (Fermentas, Waltham, MA) as described previously.10 mRNA abundance was calculated by normalization to ribosome 18S. The primers used were ADAM17: forward ACTCTGAGGACAGTTAACCAAACC, reverse AGTAAAAGGAGCCAATACCACAAG; Ribosome 18S: forward CTCTCTTCCACAGGAGGCCTACACG, reverse AGGCTATTTTCCGCCGCCCATC.
Ang II infusion and immunohistochemistry
All animal procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85-23, revised 1996) and Temple University. Eight-week-old male C57Bl/6 mice (SAGE Labs, St. Louis, MO) were infused with either saline or Ang II (1 μg/kg/minute) for 14 days by osmotic minipump. The heart, kidney, and aorta were extracted, fixed, and used for immunohistochemical studies as described previously.5,6
Statistical analysis
Data are presented as means ± SEMs. Student t test or paired t test was performed as appropriate for 2-group comparisons. Values of P < 0.05 were considered to be statistically significant.
RESULTS
To examine whether Ang II increases ADAM17 expression in VSMCs, the cells were stimulated with 100nM Ang II for 1–24 hours. As shown in Figure 1a, Ang II increased ADAM17 protein expression at 2–24 hours of stimulation. Pretreatment of AT1 receptor blocker RNH6270 (10 μM; 30 minutes) prevented ADAM17 induction by 100nM Ang II in VSMCs (Supplementary Figure S1a). Ang II also increased ADAM17 mRNA at 8 and 24 hours in VSMCs (Supplementary Figure S1b). The ADAM17 induction by Ang II was associated with enhanced ADAM17 activation as assessed by HB-EGF shedding and EGFR activation (Supplementary Figure S1c,d).
Figure 1.
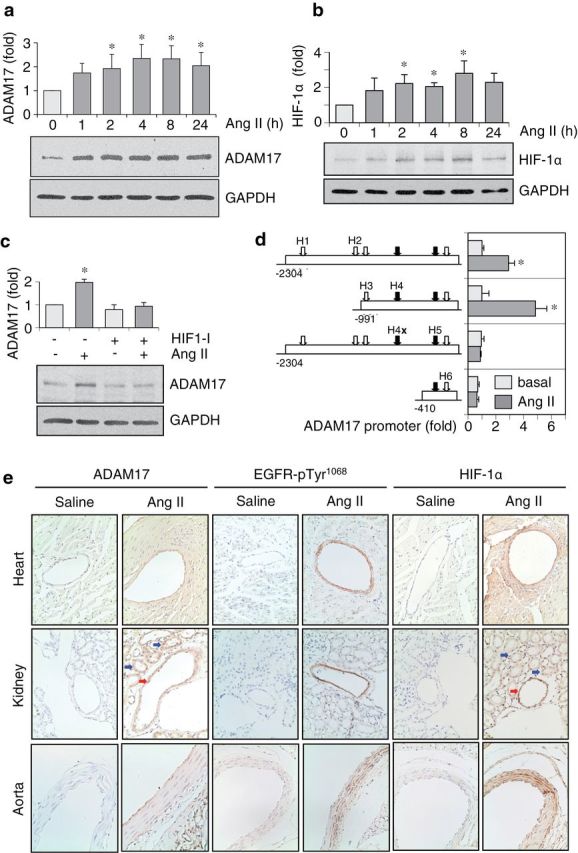
Angiotensin II (Ang II) induces a disintegrin and metalloprotease 17 (ADAM17) expression through hypoxia inducible factor 1α (HIF-1α). (a,b) Rat vascular smooth muscle cells (VSMCs) were stimulated with 100nM of Ang II for 1–24 hours, and expression of ADAM17, HIF-1α, and GAPDH was determined by immunoblotting. The ADAM17 antibody mainly detected ADAM17 precursor at just above 102kDa MW marker in whole VSMC lysates, which was quantified in the bar graph. Representative blots and densitometry analysis of the data (mean ± SEM; *P < 0.05 compared with basal control) are shown from 3 separate experiments. (c) Rat VSMCs pretreated with or without HIF-1α inhibitor CAS 934593-90-5 (20 μM) for 1 hour were stimulated with 100nM of Ang II for 4 hours, and expression of ADAM17 and GAPDH was determined by immunoblotting. Representative blots and densitometry analysis of the data (mean ± SEM; *P < 0.05 compared with basal control) are shown from 3 separate experiments. (d) Upon Amaxa transfection of ADAM17 luciferase promote–reporter constructs (−2,340 −1; and its mutants)9 for 48 hours, VSMCs were stimulated with 100nM of Ang II for 24 hours, and luciferase activity was determined (mean ± SEM; n = 3; *P < 0.05 compared with basal control). The arrows denote 6 HIF-1 binding sites, with black arrows denoting 2 hypoxia responsive elements composed of one HIF-1 binding sequence and one HIF-1 ancillary sequence. H4x: HIF-1 binding site mutation at position H4. (e) Eight-week-old C57Bl/6 mice were infused with Ang II (1 μg/kg/minute) or saline for 2 weeks. Tissues were immunostained with ADAM17 antibody, HIF-1α antibody, or EGFR-pY1068 antibody as indicated. Representative images are shown from 3 mice for each. The red arrows indicate renal arteries. The blue arrows denote renal tubules.
A recent study demonstrated that TNFα stimulated ADAM17 transcription through HIF-1α and HRE located in the ADAM17 promoter.9 Ang II increased HIF-1α protein expression in VSMC at 2–8 hours (Figure 1b). An HIF-1α inhibitor attenuated Ang II-induced ADAM17 protein expression (Figure 1c), whereas the inhibitor suppressed EGF receptor expression regardless of Ang II stimulation (Supplementary Figure S1e). To elucidate an involvement of HRE for ADAM17 induction by Ang II in VSMCs, effects of Ang II on several ADAM17 promoter luciferase constructs were examined. Ang II stimulated ADAM17 promoter activity with a 5-fold induction observed with the −991 construct. However, Ang II–stimulated ADAM17 promoter activity was dependent upon sequences in the region −991 to –410, most notably the HRE at position H4 (Figure 1d). In fact, mutation of the H4 site completely abrogated the Ang II response. To test the ability of Ang II to stimulate HRE-dependent transcription, VSMCs were transfected with a luciferase reporter gene driven by 3 repeats of the HRE retrieved from the erythropoietin gene, HRE3-Luc. Ang II increased this luciferase activity in VSMCs (Supplementary Figure S1f). These data suggest that Ang II stimulates ADAM17 transcription through HIF-1α and an HRE located within the −991 to −410 region of the ADAM17 promoter.
To examine whether Ang II induces vascular ADAM17 expression in vivo, C57Bl/6 mice were infused with Ang II (1 μg/kg/minute) or control saline for 2 weeks. We confirmed that Ang II induced end-organ damage in heart and kidney associated with vascular medial hypertrophy and perivascular fibrosis (data not shown). Enhanced ADAM17 staining was seen in coronary arteries, renal arteries, and aortae in mice infused with Ang II (Figure 1e). Marked EGF receptor Tyr1068 phosphorylation was detected in these arteries with Ang II infusion. Moreover, strong HIF-1α staining was observed in the arteries with Ang II infusion.
DISCUSSION
It has been recently reported that Ang II infusion stimulated ADAM17 induction in abdominal aorta of apoE-/- mice.11 In our study, we have demonstrated that Ang II induces ADAM17 expression in VSMCs through HIF-1α/HRE-dependent transcriptional upregulation. Although induction of HIF-1α mRNA and protein has been observed in VSMCs stimulated with Ang II12,13 and in rodent aorta14 infused with Ang II, limited information is available regarding the target gene(s) regulated by the HIF-1α induction. In aorta, Ang II appears to generate vascular endothelial growth factor through HIF-1α induction.14 Silencing VSMC HIF-1α protected against vascular remodeling in mice infused with Ang II.15 Our data indicate that ADAM17 is another HIF-1α target gene induced by Ang II in heart, kidney, and aorta, suggesting a potential contribution of HIF-1α–dependent ADAM17 induction in cardiovascular pathophysiology. Although induction of ADAM17, as well as HIF-1α, by Ang II were mainly observed in the vasculature, these proteins were also increased in cardiac myocytes and renal tubules. By contrast, the EGF receptor activation by Ang II was more prominent in the vasculature. This could be because of differences in ADAM17 activity or expressions of ADAM17 substrates and EGF receptors. However, it seems difficult to interpret the involvement of HIF-1α in Ang II–induced sustained EGFR activation in VSMCs by the HIF-1α inhibitor because the inhibitor reduced EGFR expression. Inclusion of genetic tools such as VSMC HIF-1α–deficient mice15 may solve this limitation of the study. In addition, potential contribution of hypertension in ADAM17 induction by Ang II in vivo remains to be elucidated.
The mechanism by which Ang II increases ADAM17 protein expression may not be limited to transcriptional upregulation. As noted, there is a discrepancy in the induction time course of HIF-1α and ADAM17 with Ang II stimulation. The early increase in ADAM17 protein levels could be due to effects on post-translational fate of ADAM17 protein, such as enhanced trafficking16 or suppression of degradation.17 In addition, ADAM17 3’UTR has conserved regions targeted by a microRNA, miR-145, whose expression is markedly reduced in a synthetic phenotype of VSMCs.18 Upon induction, ADAM17 activation by Ang II requires its maturation,16 as well as regulatory phosphorylation.19 Further research is required to determine whether Ang II also increases ADAM17 mRNA stability, suppresses protein degradation, and enhances its trafficking.
Our data presented here may suggest the notion that VSMC ADAM17 could provide a novel therapeutic target toward preventing cardiovascular diseases associated with the enhanced renin angiotensin system. Silencing of ADAM17 in VSMCs attenuated rapid EGF receptor transactivation by Ang II in VSMCs,19 and induction of ADAM17 by Ang II may lead to sustained EGF receptor activation. Conditional EGF receptor null mice have been used to prove the critical role of EGF receptor transactivation in hypertensive renal damage induced by Ang II infusion.20 It has been shown that EGF receptor transactivation partially contributes to HIF-1α–dependent transcription in VSMCs.13 Therefore, the induction of ADAM17 by Ang II likely promotes a feed-forward loop of ADAM17 activation and subsequent EGF receptor activation under a variety of pathophysiological conditions, including hypoxia/ischemia, leading to end-organ damage in the cardiovascular system.
SUPPLEMENTARY MATERIAL
Supplementary materials are available at American Journal of Hypertension (http://ajh.oxfordjournals.org).
DISCLOSURE
The authors declared no conflict of interest.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institute of Health grant HL076770 (to S.E.) and by American Heart Association grants 13GRNT17060036 (to S.E.) and 11POST7800000 (to A.B.). T. Obama and T. Takayanagi contributed equally to this work. T. Obama performed in vitro experiments and the revision experiments and contributed to the discussion. T. Takayanagi performed in vitro experiments and animal studies and contributed to the discussion. T. Kobayashi and A. Bourne performed animal studies. K. J. Elliott performed in vitro experiments and wrote/edited the paper. M. Charbonneau provided expertise in ADAM17 promoter and contributed to the promoter assays. C. M. Dubois provided expertise in ADAM17 promoter, contributed to the discussion, and edited the paper before submission. S. Eguchi conceived, designed, and coordinated the research plan and wrote/edited the paper.
REFERENCES
- 1. Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol 2006; 291:C1–C10. [DOI] [PubMed] [Google Scholar]
- 2. Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, Nakashima H, Eguchi K, Eguchi S. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol 2006; 26:e133–e137. [DOI] [PubMed] [Google Scholar]
- 3. Wang X, Oka T, Chow FL, Cooper SB, Odenbach J, Lopaschuk GD, Kassiri Z, Fernandez-Patron C. Tumor necrosis factor-alpha-converting enzyme is a key regulator of agonist-induced cardiac hypertrophy and fibrosis. Hypertension 2009; 54:575–582. [DOI] [PubMed] [Google Scholar]
- 4. Canault M, Peiretti F, Kopp F, Bonardo B, Bonzi MF, Coudeyre JC, Alessi MC, Juhan-Vague I, Nalbone G. The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis 2006; 187:82–91. [DOI] [PubMed] [Google Scholar]
- 5. Takayanagi T, Crawford KJ, Kobayashi T, Obama T, Tsuji T, Elliott KJ, Hashimoto T, Rizzo V, Eguchi S. Caveolin-1 is critical for abdominal aortic aneurysm formation induced by angiotensin II and inhibition of lysyl oxidase. Clin Sci 2014; 126:785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takaguri A, Kimura K, Hinoki A, Bourne AM, Autieri MV, Eguchi S. A disintegrin and metalloprotease 17 mediates neointimal hyperplasia in vasculature. Hypertension 2011; 57:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morange PE, Tregouet DA, Godefroy T, Saut N, Bickel C, Rupprecht HJ, Lackner K, Barbaux S, Poirier O, Peiretti F, Nalbone G, Juhan-Vague I, Blankenberg S, Tiret L. Polymorphisms of the tumor necrosis factor-alpha (TNF) and the TNF-alpha converting enzyme (TACE/ADAM17) genes in relation to cardiovascular mortality: the AtheroGene study. J Mol Med (Berl) 2008; 86:1153–1161. [DOI] [PubMed] [Google Scholar]
- 8. Takaguri A, Shirai H, Kimura K, Hinoki A, Eguchi K, Carlile-Klusacek M, Yang B, Rizzo V, Eguchi S. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin II. J Mol Cell Cardiol 2011; 50:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J Biol Chem 2007; 282:33714–33724. [DOI] [PubMed] [Google Scholar]
- 10. Takayanagi T, Bourne AM, Kimura K, Takaguri A, Elliott KJ, Eguchi K, Eguchi S. Constitutive stimulation of vascular smooth muscle cells by angiotensin II derived from an adenovirus encoding a furin-cleavable fusion protein. Am J Hypertens 2012; 25:280–283. [DOI] [PubMed] [Google Scholar]
- 11. Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, Dyer JS, Maegdefessel L, Dalman RL, Tsao PS. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiol Genomics 2011; 43:993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem 2000; 275:26765–26771. [DOI] [PubMed] [Google Scholar]
- 13. Lauzier MC, Page EL, Michaud MD, Richard DE. Differential regulation of hypoxia-inducible factor-1 through receptor tyrosine kinase transactivation in vascular smooth muscle cells. Endocrinology 2007; 148:4023–4031. [DOI] [PubMed] [Google Scholar]
- 14. Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension 2004; 44:264–270. [DOI] [PubMed] [Google Scholar]
- 15. Imanishi M, Tomita S, Ishizawa K, Kihira Y, Ueno M, Izawa-Ishizawa Y, Ikeda Y, Yamano N, Tsuchiya K, Tamaki T. Smooth muscle cell-specific Hif-1alpha deficiency suppresses angiotensin II-induced vascular remodelling in mice. Cardiovasc Res in press. 10.1093/cvr/cvu061. [DOI] [PubMed] [Google Scholar]
- 16. Lichtenthaler SF. Cell biology. Sheddase gets guidance. Science 2012; 335:179–180. [DOI] [PubMed] [Google Scholar]
- 17. Santiago-Josefat B, Esselens C, Bech-Serra JJ, Arribas J. Post-transcriptional up-regulation of ADAM17 upon epidermal growth factor receptor activation and in breast tumors. J Biol Chem 2007; 282:8325–8331. [DOI] [PubMed] [Google Scholar]
- 18. Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res 2009; 105:158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elliott KJ, Bourne AM, Takayanagi T, Takaguri A, Kobayashi T, Eguchi K, Eguchi S. ADAM17 silencing by adenovirus encoding miRNA-embedded siRNA revealed essential signal transduction by angiotensin II in vascular smooth muscle cells. J Mol Cell Cardiol 2013; 62:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC. EGFR signaling promotes TGFbeta-dependent renal fibrosis. J Am Soc Nephrol 2012; 23:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.