

Abstract
Non-alcoholic fatty liver disease (NAFLD) represents the most common liver disorder in western countries and it is commonly associated with obesity and progression of the metabolic syndrome. Comprehending a wide spectrum of pathologic features, it is currently well recognized that a key point for the integrity of hepatocyte functionality in NAFLD is the progression from simple steatosis to non-alcoholic steatohepatitis (NASH). Indeed, activation of the innate immune system in response to hepatic metabolic stresses represents a central process that determines the evolution and the reversibility of liver damage. Despite of the burden of studies published in recent years, it is still intriguingly unclear how accumulation of lipids in hepatocytes triggers the activation of the inflammatory response leading to the recruitment of infiltrating cells of extra-hepatic origins. In this review we offer a general view on recent advances pointing out how different classes of lipids are able to specifically affect hepatocytes functionality and survival, thus differently influencing the organization of the hepatic immune response. On the other hand, we gathered recent studies intending to illustrate the basic mechanisms through which several non-parenchymal hepatic and extra-hepatic cell populations get activated in response to lipids. Finally, we indicate latter findings proposing how the immune system majorly contributes to the progression of NASH.
Keywords: Hepatocyte, free fatty acids (FFA), lipotoxicity, toll like receptors (TLRs), lymphocytes
Introduction
The evolution of dietary habits together with drastic lifestyle modifications strongly affected modern health care of the western society during the last decades. In fact, the increasing prevalence of obesity worldwide, the metabolic syndrome, cardiovascular diseases, diabetes and related pathologies strongly correlate with the enhanced consumption of high caloric diets and sedentary life in combination with genetic factors (1). Non-alcoholic fatty liver disease (NAFLD) represents a typical feature in the pathophysiology of the metabolic syndrome and it has been demonstrated to be tightly associated with the progression of type II diabetes (2). NAFLD comprehends a wide spectrum of disorders ranging from simple steatosis, identified by an excessive accumulation of triglycerides (TG) in the liver exceeding 5% of total organ mass, to non-alcoholic steatohepatitis (NASH) often progressing to fibrosis (1) and even the development of hepatocellular carcinoma. Beyond viral hepatitis, NAFLD constitutes the second most common type of liver disease in the world (3) and it is estimated that its prevalence in the US overcomes one third of the population. Also in European countries its prevalence is rising now up to 20% and 30% of the adult population (4). Considering the central metabolic role of the liver in maintaining the energetic balance of the organism independently of its nutritional status, it is now well-established that perturbations of the balance between nutrients up-take and energy expenditure strongly affect the accumulation of lipids in hepatocytes. Therefore, in response to overfeeding and decreased physical activity, the liver, like other organs, accumulates TG. However, despite TGs represent the most abundant form of lipid energy stores, many other lipid-derivatives such as free fatty acids (FFA), diacylglycerol (DAG), cholesterol and ceramides, accumulate in hepatocytes while triggering different biological responses. Moreover, other metabolic determinants have shown to influence the lipid storage capability of hepatocytes as, for example, de novo lipogenesis induced by carbohydrates assumption or the hepatic TG efflux in form of VLDL but also from the lipid catabolic activity of hepatic mitochondria (5). As previously mentioned, simple steatosis often evolves to NASH with a pathogenetic picture characterized by diffuse hepatocyte death and inflammatory cells infiltrating from different origins. How the hepatic accumulation of TG can lead to a necro-inflammatory condition still remains unknown. In this direction, overproduction of reactive oxygen species (ROS) has been classically proposed as a potential link between fat content increase and generation of a pro-inflammatory milieu (6), but convincing evidences for an indispensable role of free radicals in this context are still missing. Furthermore, growing proofs indicate simple TG accumulation in cytosolic lipid droplets as a cellular mechanism of defense from fatty acid hepato-toxicity rather than a process directly responsible for cell damage (7). Indeed, recent in vitro and in vivo studies support the hypothesis that FFAs not esterified and not compartmentalized in lipid droplets, alone or in combination with other lipid metabolites, are able to induce irreversible cell damage and trigger pro-inflammatory signaling pathways (lipotoxicity) (8,9). Molecular mechanisms through which these different molecules modulate the expression of genes involved in cell death or cell cycle as well as of several pro-inflammatory mediators are still far from being clear. However, a key role for the innate immune system in the progression of NASH is gradually consolidating (10,11). In fact, whereas the action of toxic lipid metabolites on hepatocytes might induce cell damage, FFAs and fragments derived from injured cells are able to activate and mobilize resident liver macrophages Kupffer cells (KCs) as well as dendritic cells. This leads to further recruitment of neutrophils and lymphocytes orchestrating the development of the destructive inflammatory response.
Lipids and hepatotoxicity
Triglycerides (TG) and lipid droplets
TGs, which represent the major constituent of lipid droplets, are the most important storage of fatty acids (FAs), thereby providing energy to the liver and other tissues. They originate by two processes that depend on the esterification of FAs directly synthesized by the cell (de novo lipogenesis) or re-esterification of FAs coming from the circulation (lipid up-take) (12). Increased TG accumulation and expansion is commonly associated with metabolic disorders such as obesity, and diabetes, usually characterized by hepatic steatosis. Anyway, it is not understood whether exceeding TGs or insulin resistance develop first (13). Pharmacological or genetic inhibition of enzymes involved in TG synthesis, such as glycerol-3-phosphate acyltransferase (GPAT) or acyl-coenzymeA-diacylglycerol acyltransferase (DGAT) members, have been shown to reduce hepatic steatosis (14,15). However, despite of the classical notion that TG might represent a first “hit” triggering the initial phases of NASH, it is recently getting more evident that TG per se are not deleterious for hepatocytes. Interestingly, other in vitro and in vivo experimental studies show that inhibiting the hepatic TG synthesis results in an amelioration of hepatic steatosis but exacerbate liver cell damage by an increased intra-hepatic accumulation of FFAs (8,16). Taken together these latter observations suggest a possible protective role for TGs against FFA mediated cyto-toxicity (Figure 1). On the same line, lipid droplets biology recently caught the attention of many investigators regarding its function in NASH (17), with a particular focus on the role of lipid droplets-associated proteins with lipase activity. For instance, mutations of the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene, a lipid droplets-associated protein able to regulate TG hydrolysis, have shown to have a high correlation grade with NAFLD occurrence in humans (18). Furthermore, members of the perilipins family (PLIN), in particular PLIN1 and -2, were found to be increased with hepatic steatosis and associated with lipid droplets formation (19). Of note, PLIN-2 overexpression was reported to decrease TG hydrolysis thereby resulting in increased steatosis (20). Therefore, it could also be important to note that accumulation of DAG, has been shown to trigger PKC-dependent signal transduction pathways responsible for hepatic insulin resistance and possibly also lipotoxicity (21). This accumulation can either be the result of lipase activity or is newly generated by esterification of increased FFA intracellular levels.
Figure 1.
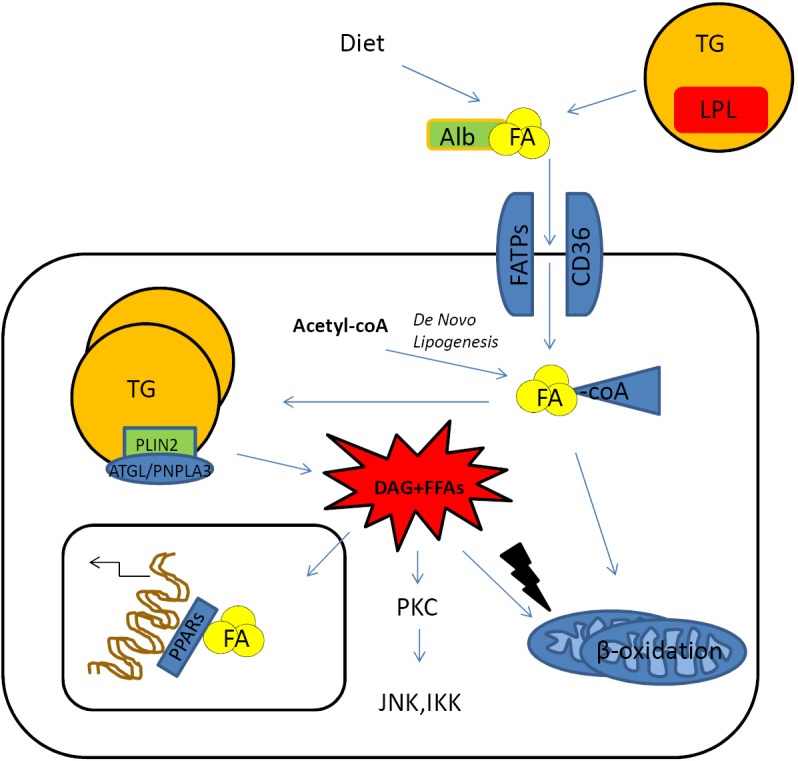
Hepatic up-take of FAs. FAs coming from diet or mobilized from peripheral storage places are usually conjugated with albumin that allows to enter the hepatocyte through specific transmembrane proteins, fatty acid transporter protein families or CD36/FAT transporter. Inside the cell, FAs originating from liponeogenesis, lipolysis or up-taken from the circulation are shuttled to several cell compartments according to the metabolic requirements. The bio-products of lipolysis such as diacylglycerol and FFAs might exert cytotoxic. Effects triggering stress signals lead to the activation of pro-inflammatory pathways (JNKs, IKKs) or increased ROS production in the mitochondrial compartment. FA, fatty acid; TG, triglycerides; PLIN, perilipins family; DAG, diacylglycerol; FFAs, free FAs; JNK, Jun-(N)-terminal kinase; ROS, reactive oxygen species.
Free fatty acids (FFAs) and lipotoxicity: saturated vs. unsaturated fatty acids (FAs)
The cytotoxic effects of circulating FFAs could be identified as the culprit of hepatocyte dysfunction and death observed during NAFLD (22). These free circulating FAs result from increased dietary intake or from mobilization of the peripheral stores in the adipose tissue, or from the partitioning activity of intracellular lipases on TG. Thus they are rather not just a merely accumulation of esterified lipids. It looks like that it is much more the quality of fatty acids than the quantity that impact on the cell functionality. A growing body of evidences indicates that saturated FAs (SFAs) (e.g., palmitate) mediate their effects directly on a cellular level by triggering pro-inflammatory pathways via toll like receptors (TLRs) or death receptor intracellular signals. Through these mechanisms they can also induce mitochondrial alterations responsible for ROS production and apoptosis (lipoapoptosis) (23-26).
Furthermore, FFAs mediate many other metabolic effects by binding nuclear receptors (e.g., PPARs) that regulate the expression of genes involved in lipid anabolism and catabolism (27). They can also indirectly modulate the inflammatory response towards lipid toxicity (28). Most of the lipotoxic activities of lipids, in particular of saturated FFAs, have been shown to develop upon binding to TLRs (mainly TLR4 and TLR2) (29) on hepatocytes and KCs and thereby activating Jun-(N)-terminal kinase (JNK)/IKK dependent pathways leading to the activation of NF-κB and AP-1 transcription factor families (30). They are the major regulators of pro-inflammatory genes, such as tumour necrosis factor-α (TNF-α), IL-1-beta and other chemoattractant mediators, such as MCP-1 (31). Palmitate was also shown to induce the release of the pro-inflammatory chemoattractant IL-8 in hepatocytes through a mechanism involving a synergistic action of NF-κB and AP-1 (32). The orchestration of so many pro-inflammatory signals finally leads to the recruitment of different cell populations that contribute to the amplification of endocrine signals involving several parts of the organism (33,34). Alternatively, FFAs could also be shuttled to the mitochondria in order to get catabolized through beta-oxidation consequently increasing the oxygen free radicals generation and contributing to spread cell damage and activation of macrophages (35). Recently, the activating effects of SFAs on the JNK pathway have been convincingly supported by in vivo investigations. These data show that the JNK cascade, triggered by the kinase MLK3 in response to a SFA enriched diet, not only contributes to the development of hepatic steatosis and insulin resistance but also induces a pro-inflammatory M1 phenotype on bone marrow-derived macrophages (36). In other in vitro studies, palmitate has been reproducibly shown to induce endoplasmatic reticulum (ER) and oxidative stress in hepatocytes (37) and to trigger inflammasome activation in macrophages through TLR2/1 dimerization (38). Interestingly, most of these molecular mechanisms have been reported to play a key role in hepatic insulin resistance usually associated with NAFLD (39). On the contrary, although their contribution to insulin resistance is still controversially debated, unsaturated fatty acids (e.g., oleate, linoleate) seem not to affect the vitality of the cell although they have an impact on TG storage (40).
Finally, FFAs, other than giving origin to DAG and TAG, are also the source of other metabolites, such as ceramides that are synthesized in the ER of hepatocytes using as substrate long-chain SFA (41). Whereas ceramides have been shown to exert a lipotoxic action on pancreatic cells and to participate in mechanisms involved in hepatic insulin resistance (42), direct evidences for a pro-apoptotic role on hepatocytes are still missing (43).
Cholesterol
On the base of clinical studies in humans (44), the supplementation of variable concentrations (0.2-2%) of cholesterol in experimental diets of murine NASH models resulted in acceleration of steatohepatitis development and increased hepatic steatotic grades (45). In fact, other than having a lipotoxic effect on hepatocytes (46), free cholesterol massively activates macrophages and hepatic stellate cells resulting in aggravation of steatohepatitis with worsening of fibrosis (47). Although cholesterol esters represent the major component of the cellular membranes and an important structural constituent of lipid droplets, thereby favoring cyto-protection and cell proliferation, free cholesterol has been shown to exert cytotoxic effects on hepatocytes. In fact, loss of the cholesterol esterifying enzyme ACAT1 was shown to induce TLR4 expression on hepatic stellate cells leading to increased TGF-beta sensitization with consequent aggravation of fibrosis (48). Moreover, cholesterol content in modified lipoproteins has been demonstrated to bind specific receptors on KCs (SR and CD36) thereby increasing the NASH-related inflammatory response (49). Cholesterol can further be stored in lysosomes of KCs and hepatocytes. It can further generate crystal precipitates typical of NASH histology responsible for inflammasome activation (50). Hence, the pharmacological reduction of free cholesterol with atorvastatin in mice resulted in reduced inflammation and liver damage without modifying levels of FFAs (51).
Toll like receptors (TLRs) and the innate immunity during NASH-pathogenesis
The worst consequence of hepatic cellular alterations directly or indirectly generated by an abnormal accumulation of fatty acids is loss of cellular functions leading to irreversible cell damage. In this context, the innate immune system has the capability to respond to specific key molecules, which are released by damaged cells, thus leading to their elimination. Thereby, those metabolic stressors or even dead cells release so-called damage-associated molecular patterns (DAMPs). Intracellular DAMPS include high-mobility group gel box 1 (HMGB1) heat shock proteins, fibrinogen and fibrinonectin and mitochondrial products such as formyl-peptides and mitochondrial DNA. Comparable mechanisms are used to eliminate certain pathogens, indicated as pathogen associated molecular patterns (PAMPs). Interestingly some of their receptors are used redundantly by both PAMPs and DAMPs. In particular, TLRs can be utilized by both pathways in response for instance to HMGB1 and lipopolysaccharides (LPS) (52-54). TLRs are widely expressed within the liver on KCs, hepatocytes, sinusoidal endothelial cells and hepatic stellate cells (55). Functionally, TLRs decode molecular patterns presented on a broad range of pathogens and host molecules. After binding with respective ligands, they trigger both antiviral and inflammatory responses. On a cellular level individual TLRs interact with different combinations of adapter proteins and activate consecutively transcription factors already discussed such as NF-κB, AP-1 (via JNK) and interferon responsive factors (IRF). The intra-cellular transduction molecule MyD88 is shared by almost all TLRs and recruits members of the IL-1 receptor associated kinase family. Therefore, TLR-signaling shows close similarities to IL-1 intracellular signaling, as common adaptor molecules are used.
Similarly, after release from necrotic cells, HMGB1 stimulates KCs and monocytes to produce pro-inflammatory mediators, thereby acting as an endogenous ligand for TLR4 giving possibly rise to the formation of inflammatory complexes with other pattern-related molecules (ssDNA, endotoxin, IL-1β, nucleosomes) (52). Other than being classically involved in acute liver injury, like hepatic ischemia-reperfusion injury (56) and alcoholic liver injury (57), TLR4 is also up-regulated in methionine and choline deficient diet (MCD) steatohepatitis (54) and fructose-induced hepatic steatosis (58). As described above, saturated FFAs represent potential TLR4 ligands (59), indicating this receptor family as an interesting therapeutic target in NASH. Indeed, deletion of either TLR4 or MD2 dampens (but does not completely abolish) necro-inflammatory activity of MCD steatohepatitis, with the most impressive effects exerted against NADPH oxidase expression and activation of inflammatory cells (60). Other studies have identified activation of TLRs 2 and 9 in various experimental models of NAFLD or NASH. In this line, TLR9-deficient mice are protected from steatohepatitis in the CDAA model (61), which is of particular interest, as TLR9 amplifies the inflammatory response of macrophages (62) indicating other potential loops for the perpetuation of inflammation in NASH. TLR5, which is not expressed in the liver, has recently been reported to influence gut flora and being related to the development of obesity, metabolic syndrome, insulin resistance and steatosis (63). Any relevance to NASH has yet to be established, although fascinating and promising indications emerged from the observation that transplanting the altered intestinal flora from TLR5-/- mice to healthy animals resulted in a similar disease phenotype, including (non-inflamed) fatty liver. An interesting, although to date not yet fully resolved, aspect is whether and how the intestinal microbiome itself can contribute to hepatotoxicity. In this regard it was shown that dietary fat intake can promote the absorption of LPS from gut flora (64). LPS translocates into the circulatory system likely either through direct diffusion via intestinal paracellular permeability or through absorption by enterocytes via chylomicron secretion (65). This process can then participate to support inflammatory conditions thus finally contributing to NASH-development.
Fatty acids (FAs)-induced ER-stress
Beyond their negative impact on mitochondrial functionality and their contribution to ROS generation, lipids have been shown to further mine hepatocyte vitality via triggering ER-stress. Accumulation of unfolded proteins within the ER is often observed in cells like hepatocytes that have high rates of protein synthesis. Usually it provokes cellular responses, known as the unfolded protein response (UPR), involving the activation of chaperones that seemingly converge in the activation of three key molecular events (NF-κB, JNK and C/EBP activation). Although detailed information about molecular players are still missing, downstream effects are the recruitment of inflammatory cells, increased phosphorylation of insulin receptor signalling intermediates (which worsens insulin resistance), enhanced lipogenesis and oxidative stress resulting in apoptosis of cells. ER stress contributes to disease progression of NASH and it has been shown that saturated FFAs directly induce a hepatocyte ER stress response, with increased levels of ER stress seen in patients with NAFLD/NASH (66). Furthermore, X-box binding protein-1 (XPB-1), a master regulator of the UPR, was shown to be over-expressed in both mice and men under NASH conditions (67,68). Interestingly, the UPR is tightly linked to the activation of oxidative stress. Nrf2-/- mice displayed more oxidative stress during high fat diet, thus provoking an enhanced UPR and pro-inflammatory environment of the liver. NRF2-dependent gene production physiologically seems to suppress lipogenesis, support mitochondrial function, increase the threshold for the UPR and inflammation and thus enables livers to adapt to dietary-induced oxidative stress (69).
Lipotoxicity and cell death
The final result of lipotoxicity is the death of individual hepatocytes, which undergo apoptosis or necrosis at the cellular level. Apoptosis represents an important connection linking liver injury and fibrosis and can thus be an important driver of inflammation and fibrosis-development during liver disease. Increased levels of apoptosis, however, are indeed a feature of NASH, as they are seen in obese patients with NASH compared with controls (70). Animal and in vitro studies have shown that FFAs sensitize hepatocytes to the cytotoxic effects of death ligands [such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)] and expression of the death receptor Fas (CD95) is higher in liver biopsies of patients with NASH compared with those from patients with simple steatosis or no disease (Figure 2). Murine studies have also demonstrated that FFAs can induce up-regulation of the p53 tumor suppression gene, expression of the TRAIL-receptor (TRAIL-R) and increased levels of JNK as an important regulator of apoptosis that has been shown to promote the development of NASH in mice (71). Another possible mechanism, recently shown to prevent apoptosis during NASH development, is based on the interaction of the hepatocyte growth factor (HGF) receptor c-Met with the FAS-receptor. The pleiotropic growth factor HGF binds to its receptor c-Met and induces its dimerization and phosphorylation. Subsequently, specific intracellular cascades activating PI3K, Ras, and ERK-dependent pathways, responsible for pro-mitogenic and anti-apoptotic events, are induced. The apoptosis-inducing death-receptor Fas is usually bound by c-Met, thus allowing a very tight regulation of apoptosis (72). Interestingly, this control mechanism was shown to be altered during NASH pathogenesis, because under this condition Fas ligand is produced in excess and the physiological inhibition through c-Met is hampered (73). The relevance of this theory could be proven experimentally by the use of mice lacking hepatocellular c-Met, which developed significantly more liver damage after MCD administration (74). In order to investigate the interplay with death receptors signaling in particular (e.g., TNF) and TRAIL, c-Met/Caspase-8 double conditional KO-mice were generated. The fact that the deleterious effects of the single mutant were repressed in c-Met/Casp8 double mutants brings further strong arguments for a relevant role of death receptor signaling during NASH-pathogenesis. Hence, on a molecular level Caspase-8 seems to be the relevant factor triggering apoptosis in hepatocytes during NASH (75,76). Recently, besides classical apoptosis, the concept of necroptosis, defined as a “programmed” necrotic cell death pathway controlled by RIP1 and RIP3 kinases, becomes of interest. In a recent paper, Gautheron et al. showed an upregulation of RIP3 in human NASH and in a dietary mouse model of steatohepatitis (77). RIP3 is known to mediate liver injury, inflammation, induction of hepatic progenitor cells/activated cholangiocytes and liver fibrosis through a pathway counteracted by Caspase-8, functions that have been shown to be mediated by a positive feedback loop involving activation of JNK. Furthermore, RIP3-dependent JNK activation promotes the release of pro-inflammatory mediators like MCP-1, thereby attracting macrophages to the injured liver and further augmenting RIP3-dependent signaling, cell death, and liver fibrosis. This pathway might represent a novel and specific target for pharmacological strategies in patients with NASH. Caspase-3, an important mediator of the intrinsic apoptotic pathway also seems to be of importance for NASH-pathogenesis. Accordingly, apoptosis was reduced in mice deficient in Caspase-3 (Casp3-/-) with a marked reduction in the expression of cytokines involved in inflammatory signaling and of profibrotic genes which resulted in reduced hepatic collagen deposition (78). Nevertheless, serum aminotransferase levels and NAFLD activity scores (NAS) were similar to WT MCD-fed mice.
Figure 2.
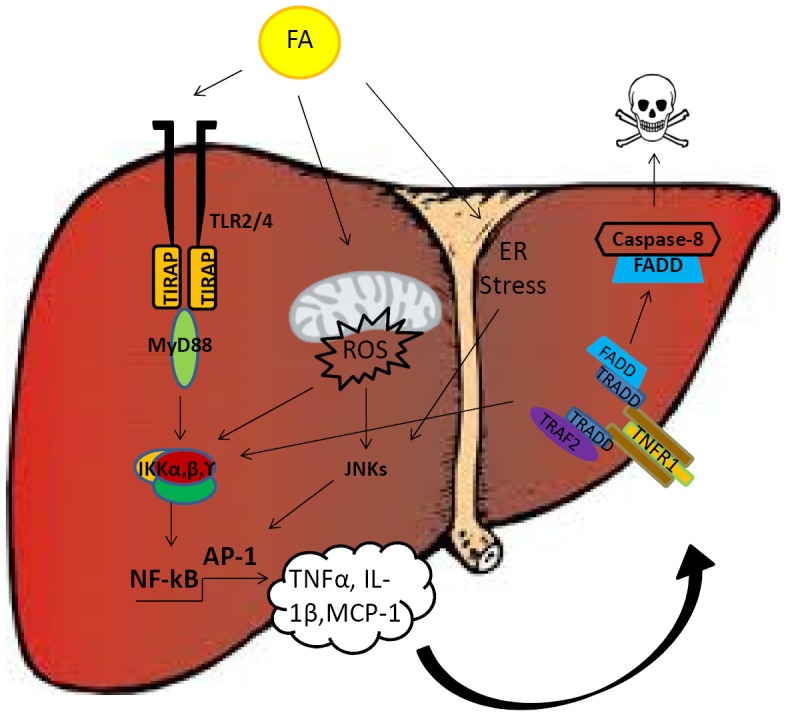
Hepatotoxic effects of FAs. FAs have been shown to induce cytotoxicity and hepatic damage through four main mechanisms: (I) activation of inflammatory signals via direct TLR2/4 binding; (II) mitochondrial dysfunction and increased oxidative stress; (III) induction of ER stress with consequent JNK cascade activation; (IV) indirect activation of cell death signals depending on the release of specific inflammatory mediators such as TNF-α. FA, fatty acid; TIR, toll like receptor; TNF, tumour necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand.
The inflammasome
The inflammasome is a larger multiprotein oligomer involved in inflammatory activation that regulates caspase-1 activation. The NLRP3 (nucleotide-binding domain, leucine-rich repeat containing, also known as NALP3) inflammasome is expressed in myeloid cells and up-regulated by PAMPs. Once all the components of the NALP3-inflammasome are assembled in the cytosol, caspase 1 gets released and promotes the cleavage and maturation of pro-inflammatory cytokines (pro-IL-1β, pro-IL-18, and IL33) to trigger inflammation. The inflammasome is typically activated in experimental alcohol- induced liver injury and in mice fed the MCD diet, but not during simple HF diet-induced steatosis (24). In vivo, it seems likely that hepatocytes challenged with SFAs release undefined “danger signals” triggering the activation of the inflammasome also in hepatic lymphocytes and macrophages. They are thereby amplified through the release of inflammatory mediators such as IL-1β and TNF-α. Experiments using loss of function tamoxifen-inducible Nlrp3 knock-out mice unraveled that those are protected from diet-induced steatohepatitis and show a reduced activation of macrophages (79). Also bone-marrow derived cells might contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis (80).
Hepatic immune cells in NASH development
Kupffer cells (KCs)
KCs, as specialized hepatic tissue macrophages, mediate the inflammatory response in many liver diseases (Figure 3). Gut-derived endotoxins are able to stimulate their phagocytic capacity through binding CD14, TLRs 2 and 4 and triggering adapter proteins such as MD2/MyD88 leading to NF-κB activation. KC is also a major source of IFN-γ, which is an important cytokine for lymphocyte recruitment. Specific KC depletion (e.g., by gadolinium or clodronate liposomes) was shown to produce beneficial effects in experimental models of alcohol-induced steatohepatitis by reducing hepatic damage and inflammation (81). Similarly, KC ablation by liposomal clodronate resulted in an inhibition of inflammation and fibrosis in a CDAA model of steatohepatitis (82). Moreover, employment of an analogue depletive strategy resulted in reduction of hepatic TG accumulation in a murine model of diet induced obesity. In this specific case, a diminished NF-κB activity turned out to enhance PPAR-α-dependent lipid oxidative catabolism (83).
Figure 3.

Cellular interactions during NASH-pathogenesis. Development of hepatic steatosis is resulting from a complex interplay of involved organs and different cell types. Gut-derived endotoxins and white fat tissue produced FFAs together with pro-inflammatory signals trigger an intrahepatic inflammatory response. This involves activation of liver resident macrophages (Kupffer-cells) and other hepatic immune cells and the subsequent infiltration of immune cells into the liver, which then release cytokines, chemokines and other mediators. In term hepatocytes start to accumulate intracellular fat, inflammation sustains and eventually liver fibrosis can develop. TNF, tumour necrosis factor; FFAs, free fatty acids; KC, Kupffer cells; LPS, lipopolysaccharides; NASH, non-alcoholic steatohepatitis.
Mechanistically, depletion of KCs leads to reduced IL-1b expression, which in term reduces the level of inflammation (84). Another way through which KCs contribute to proinflammatory hepatic conditions is their response to an overload with toxic lipids, especially in the initial phase of NASH-development. Interestingly, in their study Leroux and colleagues could show that the total amount of KCs was not changed in steatotic livers in spite of a massive overload with enlarged lipid droplets. Functional assays then revealed that those KCs were able to recruit significantly more inflammatory cells and expressed more pro-inflammatory chemokines and cytokines, such as IFN-γ, IL1b, CCL5 or -2 (85).
An interesting and obviously clinically relevant mechanism of NASH-pathogenesis is the infiltration of macrophages into the hepatic tissue. Those macrophages expressed predominantly CD68, CCR2 and Ly6C. CCR2 knockout mice developed less steatosis, less cell infiltration and even less fibrosis in an experimental model of NASH (86). The inhibition of this receptor also seems to be a clinically promising approach, as first phase 1 and 2 studies using CCR2 (BMS-741672) or combined CCR5/2 inhibitors (cenicriviroc) have been undertaken in patients with diabetes and diabetic complications (clinicaltrials.gov identifier: NCT00699790 and NCT01712061).
Neutrophils
The presence of neutrophils [polymorphonuclear neutrophils (PMNs)] among the liver inflammatory infiltrate of alcoholic steatohepatitis has long been acknowledged. Neutrophils are also present in NASH where their possible pathogenic significance remains obscure (87). In a mouse model of NASH development it was recently shown that neutrophil-derived myeloperoxidase aggravates the severity of the liver phenotype (88). Accordingly, another report remarked a contribution of neutrophils in NASH by feeding transgenic mice over-expressing the human neutrophils peptide (HNP1) a CDAA diet (89). In further clinical studies, a large NASH patient cohort was analysed for their neutrophil to lymphocyte ratio. The authors observed a higher N/L ratio in patients with NASH and advanced fibrosis suggesting this ratio as a novel non-invasive diagnostic marker to predict advanced disease (90). From a clinical point of view attempts have been made to find methods to better predict the course of NASH. In this regard it was found that serum levels of the apoptotic marker M30 and full-length CK8/18 (necrosis) are increased in patients with the predominantly inflammatory form of liver steatosis, indicating that both apoptosis and necrosis occur in humans with the inflammatory form of NAFLD (91).
Lymphocytes and dendritic cells
Lymphocytes are abundantly present in the liver and have been shown to accumulate in NASH livers, but individual subpopulations and their pathogenic roles during injury and inflammation are not functionally characterized. It was recently described that during all stages of the disease portal tracts were more densely populated and dominated by CD68+ macrophages and CD8+ lymphocytes (10). An increase in portal macrophages in NAFLD patients with simple steatosis represented an early detectable change, even before an enhanced expression of proinflammatory cytokines (IL1b, TNF-α) became evident. Portal and periductal accumulation of other cell types examined most significantly occurred in advanced stages of NASH. For intrahepatic dendritic cells it was shown that they expand in NASH livers and presumably mature there into an activated immune phenotype (92). Hence they can limit sterile inflammation through the clearance of apoptotic cells and necrotic debris and they seem to play an important immune-modulatory role on CD4+T-cell functions. However, further experimental studies are required to establish a therapeutic approach aiming at the modulation of dendritic cells activity in NASH progression.
Interestingly a significant accumulation of natural killer T (NKT) cells could be detected in the MCD dietary mouse model contributing to the development of NASH (93,94). Indeed, several research groups demonstrated a significant decrease of CD4+ T regulatory cells (Tregs) in a high fat diet mouse model (95,96). Moreover, a Th17 response seems to be important for the progression from simple steatosis to steatohepatitis (97). Sutti et al. could even show that oxidative stress leads to the induction of the oxidative stress response, thereby triggering cellular immunity leading to a more severe development of NASH (35). Finally, a study by Miyagi et al. (98) demonstrated a protective role of invariant NKT cells for progression from inflammation to fibrosis, not altering steatosis in a HF mouse model whereas Gadd et al. (10) described that the presence of an enriched portal infiltrate (CD3+, CD4+, CD8+, CD20+ and CD68+ cells) in human NAFLD strongly correlates with the fibrosis score.
Conclusive remarks
Decades of studies on clinical and experimental models of steatohepatitis gave light on novel aspects of NAFLD pathophysiology. The dissection of metabolic pathways regulating lipid turn-over in the liver enabled the comprehension of mechanisms through which hepatocytes can adapt and re-organize their metabolic activity once challenged by stressful conditions. On the other hand, specific lipid metabolites, endogenous or exogenous, turned out to activate pathways that irreversibly alter the cell functionality in a manner that might not necessarily depend on their amount.
A growing body of evidence indicates that such metabolic alterations of the hepatocyte directly or indirectly trigger a plethora of defensive mechanisms that not only involve the hepatic innate immune system but also result in an inter-organ communication process involving a wide variety of cell populations and molecular mediators. The importance of identifying and unravelling this fine communicative network will offer new keys of lecture for the development of therapeutic and diagnostic strategies for the management of fatty liver diseases.
Acknowledgements
Funding: This work is supported by a grant (SFB TRR57 p22) of the Deutsche Forschungsgemeinschaft (DFG).
Disclosure: The authors declare no conflict of interest.
References
- 1.Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol 2013;10:645-55. [DOI] [PubMed] [Google Scholar]
- 2.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013;10:330-44. [DOI] [PubMed] [Google Scholar]
- 3.Yeh MM, Brunt EM. Pathological features of fatty liver disease. Gastroenterology 2014;147:754-64. [DOI] [PubMed] [Google Scholar]
- 4.Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686-90. [DOI] [PubMed] [Google Scholar]
- 5.Trauner M, Arrese M, Wagner M. Fatty liver and lipotoxicity. Biochim Biophys Acta 2010;1801:299-310. [DOI] [PubMed]
- 6.Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology 1998;114:842-5. [DOI] [PubMed] [Google Scholar]
- 7.Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol 2008;19:295-300. [DOI] [PubMed] [Google Scholar]
- 8.Mantzaris MD, Tsianos EV, Galaris D. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS J 2011;278:519-30. [DOI] [PubMed] [Google Scholar]
- 9.Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 2003;100:3077-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadd VL, Skoien R, Powell EE, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393-405. [DOI] [PubMed] [Google Scholar]
- 11.Harley IT, Stankiewicz TE, Giles DA, et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014;59:1830-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradbury MW. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: possible role in steatosis. Am J Physiol Gastrointest Liver Physiol 2006;290:G194-8. [DOI] [PubMed] [Google Scholar]
- 13.Nagle CA, Klett EL, Coleman RA. Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res 2009;50Suppl:S74-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu H, Wilcox D, Nguyen P, et al. Hepatic knockdown of mitochondrial GPAT1 in ob/ob mice improves metabolic profile. Biochem Biophys Res Commun 2006;349:439-48. [DOI] [PubMed] [Google Scholar]
- 15.Villanueva CJ, Monetti M, Shih M, et al. Specific role for acyl CoA:Diacylglycerol acyltransferase 1 (Dgat1) in hepatic steatosis due to exogenous fatty acids. Hepatology 2009;50:434-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007;45:1366-74. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg AS, Coleman RA, Kraemer FB, et al. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest 2011;121:2102-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Straub BK, Stoeffel P, Heid H, et al. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 2008;47:1936-46. [DOI] [PubMed] [Google Scholar]
- 20.Sapiro JM, Mashek MT, Greenberg AS, et al. Hepatic triacylglycerol hydrolysis regulates peroxisome proliferator-activated receptor alpha activity. J Lipid Res 2009;50:1621-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miura K, Seki E, Ohnishi H, et al. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol Res Pract 2010;2010:362847. [DOI] [PMC free article] [PubMed]
- 24.Csak T, Ganz M, Pespisa J, et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011;54:133-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo TF, Tatsukawa H, Matsuura T, et al. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J Cell Physiol 2012;227:1130-7. [DOI] [PubMed] [Google Scholar]
- 26.Jun DW, Cho WK, Jun JH, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by carnitine via reversal of mitochondrial dysfunction. Liver Int 2011;31:1315-24. [DOI] [PubMed] [Google Scholar]
- 27.Latruffe N, Cherkaoui Malki M, Nicolas-Frances V, et al. Peroxisome-proliferator-activated receptors as physiological sensors of fatty acid metabolism: molecular regulation in peroxisomes. Biochem Soc Trans 2001;29:305-9. [DOI] [PubMed] [Google Scholar]
- 28.Romics L, Jr, Kodys K, Dolganiuc A, et al. Diverse regulation of NF-kappaB and peroxisome proliferator-activated receptors in murine nonalcoholic fatty liver. Hepatology 2004;40:376-85. [DOI] [PubMed] [Google Scholar]
- 29.Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through Toll-like receptors. Mol Cells 2006;21:174-85. [PubMed] [Google Scholar]
- 30.Videla LA, Tapia G, Rodrigo R, et al. Liver NF-kappaB and AP-1 DNA binding in obese patients. Obesity (Silver Spring) 2009;17:973-9. [DOI] [PubMed] [Google Scholar]
- 31.Bertola A, Bonnafous S, Anty R, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One 2010;5:e13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joshi-Barve S, Barve SS, Amancherla K, et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007;46:823-30. [DOI] [PubMed] [Google Scholar]
- 33.Haukeland JW, Damås JK, Konopski Z, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol 2006;44:1167-74. [DOI] [PubMed] [Google Scholar]
- 34.Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutti S, Jindal A, Locatelli I, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014;59:886-97. [DOI] [PubMed] [Google Scholar]
- 36.Gadang V, Kohli R, Myronovych A, et al. MLK3 promotes metabolic dysfunction induced by saturated fatty acid-enriched diet. Am J Physiol Endocrinol Metab 2013;305:E549-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leamy AK, Egnatchik RA, Shiota M, et al. Enhanced synthesis of saturated phospholipids is associated with ER stress and lipotoxicity in palmitate treated hepatic cells. J Lipid Res 2014;55:1478-1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snodgrass RG, Huang S, Choi IW, et al. Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol 2013;191:4337-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueki K, Kondo T, Tseng YH, et al. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci U S A 2004;101:10422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das SK, Mondal AK, Elbein SC. Distinct gene expression profiles characterize cellular responses to palmitate and oleate. J Lipid Res 2010;51:2121-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang G, Badeanlou L, Bielawski J, et al. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab 2009;297:E211-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ussher JR, Koves TR, Cadete VJ, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 2010;59:2453-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei Y, Wang D, Topczewski F, et al. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 2006;291:E275-81. [DOI] [PubMed] [Google Scholar]
- 44.Caballero F, Fernández A, De Lacy AM, et al. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 2009;50:789-96. [DOI] [PubMed] [Google Scholar]
- 45.Ray K.Liver: Dietary cholesterol and fat synergistically fuel the development of NASH in experimental models. Nat Rev Gastroenterol Hepatol 2012;9:303. [DOI] [PubMed] [Google Scholar]
- 46.Gan LT, Van Rooyen DM, Koina ME, et al. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J Hepatol 2014;61:1376-84. [DOI] [PubMed] [Google Scholar]
- 47.Teratani T, Tomita K, Suzuki T, et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012;142:152-164.e10. [DOI] [PubMed]
- 48.Tomita K, Teratani T, Suzuki T, et al. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol 2014;61:98-106. [DOI] [PubMed] [Google Scholar]
- 49.Bieghs V, Verheyen F, van Gorp PJ, et al. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS One 2012;7:e34378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ioannou GN, Haigh WG, Thorning D, et al. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res 2013;54:1326-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Rooyen DM, Gan LT, Yeh MM, et al. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J Hepatol 2013;59:144-52. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464:104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bianchi ME. HMGB1 loves company. J Leukoc Biol 2009;86:573-6. [DOI] [PubMed] [Google Scholar]
- 54.Rivera CA, Adegboyega P, van Rooijen N, et al. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 2007;47:571-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology 2006;130:1886-900. [DOI] [PubMed] [Google Scholar]
- 56.Ellett JD, Evans ZP, Atkinson C, et al. Toll-like receptor 4 is a key mediator of murine steatotic liver warm ischemia/reperfusion injury. Liver Transpl 2009;15:1101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szabo G, Velayudham A, Romics L, Jr, et al. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res 2005;29:140S-145S. [DOI] [PubMed] [Google Scholar]
- 58.Spruss A, Kanuri G, Wagnerberger S, et al. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009;50:1094-104. [DOI] [PubMed] [Google Scholar]
- 59.Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006;116:3015-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Csak T, Velayudham A, Hritz I, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol 2011;300:G433-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010;139:323-34.e7. [DOI] [PMC free article] [PubMed]
- 62.De Nardo D, De Nardo CM, Nguyen T, et al. Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J Immunol 2009;183:8110-8. [DOI] [PubMed] [Google Scholar]
- 63.Vijay-Kumar M, Aitken JD, Carvalho FA, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010;328:228-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghoshal S, Witta J, Zhong J, et al. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 2009;50:90-7. [DOI] [PubMed] [Google Scholar]
- 65.Moreira AP, Texeira TF, Ferreira AB, et al. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 2012;108:801-9. [DOI] [PubMed] [Google Scholar]
- 66.Gregor MF, Yang L, Fabbrini E, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 2009;58:693-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ye D, Li FY, Lam KS, et al. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 2012;61:1058-67. [DOI] [PubMed] [Google Scholar]
- 68.Lake AD, Novak P, Hardwick RN, et al. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci 2014;137:26-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meakin PJ, Chowdhry S, Sharma RS, et al. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol 2014;34:3305-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol 2011;5:201-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brenner C, Galluzzi L, Kepp O, et al. Decoding cell death signals in liver inflammation. J Hepatol 2013;59:583-94. [DOI] [PubMed] [Google Scholar]
- 72.Wang X, DeFrances MC, Dai Y, et al. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Mol Cell 2002;9:411-21. [DOI] [PubMed] [Google Scholar]
- 73.Zou C, Ma J, Wang X, et al. Lack of Fas antagonism by Met in human fatty liver disease. Nat Med 2007;13:1078-85. [DOI] [PubMed] [Google Scholar]
- 74.Kroy DC, Schumacher F, Ramadori P, et al. Hepatocyte specific deletion of c-Met leads to the development of severe non-alcoholic steatohepatitis in mice. J Hepatol 2014;61:883-90. [DOI] [PubMed] [Google Scholar]
- 75.Hatting M, Zhao G, Schumacher F, et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology 2013;57:2189-201. [DOI] [PubMed] [Google Scholar]
- 76.Liedtke C, Bangen JM, Freimuth J, et al. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology 2011;141:2176-87. [DOI] [PubMed] [Google Scholar]
- 77.Gautheron J, Vucur M, Reisinger F, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thapaliya S, Wree A, Povero D, et al. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig Dis Sci 2014;59:1197-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wree A, McGeough MD, Peña CA, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 2014;92:1069-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Csak T, Pillai A, Ganz M, et al. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int 2014;34:1402-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Owumi SE, Corthals SM, Uwaifo AO, et al. Depletion of Kupffer cells modulates ethanol-induced hepatocyte DNA synthesis in C57Bl/6 mice. Environ Toxicol 2014;29:867-75. [DOI] [PubMed] [Google Scholar]
- 82.Miura K, Yang L, van Rooijen N, et al. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 2012;302:G1310-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stienstra R, Saudale F, Duval C, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010;51:511-22. [DOI] [PubMed] [Google Scholar]
- 84.Negrin KA, Roth Flach RJ, DiStefano MT, et al. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS One 2014;9:e107265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leroux A, Ferrere G, Godie V, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol 2012;57:141-9. [DOI] [PubMed] [Google Scholar]
- 86.Miura K, Yang L, van Rooijen N, et al. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 2012;302:G1310-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brunt EM, Janney CG, Di Bisceglie AM, et al. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467-74. [DOI] [PubMed] [Google Scholar]
- 88.Rensen SS, Bieghs V, Xanthoulea S, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS One 2012;7:e52411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ibusuki R, Uto H, Arima S, et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, L-amino acid-defined diet. Liver Int 2013;33:1549-56. [DOI] [PubMed] [Google Scholar]
- 90.Alkhouri N, Morris-Stiff G, Campbell C, et al. Neutrophil to lymphocyte ratio: a new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2012;32:297-302. [DOI] [PubMed] [Google Scholar]
- 91.Joka D, Wahl K, Moeller S, et al. Prospective biopsy-controlled evaluation of cell death biomarkers for prediction of liver fibrosis and nonalcoholic steatohepatitis. Hepatology 2012;55:455-64. [DOI] [PubMed] [Google Scholar]
- 92.Henning JR, Graffeo CS, Rehman A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013;58:589-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Syn WK, Oo YH, Pereira TA, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010;51:1998-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kroy DC, Beraza N, Tschaharganeh DF, et al. Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatology 2010;51:463-73. [DOI] [PubMed] [Google Scholar]
- 95.Syn WK, Agboola KM, Swiderska M, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012;61:1323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Locatelli I, Sutti S, Vacchiano M, et al. NF-κB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin Sci (Lond) 2013;124:279-87. [DOI] [PubMed] [Google Scholar]
- 97.Tang Y, Bian Z, Zhao L, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol 2011;166:281-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miyagi T, Takehara T, Uemura A, et al. Absence of invariant natural killer T cells deteriorates liver inflammation and fibrosis in mice fed high-fat diet. J Gastroenterol 2010;45:1247-54. [DOI] [PubMed] [Google Scholar]