

Background: MeCP2 is a multifunctional protein whose full spectrum of activities remains enigmatic.
Results: MeCP2 localizes at the centrosome and at the mitotic spindle. Its loss causes deficient spindle morphology and microtubule nucleation and prolonged mitosis.
Conclusion: Through its centrosomal localization, MeCP2 regulates cell growth and cytoskeleton stability.
Significance: This novel MeCP2 function may improve our comprehension of MeCP2 under physiological and pathological conditions.
Keywords: Cell Cycle, Cell Proliferation, Centrosome, Microtubule, Mitotic Spindle, Protein Phosphorylation, MeCP2
Abstract
Mutations in MECP2 cause a broad spectrum of neuropsychiatric disorders of which Rett syndrome represents the best defined condition. Both neuronal and non-neuronal functions of the methyl-binding protein underlie the related pathologies. Nowadays MeCP2 is recognized as a multifunctional protein that modulates its activity depending on its protein partners and posttranslational modifications. However, we are still missing a comprehensive understanding of all MeCP2 functions and their involvement in the related pathologies. The study of human mutations often offers the possibility of clarifying the functions of a protein. Therefore, we decided to characterize a novel MeCP2 phospho-isoform (Tyr-120) whose relevance was suggested by a Rett syndrome patient carrying a Y120D substitution possibly mimicking a constitutively phosphorylated state. Unexpectedly, we found MeCP2 and its Tyr-120 phospho-isoform enriched at the centrosome both in dividing and postmitotic cells. The molecular and functional connection of MeCP2 to the centrosome was further reinforced through cellular and biochemical approaches. We show that, similar to many centrosomal proteins, MeCP2 deficiency causes aberrant spindle geometry, prolonged mitosis, and defects in microtubule nucleation. Collectively, our data indicate a novel function of MeCP2 that might reconcile previous data regarding the role of MeCP2 in cell growth and cytoskeleton stability and that might be relevant to understand some aspects of MeCP2-related conditions. Furthermore, they link the Tyr-120 residue and its phosphorylation to cell division, prompting future studies on the relevance of Tyr-120 for cortical development.
Introduction
Methyl-CpG binding protein 2 (MeCP2) was first described as an epigenetic factor that binds specifically to methylated DNA and represses transcription through the recruitment of chromatin remodeling complexes containing histone deacetylase activities (1, 2). Mutations in the X-linked MECP2 gene were later found in several patients affected by Rett syndrome (RTT,4 OMIM no. 312750), a devastating neuronal disease that, because of its incidence, is considered to be one of the main causes of severe intellectual disabilities in girls (3). Since then, hundreds of different mutations in MECP2 have been associated with RTT or, less frequently, with other forms of intellectual disability.
Although MECP2 mutations have profound effects on brain functions, several recent studies have demonstrated that RTT is not an irreversible condition in mice because phenotypic rescue is possible (4). MeCP2 studies have therefore been boosted dramatically, leading to a progressive expansion of MeCP2 functions beyond the original role of the protein in transcriptional repression through the recruitment of chromatin remodeling complexes (5). In 2003, Georgel et al. (6) proposed that MeCP2, when highly abundant, might work directly (without other corepressors or enzymatic activities) as a potent chromatin condensing factor. Accordingly, Skene et al. (7) have demonstrated that, in mature neurons where MeCP2 levels are sufficiently high to saturate methylated DNA but not in non-neuronal cells characterized by 10–30 times less MeCP2, the protein can substitute histone H1 and function as a global architectural factor. The effect on the genomic architecture is outlined by a selective increase in histone acetylation, H1 levels, and transcription of repetitive elements and L1 retrotransposons in Mecp2-null neurons but not in glia (7, 8). These data indicate that MeCP2 functions may change in accordance with its abundance. The capability of the protein to work as a transcriptional activator, possibly through its interaction with the transcriptional activator cAMP response element-binding protein, has also been suggested (9, 10). Fittingly, MeCP2 has been reported recently as the major 5-hydroxymethylcytosine binding protein in the brain, an epigenetic signal enriched in the body of active genes (11). Because MeCP2 can interact with YB1, a functional link between the methyl-binding protein and mRNA splicing has also been proposed (12). Eventually, a direct or indirect role of MeCP2 in regulating the AKT/mammalian target of rapamycin signaling pathway and, therefore, protein synthesis and cell homeostasis has been demonstrated (13).
Together, these data imply that MeCP2 is a multifunctional protein whose full activities might still have to be unraveled. Differential posttranslational modifications (PTMs) may participate in the regulation of these functions. In fact, several recent papers have found that MeCP2 activities might be regulated through its PTMs. A number of residues have been found differentially phosphorylated in the brain, neurons, or cultured cells in response to extracellular cues. Diverse phosphorylation events appear to affect the affinity of MeCP2 for chromatin (14, 15). These data, together with those demonstrating that PTMs other than phosphorylation occur on MeCP2, imply that a complex pattern of PTMs affects the capability of the protein to interact with chromatin and protein partners and regulates its activities (16).
One of the most interesting residues of MeCP2 that may be affected by phosphorylation-mediated regulation is Tyr-120, which was connected to Rett syndrome in 2001. Indeed, a patient affected by a variant form of Rett syndrome was found to harbor a MECP2 mutation, resulting in the substitution of Tyr-120 with aspartic acid (Y120D) (17), possibly mimicking a constitutively phosphorylated state. Tyr-120 is contained within the methyl-DNA binding domain of MeCP2 and is highly conserved in all mammals. Functional studies of the same mutation showed a decreased affinity of the pathogenic mutant for chromocenters (18, 19).
Therefore, we embarked on a study to determine the intracellular localization and the function of this specific phospho-isoform. These studies led us to discover that MeCP2 localizes in the centrosomes of both dividing and non-proliferating cells. The lack of MeCP2 causes several phenotypes that can be related to centrosome functions, such as a prolonged timing of mitosis, abnormal cell cycle and/or mitotic spindle geometry, and defects in microtubule nucleation.
EXPERIMENTAL PROCEDURES
Plasmids
Human MeCP2E1 was amplified by PCR and cloned into the BamHI site of pEGFPC1 (Clontech) and pCMV-FLAG-2B (Stratagene). MeCP2-Y120D and Y120F mutants were obtained by site-directed mutagenesis using the QuikChange XL site-directed mutagenesis kit (Stratagene) following the protocol of the manufacturer. The PCR-amplified regions were verified by sequencing. pEGFPC1-Centrin2 was provided by M. Stiess (Max Planck Institute of Neurobiology, Germany).
To silence MeCP2, cells were transfected with siMeCP2#1 (sense, 5′-GGAAAGGACUGAAGACCUGUU-3′) or, as corresponding control, a scrambled siRNA (sense, 5′-UAGCGACUAAACACAUCAA-3′), both purchased from Dharmacon. A second couple of oligos was purchased from Sigma-Aldrich, siMeCP2#2 (Mission esiRNA human MECP2, catalog no. EHU030271) and control#2 (Mission esiRNA targeting RLUC, catalog no. EHURLUC).
Antibodies
The anti-MeCP2 Tyr-120 phospho-site-specific antibody was generated by Covance, Inc. Rabbits were immunized with the peptide [NH2]-RKSGRSAGK-pY-DVYLINPQGK-[COOH] corresponding to amino acids 111–130 of human MeCP2. To purify the antibody, the antiserum was first passed on a column containing the non-phosphorylated peptide, and then the flow-through was applied to a second column that was conjugated to the phosphorylated peptide. The affinity-purified anti-MeCP2 Tyr(P)-120 antibody was used at 1:1000 for Western blotting and 1:100 for immunofluorescence.
Antibodies used were anti-β-actin (Sigma-Aldrich, catalog no. A5441), anti-γ-tubulin (Sigma-Aldrich, catalog no. T5326), anti-pericentrin (BD Transduction Laboratories, catalog no. 611814), anti-α-tubulin (Sigma-Aldrich, catalog no. T6074), anti-cleaved caspase-3 (Asp-175, Cell Signaling Technology, catalog no. 9661), anti-histone H3-Ser(P)-10 (Abcam, catalog no. ab1791), anti-MeCP2 (Sigma-Aldrich, catalog no. M9317), anti-phospho-p42/44 MAPK (Thr-202/Tyr-204, Cell Signaling Technology, catalog no. 9101), anti-phospho-Aurora A/B/C (Cell Signaling Technology, catalog no. 2914), anti-FLAG (Sigma-Aldrich, catalog no. F3165), anti-GFP (Roche, catalog no. 1814460), anti-Tyr(P) (Cell Signaling Technology, catalog no. 9411), and anti-Cyclin B1 (Abcam, catalog no. ab72). HRP-conjugated goat anti-mouse or anti-rabbit secondary antibodies for Western blotting were purchased from Thermo Scientific. DAPI and secondary Alexa Fluor anti-rabbit and anti-mouse antibodies for immunofluorescence experiments were obtained from Invitrogen.
Cell Cultures and Transfections
HeLa, MRC-5, HEK293, and COS-7 cells were maintained in DMEM (Sigma-Aldrich) supplemented with 10% FBS, l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C with 5% CO2. Astroglial cell cultures and primary mouse cortical neurons were prepared and cultured as described previously (20). Primary mouse embryonic fibroblast (MEF) cultures from WT and Mecp2-null mice (21) were prepared from day 14.5 mouse embryos from a pregnant heterozygous female (Mecp2+/−). Briefly, the tails of the embryos were removed and transferred to sterile tubes for genotyping. For each embryo, all “red” tissue in the body cavity was removed, and the remainder of the embryo was isolated for MEF preparation. Each embryo was minced with forceps and incubated at 37 °C for 30 min in 500 μl of trypsin/EDTA 0.25% in PBS (Sigma-Aldrich). After removal of the trypsin, the tissues were resuspended in DMEM with 10% FBS and antibiotics by gentle trituration with a Pasteur pipette, and the supernatant was transferred to T25 flasks. Cell cultures were enriched in mitosis by treating cells with 0.1 μg/ml nocodazole (Sigma-Aldrich) for 24 h.
For transfection, cells were plated and cultured in 6- or 12-well dishes for 20 h and transfected with plasmids using LipofectamineTM 2000 (Invitrogen) following the protocol of the manufacturer. Cells were collected or fixed 24 h post-transfection. For siRNA transfection, 20 nm siRNA oligonucleotide targeting MeCP2 or a control siRNA were transfected into HeLa or MRC-5 cells using LipofectamineTM RNAiMAX (Invitrogen). Silenced cells were processed 96 h post-transfection.
Centrosomal Fractionation
Centrosome fractionation was performed as described previously (22). In brief, this method involves treating exponentially growing cells with 10 μg/ml nocodazole (Sigma-Aldrich) and 5 μg/ml Cytochalasin B (Sigma-Aldrich) for 90 min, followed by hypotonic lysis. Centrosomes are harvested by centrifugation onto a 20% Ficoll cushion and further purified by centrifugation through a discontinuous (70, 50, and 40%) sucrose gradient. Fractions of 0.3 ml were collected and analyzed.
Microtubule Nucleation Assay
Microtubules were disrupted by incubating MRC-5 cells with 10 μg/ml nocodazole for 3 h. To assess microtubule regrowth, cells were washed twice with warm media to remove nocodazole and then incubated at 37 °C for different times. Fixed cells were stained with anti α-tubulin antibody and analyzed for nucleation capacity and aster size. For determining aster size, images were taken from at least 60 cells from three independent experiments, and each aster was treated independently. Quantification of the aster area was obtained using ImageJ software by drawing a region of interest around the aster and measuring the area of this region. The areas occupied by centrosomes were blackened previously to obtain only the real measure of the aster.
FACS Analysis
Asynchronously growing MRC-5 cells were collected 4 days after silencing of MeCP2 and fixed with 70% ethanol. Then cells were rehydrated with PBS/3% BSA, centrifuged, and stained with propidium iodide staining solution (10 μg/ml RNase A and 5 μg/ml propidium iodide in PBS). Samples were analyzed on a BD FACScan using the CellQuest (BD Biosciences) software.
Western Blotting
Cells were washed twice with PBS and lysed with lysis buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Triton-X-100, 1 mm EDTA, 1 mm DTT, protease inhibitor mixture (Sigma-Aldrich), and PhosSTOP (Roche)). After 30 min on ice, the lysates were clarified by centrifugation, and the supernatants were collected.
Samples were loaded onto either 10% or 8% SDS-PAGE gels, transferred to nitrocellulose membranes, and blocked in 5% nonfat milk in TBS (20 mm Tris-HCl (pH 7.5) and 150 mm NaCl) with 0.2% Tween 20. Blots were incubated with primary antibody overnight at 4 °C, washed in TBS with 0.2% Tween 20, and incubated with the appropriate secondary antibody for 1 h at room temperature. Blots were washed extensively and then developed with either a West PICO chemiluminescence kit (Pierce) or, to detect phosphorylated MeCP2, with ECL Prime Western blotting detection reagent (GE Healthcare Life Sciences).
To treat nitrocellulose membranes with λ-phosphatase, the membrane was incubated overnight at 4 °C in 1% BSA in TBS/0.1% Triton-X-100 with or without the addition of λ protein phosphatase (400 units/ml, New England Biolabs). Membranes were then processed as described above.
Immunoprecipitations
Asynchronous HeLa cells were lysed in 50 mm Tri-HCl (pH 8.0), 150 mm NaCl, 1% Nonidet P-40, 10% glycerol, 0.5% sodium deoxycholate, protease inhibitor mixture (Sigma-Aldrich), and PhosSTOP (Roche). The cleared extract was incubated for 3 h with anti-MeCP2 Tyr(P)-120 antibodies and overnight with protein G-agarose (Invitrogen). HEK293 cells were collected 24 h after transfection of FLAG constructs, as described above, and lysed in 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% Triton-X-100, protease inhibitor mixture, and PhosSTOP. The extract was precleared for 1 h with protein G-agarose and then incubated with anti-FLAG antibody and beads for 3 h. In both experiments, the beads were collected, washed three times with lysis buffer without glycerol and twice with TBS, and then eluted by addition of SDS sample buffer and heating.
Immunofluorescence Microscopy
To costain for total endogenous MeCP2 (or the Tyr(P)-120 isoform) and γ-tubulin or pericentrin, cells were extracted in PHEM (60 mm Pipes, 25 mm Hepes, 10 mm EGTA, and 2 mm MgCl2) with 0.5% Triton X-100 for 2 min and then fixed in 4% paraformaldehyde. For all the other antibodies, cells were washed twice in TBS and fixed in 4% paraformaldehyde.
For immunostaining, blocking solution (TBS, 5% horse serum, and 0.2% Triton X-100) was added for 1 h at room temperature, and the appropriate primary antibodies were added overnight at 4 °C. Slides were incubated with the designated secondary antibodies for 1 h at room temperature and washed with TBS. DNA was counterstained with DAPI (Invitrogen), and slides were mounted with ProLong Gold antifade reagent (Invitrogen). The analysis was performed with a NikonEclipse Ni upright microscope.
Time-lapse Microscopy Imaging
Live-cell microscopy of HeLa cells was performed on the Zeiss Axio Observer Z1 microscope using an LD ×20 numerical aperture 0.4/Ph2 Plan NEOFLUAR objective. Images were taken every 5 min from 80–96 h post-transfection. Cells were incubated on the microscope in RPMI medium/10% FCS at 37 °C in a 5% CO2-containing atmosphere.
For live microscopy of α-tubulin-GFP in HeLa cells, images (typically 0.25 × 0.25 × 1 μm3 voxel size) were recorded every 5 min 80–96 h post-transfection using a Leica TCSSP5-AOBS five-channel confocal system (Leica Microsystems). Cells were incubated on the microscope in DMEM/10% FCS at 37 °C in a 5% CO2-containing atmosphere.
Time-lapse imaging of MEFs was performed with a Zeiss Axiovert S100 TV2 microscope using an LD Achrostigmat ×20 (numerical aperture 043) Dry Ph1 objective. Cells (second passage) were plated in a 6-multiwell plate (50,000 cells/well) in complete medium and analyzed after 2 days in culture while being maintained at 37 °C and 5% CO2. Images were taken every 10 min for 48 h.
Analysis of Mitosis Duration
Individual cells were tracked for at least 18 h to measure the duration of mitosis (from DNA condensation to anaphase). To calculate mitosis length, t = 0 was determined at the first sign of DNA condensation and/or cell rounding, and frames were counted until chromosomes began to segregate. The time was calculated accordingly with the frame rate (5 min).
RESULTS
MeCP2 Phosphorylated at Tyr-120 Colocalizes with Centrosomes in Dividing Cells
Several recent works have revealed that MeCP2 is phosphorylated at various serine or threonine residues in cultured cell lines and primary neurons (23, 24). In addition, phosphorylation of Tyr-120 has been reported in phosphoproteomic mapping of HeLa cells by Dephoure et al. (25). To confirm that Tyr-120 can also be phosphorylated in other contexts, we resorted to HEK293 cells in which we expressed epitope tagged WT or Tyr-120-mutated MeCP2. FLAG-MeCP2 or its phospho-defective (Y120F) or mimetic derivatives (Y120D) were immunopurified from transfected cells and probed with two commercial polyclonal sera recognizing Tyr(P) residues and MeCP2, respectively. Although the obtained Western blot confirmed that all the expressed derivatives were immunoprecipitated efficiently (Fig. 1A, bottom panel), only the wild-type methyl-binding protein was well recognized by the Tyr(P) antiserum, therefore suggesting that tyrosines within MeCP2 can be phosphorylated and that Tyr-120 is the main or exclusive target (Fig. 1A, top panel). To investigate the effect of Tyr-120 phosphorylation on MeCP2 localization and functions, we raised an anti-MeCP2 Tyr-120 phospho-site-specific antibody (anti-Tyr(P)-120). We started analyzing the specificity of the purified antibody through immunoprecipitations and demonstrated that the anti-Tyr(P)-120 antibody, but not IgG, immunoprecipitates a protein specifically recognized by a commercial polyclonal serum against MeCP2 from a HeLa cell extract (Fig. 1B). The specificity for MeCP2 was further confirmed by depleting MeCP2 through siRNA (Fig. 1C). To demonstrate the phospho-specificity of the antibody, we overexpressed WT MeCP2 or its phosphomimetic and defective derivatives in HeLa cells. As shown in Fig. 1D, the antibody recognizes exogenously expressed MeCP2 exclusively when the Tyr residue mimics the phosphorylated state. This result proves that the antibody is unable to recognize the unmodified methyl-binding protein and suggests that an undetectable amount of overexpressed protein, if any, is phosphorylated in transfected cells. As a second approach, we prepared two identical filters blotted with asynchronous and mitotic HeLa cell extracts obtained in the absence or presence of phosphatase inhibitors and treated them with λ phosphatase (λ-PPase) or, as control, with buffer only. The anti-Tyr(P)-120 antibody effectively recognizes a band migrating like MeCP2 in mitotic extracts prepared in the presence of phosphatase inhibitors, but not in their absence, therefore confirming the proteomic data suggesting that this phospho-isoform of MeCP2 is enriched during mitosis (Fig. 1E, left panels) (25). On the λ-PPase treated membrane, both the anti-Tyr(P)-120 antibody and the commercial antibody against activated ERK, used as control of the dephosphorylation, did not detect any signal, whereas the antibody recognizing total MeCP2 maintained its reactivity (Fig. 1E, right panels). Interestingly, we noticed that, in mitotic extracts, MeCP2 has a visibly reduced electrophoretic mobility. We demonstrate that this effect is given by phosphorylation of sites other than Tyr-120 and whose characterization is not the focus of this publication.5
FIGURE 1.

MeCP2 phosphorylated at Tyr-120 is localized at the centrosome. A, exogenously expressed FLAG-tagged MeCP2 or the Y120F and Y120D derivatives were immunoprecipitated (IP) from HEK293 cell extracts with anti-FLAG antibodies, and the immunocomplexes analyzed by WB for phosphorylated tyrosine (P-Tyr) and MeCP2. Expression of the empty FLAG vector was used as negative control (n = 3). B, a HeLa whole cell extract was used for immunoprecipitation with the phospho-specific MeCP2 Tyr(P)-120 antibody or, as a negative control, with rabbit IgG. The presence of MeCP2 or MeCP2 Tyr(P)-120 in the immunocomplexes was then confirmed by WB with the indicated antibodies. The input corresponds to 0.5% of the total extract used for the immunoprecipitation. (n = 2). C, HeLa cells were transfected with siMeCP2#1 or with the corresponding control siRNA. Four days post-transfection, the levels of MeCP2 and Tyr(P)-120 were analyzed by WB, using actin as loading control (n = 3). D, wild-type MeCP2 or the Y120F and Y120D derivatives were expressed in HeLa cells as GFP fusion proteins and analyzed by WB with antibodies against MeCP2 Tyr(P)-120 or total MeCP2. Actin was used as loading control. E, extracts were prepared from asynchronous (Asyn) or mitotic (Mit) HeLa cells with or without phosphatase inhibitors (PPase-inhib.) and separated by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane that was treated with λ-PPase for 16 h or left untreated. WB was performed with the indicated antibodies, using anti-p42/44P (pERK) as a control for the PPase treatment. The asterisk indicates a nonspecific band. F, HeLa cells were grown on coverslips, fixed, and treated with λ-PPase for 1 h or left untreated before immunostaining with antibodies against MeCP2 Tyr(P)-120 (green) and γ-tubulin (red). DNA was stained with DAPI (blue) (n = 3). Scale bar = 10 μm.
Strikingly, when the Tyr(P)-120 antibody was used in immunofluorescence analyses of cycling HeLa cells, we observed that MeCP2 phosphorylated at Tyr-120 is strongly enriched at the spindle poles of mitotic cells, where it colocalizes with the centrosomal marker γ-tubulin (Fig. 1F). The phosphospecificity of the obtained signal was again confirmed by treating the coverslips with λ-PPase.
MeCP2 and Its Phospho-isoform Tyr-120 Are Targeted to the Centrosome throughout the Cell Cycle
Because the anti-Tyr(P)-120 antibody permitted us to reveal a hitherto unrecognized association of MeCP2 with centrosomes, we investigated the subcellular localization pattern of MeCP2 Tyr(P)-120 during the different phases of the cell cycle (Fig. 2A) and compared it with that of MeCP2, regardless of its phosphorylation state (Fig. 2B). During mitosis, progression into prophase is marked by an increase in the centrosomal MeCP2 Tyr(P)-120 signal up to metaphase, whereas, during anaphase, the signal decreases dramatically, becoming undetectable in telophase. In interphase nuclei, the MeCP2 Tyr(P)-120 antibody prominently decorates nuclear puncta whose identity and specificity remain to be demonstrated, making it challenging to demonstrate a specific colocalization of the signal with the γ-tubulin-positive centrosome. However, a colocalization between γ-tubulin and some of these dots was always observed, suggesting that MeCP2 Tyr(P)-120 is localized at the centrosomes even before the initiation of mitosis.
FIGURE 2.

Localization of MeCP2 during the cell cycle. A and B, HeLa cells were grown on coverslips, fixed, and costained with antibodies against MeCP2 Tyr(P)-120 or total MeCP2 (A and B, respectively, both in green) and γ-tubulin (γ-tub, red) and with DAPI (blue) to visualize DNA. Arrows indicate centrosomal MeCP2 in metaphase cells (n > 3). Scale bars = 10 μm.
By using a commercial antibody against total MeCP2, we found that the expected high MeCP2 nuclear signal often masks a possible colocalization of the protein with the centrosome. However, we could confirm that MeCP2 and γ-tubulin colocalize when the centrosome does not overlap the nucleus. The colocalization was also evident during metaphase (Fig. 2B).
To confirm these results and measure the frequency of the association of MeCP2 with centrosomes, we exploited different cell lines. In particular, we selected cell lines that are characterized by a localization of the centrosome outside of the nuclear area. Fig. 3A demonstrates that MeCP2 colocalizes with γ-tubulin both in human fetal lung MRC-5 cells and in monkey fibroblast COS-7 cells. This result was obtained for virtually all cells and was further supported by the data in Fig. 3B, demonstrating that, in MRC-5 and mouse glial cells, MeCP2 colocalizes with two other centrosomal markers, such as centrin and pericentrin. Furthermore, Tyr(P)-120 MeCP2 colocalizes with γ-tubulin in MRC-5 and mouse glial cells (Fig. 3C), and MeCP2 and its phospho-isoform colocalize with pericentrin in postmitotic primary cortical neurons (Fig. 3D). Although two different antibodies (anti-Tyr(P)-120 and anti-MeCP2) were used to show the association of MeCP2 with the centrosome, we excluded the possibility of a cross-reaction by using Mecp2-null MEFs (Fig. 3E), obtained from the Mecp2 bird mouse model of RTT (21), as a negative control. As can be seen in Fig. 3, E and F, we often observed multiple centrosomes in Mecp2-null MEFs.
FIGURE 3.

MeCP2 is targeted to the centrosome in different cell lines. A, MRC-5 and COS-7 cells were immunostained with anti-MeCP2 (green) and anti-γ-tubulin (γ-tub, red) antibodies (n > 3). B, MRC-5 cells (top panels) expressing an exogenous GFP-centrin (GFP-centr.) fusion protein (green) were stained with anti-MeCP2 (red). Mouse glial cells (bottom panels) were costained with anti-MeCP2 (red) and anti-pericentrin (Peric., green) antibodies (n = 3). C, MRC-5 and mouse glial cells were stained with anti-Tyr(P)-120 (green) and anti-γ-tubulin (red) antibodies (n = 3). D, primary cortical neurons were immunostained with anti-MeCP2 or anti-Tyr(P)-120 (green) together with anti-pericentrin (red) (n = 4). E, MEFs from normal or Mecp2-null mice were stained for MeCP2 (green) and γ-tubulin (red) (n = 3). The insets in A–E show the magnified centrosomes. Scale bars = 10 μm. F, -fold increase of Mecp2-null MEFs with centrosome amplification (number of centrosomes >2) with respect to WT cells (n = 4, >500 cells, mean ± S.E.). **, p < 0.01, unpaired Student's t test.
To obtain biochemical evidence in support of our immunocytochemical observations, we made use of an established protocol to purify centrosomes from nocodazole/cytochalasin B-treated HeLa cells (see “Experimental Procedures” and Ref. 22). To validate the purification procedure, fractions from the final sucrose gradients were analyzed by Western blotting (Fig. 4, A, B, and D) and immunofluorescence (Fig. 4C) using γ-tubulin as marker to detect the fractions enriched in centrosomal proteins. The results obtained analyzing the endogenous methyl-binding protein clearly show that both the antibody against MeCP2 as well as the one specifically recognizing MeCP2 Tyr(P)-120 immunodecorate a band whose distribution parallels that of γ-tubulin (Fig. 4, A and B). The specificity of the result is demonstrated by the absence of any detectable signal in cells specifically depleted for MeCP2 (Fig. 4A). In addition to endogenous MeCP2, we found that exogenously expressed GFP-MeCP2, detected with anti-GFP antibodies, peaks together with γ-tubulin in the sucrose gradient (Fig. 4D). On the contrary, the highly abundant and ubiquitously localized GFP, which we used as a control, is present in the sucrose fractions but is always distributed differently from γ-tubulin, therefore strengthening the significance of the cofractionation of MeCP2 with the centrosomal marker. It is important to mention that, in this assay, the vast majority of MeCP2 is discarded with the nuclear extract. By comparing the GFP-MeCP2 input signal corresponding to 0.6% of the extract loaded on the sucrose gradient with the signal in fractions 3 and 4, we can state that centrosomal MeCP2 represents a minor (less than 1%) but biochemically detectable fraction of the total cellular protein. We used the same fractionation protocol to address whether the exogenously expressed pathogenic Y120D mutant shows any alteration in the fractionation. By comparing the amount of exogenously expressed MeCP2 (input) to the quantity of protein cosedimenting with γ-tubulin, we observed that the pathogenic derivative tends to cofractionate more efficiently with the centrosome than the wild-type protein, suggesting that it might have a stronger affinity for and/or a longer residency at the centrosome.
FIGURE 4.

MeCP2 is a stable component of the centrosome. A, HeLa cells were transfected with siMeCP2#1 or with the corresponding control siRNA. Four days post-transfection, centrosomes were purified, and the obtained fractions were analyzed by WB with anti-MeCP2 and anti-γ-tubulin (γ-tub.). Input (Inp.) corresponds to ∼0.6% of the whole cell extract before fractionation. Fractions 1 and 5 are the bottom and top ones, respectively (n = 3). B, Western blotting of purified centrosomes using first antibodies against MeCP2 Y120P followed by stripping of the membrane and reprobing with total anti-MeCP2. γ-tubulin was used as centrosome marker. Fractions 1 and 8 are the bottom and top rows, respectively (n = 2). C, the presence of centrosomes in fraction 6 was verified by immunostaining with γ-tubulin. D, centrosomes were purified from HeLa cells expressing exogenous GFP-MeCP2, GFP, or GFP-MeCP2-Y120D, and the obtained fractions were analyzed by WB with anti-GFP and anti-γ-tubulin. Input corresponds to 0.6% of the extract before fractionation. As above, fractions 1 and 6 are the bottom and top rows, respectively (n = 3). E and F, MRC-5 cells were treated with 10 μg/ml nocodazole for 1 h to depolymerize microtubules, fixed, and stained with antibodies against MeCP2 (E, green), Tyr(P)-120 (F, green), and γ-tubulin (red) (n = 3). Scale bars = 10 μm. The insets show the magnified centrosomes.
In interphase, the centrosome consists of at least 100 proteins. The recruitment of proteins to this organelle can depend on dynein and intact microtubules or it can be mediated by specific localization motifs (26). Some proteins reach the centrosome in a microtubule-independent manner, although specific localization domains have not yet been identified (27). Therefore, we addressed the dependence of the centrosomal localization of MeCP2 on microtubules. For this purpose, MeCP2 (Fig. 4E) and its Tyr-120 phospho-isoform (Fig. 4F) were immunolocalized in MRC-5 cells after microtubule depolymerization through nocodazole treatment. In both cases, MeCP2 remains colocalized with γ-tubulin.
MeCP2 Deprivation Slows Down Cell Proliferation by Perturbing Mitotic Spindle Geometry
It is well accepted that the centrosome plays an essential role in cell cycle progression and cell polarity, organizing the microtubule network in interphase and forming the bipolar spindle in mitosis. To achieve this, the composition of the centrosome as well as its duplication, maturation, and division are tightly regulated. Dysregulation of this organelle, both at the structural or signaling level, often leads to defective proliferation and is also considered a prominent event in tumorigenesis (28).
The recruitment of MeCP2 to centrosomes raised the possibility that it might affect the functions of this organelle. To test this hypothesis, we started investigating the influence of MeCP2 on cell growth. To this purpose we exploited Mecp2-null MEFs and MRC-5 cells in which endogenous MeCP2 was suppressed by siRNA (Fig. 5, A and B, respectively). As shown in Fig. 5, A and B, and already suggested by other authors (29–31), the lack of MeCP2 reduces cell growth. Indeed, in Mecp2-null MEFs, cell growth was attenuated by 70%, whereas the proliferation curve of MeCP2-ablated MRC-5 cells (through two independent siRNAs) demonstrated significantly reduced growth in the analyzed time frame with respect to the control. To further confirm that the latter phenotype is not caused by off-target effects, we performed a rescue experiment in which a GFP-MeCP2-expressing vector, permitting to express MeCP2 despite the interfering protocol, was added to the depleted MRC-5 cells (Fig. 5C). Cells that re-express MeCP2 ameliorate the proliferation rate, whereas transfection of a GFP-expressing control vector does not rescue cell growth. Interestingly, cells expressing the pathogenic mutant Y120D maintain the growth defect associated with the loss of MeCP2, therefore demonstrating that an intact Tyr-120 residue is essential for the proliferative function of MeCP2.
FIGURE 5.

MeCP2 deficiency affects cell proliferation. A, MEFs from WT or Mecp2-null animals were plated (30,000 cells) and counted at the indicated time points (n = 4). The lack of MeCP2 in the null cells was verified by WB. B, MRC-5 cells (150,000 cells) were transfected with two different siRNAs against MeCP2 or the corresponding controls (Ctr.). Cell numbers were analyzed at the indicated time points starting at 4 days post-transfection (0 h) when MeCP2 expression was efficiently silenced, as verified by WB. Because of the proliferation defect determined by the deficiency in MeCP2, at time 0 the number of cells is different between the test and the control samples (n = 3). C, MeCP2 or the Y120D derivative fused to GFP was introduced into MRC-5 cells by transfection 4 days post-silencing, and cell numbers were determined after 48 h. The increase in cell numbers was calculated with respect to the number at the time of transfection. The graph integrates the results obtained from four independent experiments (mean ± S.E.). **, p < 0.01, unpaired Student's t test. The Western blot below the graph shows one representative experiment of exogenously expressed GFP-MeCP2 and the mutant derivative, using actin as loading control.
To confirm and further define the growth defect associated with the lack of MeCP2, we analyzed cellular DNA content by flow cytometric analysis. As shown in Fig. 6A, the depletion of MeCP2 increases the fraction of G2/M cells, which have a duplicated complement (4N) of DNA, suggesting that depleted cells are delayed or arrested in mitosis or display a cytokinesis phenotype. Furthermore, an increase of the sub-G1 population was observed, indicating an increase of dead cells. In this regard, it has already been reported that altered MeCP2 levels affect cell death by inducing apoptosis and senescence (32, 33). Accordingly, by staining for activated caspase, we observed some increase of apoptosis in MeCP2-ablated cells compared with the control group (Fig. 6B).
FIGURE 6.

The loss of MeCP2 alters progression through the cell cycle. A, the DNA content of MRC-5 cells silenced for 4 days with siMeCP2#1 or with the corresponding control siRNA (right and left panels, respectively) was determined by propidium iodide staining. A representative FACS analysis is shown for each condition, with the small insets demonstrating MeCP2 levels at the time of analysis. The right panel shows the numbers of cells in the different phases of the cell cycle. The data are representative of three separate experiments. B and C, asynchronously growing MRC-5 cells were stained for caspase-3 or phosphorylated histone H3 (Ser-10) and DAPI 4 days post-transfection with siMeCP2#1or a control siRNA (Ctr.), and the percentage of cleaved caspase-3- and phospho-H3-positive cells was calculated. Approximately 2000 cells were counted for each point in three independent experiments (mean ± S.E.). *, p < 0.05, unpaired Student's t test. D, Western blot of mouse embryonic fibroblasts from wild-type or Mecp2-null embryos with antibodies against MeCP2, Cyclin B, or, as a loading control, α-tubulin (n = 2). E, control MRC-5 cells or cells silenced for MeCP2 were stained with α-tubulin and DAPI, and the number of binucleated cells was determined. The graph shows the average of two experiments counting ≥1000 cells (mean ± S.E.). *, p < 0.05; unpaired Student's t test. F, selected frames from live-cell time-lapse microscopy of HeLa cells 80 h after transfection with a control siRNA or siMeCP2#1. Time is shown in minutes:seconds. Scale bar = 10 μm. G, quantification of duration of mitosis of HeLa cells transfected with control (n = 50) or MeCP2 (n = 150) siRNA. Means ± S.E. are shown. siMeCP2 cells exhibited a near 2-fold mitosis duration (mean ± S.E.). ***, p < 0.001; unpaired Student's t test (n = 3).
The increase in G2/M cells was further supported by measuring the proportion of cells positive for mitosis-specific markers such as histone H3 phosphorylated on serine 10 (Fig. 6C, P-H3) in MeCP2-depleted cells and cyclin B in Mecp2-null MEFs (Fig. 6D). In both cases, a significant increase of these markers was demonstrated. Moreover, upon MeCP2 RNAi treatment, the number of binucleated cells increased approximately 2-fold with respect to control cells (Fig. 6E). To better understand the nature of the proliferative defect induced by MeCP2 depletion, we analyzed asynchronous HeLa cells by time-lapse microscopy (Fig. 6F and supplemental Movie 1). Interestingly, this analysis revealed a significant increase in the duration of mitosis in MeCP2-depleted cells, which was doubled compared with control cells. No cytokinesis defects were observed (Fig. 6, F and G, and supplemental Movie 2). The observed mitotic defect, together with the finding that MeCP2 localizes to centrosomes, prompted us to study whether it can affect mitotic spindle assembly, which, in animal cells, is orchestrated by centrosomes. As above, MRC-5 cells were transfected with two independent MeCP2 siRNAs and their respective controls, and spindle morphology was analyzed by immunostaining mitotic spindles with an antibody recognizing phosphorylated Aurora A, B, and C kinase. DNA was visualized through DAPI staining (Fig. 7A). As expected, we found that most of the control cells have normal mitotic bipolar spindles, with DNA aligned at the metaphase plate. On the contrary, although bipolar spindles exist in the MeCP2-depleted cells, a large fraction of them are characterized by abnormal morphology. In fact, as represented in Fig. 7A and quantified in Fig. 7B, mitotic MeCP2 RNAi-treated cells show a significantly higher percentage of monopolar and an increased tendency of multipolar spindles with respect to control cells (Fig. 7B). In addition, as shown in Fig. 7C, when the cells displayed bipolar metaphases, the distance between the spindle poles was abnormally short in the MeCP2-depleted group with respect to the control. Complementarily, we assessed the consequences of MeCP2 depletion on cell cycle progression using time-lapse imaging of HeLa cells stably expressing GFP-tagged α-tubulin (Fig. 8A and supplemental Movie 3). In accordance with the above results, the analyses confirmed an increased length of mitosis and three types of abnormal spindle organization. In the first case, microtubules formed multiple clusters that ultimately coalesced to form bipolar spindles, leading to morphologically normal anaphase and telophase (Fig. 8A, siMeCP2#1, top panel, and supplemental Movie 4). In the second case, the tubulin clusters produced multipolar spindles that led to abnormal anaphases, often ending with the formation of binucleated cells (Fig. 8A, siMeCP2#1, center panel, and supplemental Movie 5). In the third case, cells formed bipolar spindles, but the mitotic spindle angle to the substratum plane was more variable during mitosis. Indeed, it tilted several times before cell division (Fig. 8A, siMeCP2#1, bottom panel, and supplemental Movie 6). The quantification of these phenotypes is shown in Fig. 8B. The mitotic consequences of the lack of MeCP2 were also addressed in Mecp2-null MEFs that are genetically homogeneous. Time-lapse microscopy imaging (Fig. 8C) revealed misoriented cell divisions, characterized by one daughter cell dividing outside the plane of the substratum. This phenotype, probably caused by the spindle misorientation described above (34), permits cells to progress through division, although a tendency of a prolonged mitosis was often observed. Fig. 8D shows a quantification of the observed misoriented division plane.
FIGURE 7.
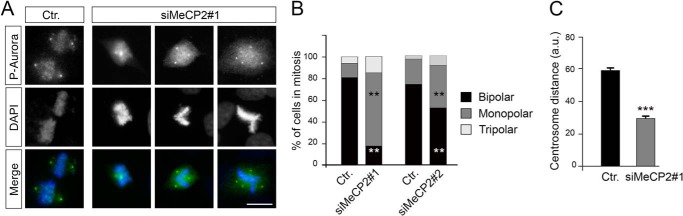
MeCP2 depletion causes defects in spindle pole geometry. A, spindle pole formation was analyzed in MRC-5 cells depleted or not depleted for MeCP2 by staining against phosphorylated aurora A, B, and C kinases (green) and with DAPI (blue) (n > 3). Ctr., control. B, the presence of mono-, bi-, and tripolar spindles was quantified from cells treated as in A. The graph shows the average of three independent experiments counting ≥180 cells. **, p < 0.01 for mono- and bipolar spindles with both siMeCP2#1 and #2 (mean ± S.E., unpaired Student's t test). C, mitotic MRC-5 cells silenced or not silenced for MeCP2 were stained with an anti-γ-tubulin antibody and DAPI. The distance between the centrosomes in cells with bipolar spindles was analyzed with ImageJ. The graph represents the average of three independent experiments counting ≥80 cells (mean ± S.E.). ***, p < 0.001; unpaired Student's t test). Scale bar = 10 μm.
FIGURE 8.

In the absence of MeCP2, several spindle aberrations are observed. A, selected frames of different classes of mitotic defects that were analyzed from live-cell time-lapse microscopy of HeLa cells stably expressing α-tubulin-GFP 80 h after treatments with control (Ctr.) or siMeCP2#1. Time is shown in minutes:seconds. Defects include ectopic MT clusters, multipolar spindle, and spindle tilt (spindle poles in different focal planes), as shown in representative x-y (maximum projection) and x-z (single plane) confocal images (bottom panel) (n = 3). Scale bar = 10 μm. B, manual quantification of mitotic defects from movies of α-tubulin-GFP-expressing HeLa cells transfected with control (n = 75) or MeCP2 (n = 234) siRNA. Means and S.E. are shown. *, p < 0.05; ***, p < 0.001; unpaired Student's t test). C, time-lapse images show uneven timing of daughter cell flattening onto the substrate after mitosis (misoriented cell division) in Mecp2-null MEFs compared with the wild type. Images represent MEFs from two different WT and null embryos, respectively. Time is shown in minutes. Scale bars = 10 μm, n = 2. D, manual quantification of mitotic defects from movies of WT (n = 50) and Mecp2-null MEFs (n = 60). Means ± S.E. are shown. *, p < 0.05; unpaired Student's t test).
Together, these observations suggest that MeCP2 deficiency does not influence cytokinesis but, rather, that it causes prolonged and/or aberrant mitosis.
Centrosomal Microtubule Nucleation Is Facilitated by MeCP2
As mentioned, centrosomes play a key role in mitotic spindle assembly by serving as the primary site of microtubule nucleation. Because our data demonstrate that loss of function of MeCP2 leads to various defects in centrosome maturation and bipolar spindle assembly, we investigated whether the microtubule nucleating potential of MeCP2-deficient centrosomes was compromised. Therefore, MRC-5 cells transfected with control or MeCP2 siRNAs were treated with nocodazole to depolymerize microtubules. The drug was then washed out, and microtubule regrowth was allowed for 2, 5, and 10 min (Fig. 9). In control cells, the microtubule-organizing centers nucleated microtubules in the first minute (Fig. 9A, Control). By contrast, in MeCP2-depleted cells, few short microtubules were visible after 5 min of recovery, whereas they were almost undetectable after 2 min (Fig. 9A, siMeCP2#1 and siMeCP2#2). Quantification of the results shown in the left panels (Fig. 9B, siMeCP2#1) demonstrates that, 10 min after nocodazole removal, less than 30% of MeCP2-deficient cells had initiated the nucleation process from centrosomes, whereas almost 80% of the control cells showed regrowing microtubules. Furthermore, cytoplasmic asters were dramatically smaller in MeCP2-interfered cells compared with control cells (Fig. 9C). Therefore, in MeCP2 knockdown cells, the microtubule nucleation potential of the centrosome is defective, indicating a role of MeCP2 in organizing nascent microtubules.
FIGURE 9.

MeCP2 deficiency affects microtubule nucleation. A, MRC-5 cells were transfected with siMeCP2#1 or #2 and their respective control (Ctr.) siRNAs, and, after 4 days of silencing, microtubules were depolymerized by adding 10 μg/ml nocodazole for 1 h. Microtubule regrowth was tested by releasing the cells in fresh medium for 2 or 5 min, and then they were fixed and stained with anti α-tubulin (red). Nuclei were stained with DAPI (blue). The silencing of MeCP2 was verified by Western blotting, using actin as loading control (n = 3 for each siRNA). Scale bars = 10 μm. B, microtubule nucleation capacity of the centrosome in cells silenced or not silenced for MeCP2 in one representative experiment of three independent experiments showing the same tendency. After nocodazole treatment, the cells were released for 2, 5, and 10 min (n = 300 cells). C, aster area in cells after 5-min release upon nocodazole treatment (n = 70 cells in three independent experiments, mean ± S.E.). ***, p < 0.001; unpaired Student's t test.
DISCUSSION
Although the best defined MeCP2-related condition is Rett syndrome, MECP2 mutations have been associated with a broad spectrum of neuropsychiatric disorders. Several studies supported the idea that MECP2-related disorders are exclusively caused by a deficiency of the methyl-binding protein in neurons. However, MeCP2 is present in glia, where it exerts biologically relevant functions (20, 35). Accordingly, Mecp2 reactivation in null astrocytes significantly improves several symptoms of the null mice. Further, the presence of wild-type microglia in Mecp2-null mice improves life span, breathing patterns, and locomotor activity (36). A role of MeCP2 for heart and skeletal development has also been proposed (37), whereas genetic polymorphisms in the MECP2 locus have been associated with increased susceptibility to the autoimmune disease lupus erythematosus (38). Eventually, a link between MeCP2 and the growth control of normal and cancer cells has been suggested by few publications (see Refs. 29–32 and the references therein).
Although these data indicate a role of MECP2 as a key gene, we are still missing a comprehensive understanding of which of its functions are involved in the observed MeCP2-associated phenotypes. In fact, as already described, MeCP2 appears as a multifunctional protein that changes its activity in function of its protein partners. Accordingly, MeCP2 is capable of several labile protein-protein interactions (39). Two molecular mechanisms might explain this versatility. First, MeCP2 is an intrinsically disordered protein (5, 40), and it is well known that the function of intrinsically disordered proteins is generally coupled to the acquisition of local secondary structures upon binding to other macromolecules (41). Second, a number of different posttranslational modifications of MeCP2 have been discovered recently. It is generally assumed that, depending on specific PTMs, the protein changes structure and partners, therefore modulating its biological functions (16).
The relevance of MeCP2 PTMs as fine-tuners of its functions appears from mouse genetics. Indeed, S80 or S421 phosphodefective knockin mice display subtle but consistent neurological symptoms (14, 15). Surprisingly, to the best of our knowledge, no human MECP2 point mutation that directly involves phosphorylation targets has ever been reported. However, the pathogenic R306C mutation disrupts the MeCP2/NCoR interaction (42), which is also negatively regulated by Thr-308 phosphorylation (43). Phosphodefective Mecp2T308A/y knockin mice show severe RTT-like symptoms, suggesting the relevance of this phospho-site for RTT onset.
Therefore, starting from the firm belief that the identification and characterization of human pathogenic mutations have the potential to identify important functional motifs and mechanisms of action of MeCP2, we decided to characterize the novel Tyr-120 phospho-isoform of MeCP2. The relevance of this PTM was suggested by its conservation through evolution and, more importantly, by a report in which a patient affected by a variant form of RTT carries a so-called phosphomimetic mutation (17). These studies permitted us to reveal the unexpected association of MeCP2 with the centrosome, indicating yet another function of the methyl-binding protein and possibly explaining its link to cell growth. Our major conclusions are that MeCP2 and its Tyr-120 phospho-isoform localize at the centrosome in dividing and postmitotic cells; that, in accordance with its novel localization, MeCP2 affects cell growth, spindle geometry, and microtubule nucleation; and that the Tyr(P)-120 isoform of MeCP2 appears to be particularly linked to the cell cycle.
MeCP2 and Its Tyr-120 Phospho-isoform Localize at the Centrosome in Dividing and Postmitotic Cells
By exploiting a phospho-specific Tyr-120 antiserum, the MeCP2 Tyr-120 phospho-isoform was found to accumulate on the mitotic spindle of cultured cells. The molecular connection of MeCP2 to the centrosome and the spindle was reinforced using largely validated commercial polyclonal antibodies and cellular and biochemical approaches. Of note, our fractionation experiments, performed using overexpressed derivatives of MeCP2, seem to indicate that the phosphorylation of Tyr-120 favors, but is not strictly required for, the association of MeCP2 with the centrosome. Furthermore, we believe that the high abundance of MeCP2 in the nucleus has so far hindered the possibility of its detection at the centrosome in cultured cells. In fact, the centrosomal localization was only detected in cells that are generally characterized by centrosomes scarcely overlapping with nuclei. Accordingly, the biochemical purification of centrosomal proteins has demonstrated that a minor fraction of the methyl-binding protein (less than 1%) cofractionates with the centrosome, therefore supporting the difficulty in its detection.
MeCP2 Affects Cell Growth, Spindle Geometry, and Microtubule Nucleation
The colocalization of MeCP2 with the centrosome encouraged us to study the possible functional connection of the methyl-binding protein with this cellular organelle in cycling cells. We propose, for the first time, that MeCP2, in addition to its activity in the nucleus, is functionally associated with centrosomes, therefore suggesting another mechanism by which MeCP2 can affect cellular processes. In fact, similar to other centrosomal proteins, MeCP2 affects spindle formation and/or cell plane division. The existence of spindle aberrations in MeCP2-deficient cells is confirmed by the observed increase in multinucleated cells. We also observed a less significant increase in the number of micronuclei (data not shown). Together, these findings support a role of MeCP2 in cell proliferation and genetic stability. Importantly, similar functions have already been linked to the methyl-binding protein. Our findings expand these previous publications and provide a novel avenue by which MeCP2 may regulate them. Therefore, future studies should address how MeCP2 and the Tyr-120 residue act on centrosomes, analyzing the composition and the recruitment of proteins to centrosomes and mitotic spindles.
The functional connection of MeCP2 with the centrosome has been further reinforced by the impaired microtubule nucleation in MeCP2-deficient cells. Again, these results extend a previous publication describing a defect in microtubule polymerization in RTT patient fibroblasts (44). Microtubules and centrosomes play an important role in cell migration, a finely tuned process of high relevance for tissue development, chemotaxis, and wound healing. Microtubules are also essential for vesicle transport. Interestingly, a previous publication demonstrated a defect in wound healing in cells devoid of MeCP2 (45). Although the authors tried to reconcile the obtained data with the transcriptional functions of MeCP2, we suggest that its effect on centrosome activity has to be considered. Similarly, a role of MeCP2 on axonal transport of vesicles has been described, possibly mediated through the Huntingtin/Hpa1 pathway (46). In the future, it will be interesting to address whether the described novel centrosomal function of MeCP2 described here contributes to the observed phenotype.
The Tyr(P)-120 Isoform of MeCP2 Appears to Be Particularly Linked to the Cell Cycle
Our phospho-specific antiserum showed, in accordance with Dephoure et al. (25), an enrichment of MeCP2 Tyr(P)-120 in dividing cells, therefore suggesting a role of this phospho-isoform in cell division. Accordingly, the proliferation defects associated with the lack of MeCP2 can be rescued by the WT protein but not its Y120D derivative. We hypothesize that the capability of this residue to affect cell proliferation might occur through the centrosomal localization of MeCP2. This hypothesis, however, is not mutually exclusive, with an effect on chromatin affinity and, therefore, on transcription. In fact, results published previously and our unpublished results5 suggest that the pathogenic Y120D derivative of MeCP2 has a reduced affinity for heterochromatin (18, 19).
Importantly, while revising the paper, Li et al. (47) have demonstrated that MeCP2 Ser-421 phosphorylation is linked to the cell cycle of adult hippocampal neuronal progenitor cells, in which it affects the balance between proliferation and neural differentiation. In adult hippocampal neuronal progenitor cells, the centrosomal Aurora kinase B interacts with and directly phosphorylates MeCP2 Ser-421. As further support of our work, the authors have hypothesized that MeCP2 phosphorylation is regulated by diverse stimuli and signaling pathways and that it might lead to different functional outcomes depending on cell type. In this regard, we want to underline that, because the hallmark of Rett syndrome is the regression phase that leads to the dramatic loss of early acquired developmental skills, MECP2 has been generally considered to be the cause of neurodevelopmental disorders, and most attention has been paid to its role in mature neurons. Therefore, its function in neuronal progenitor proliferation and differentiation remains largely unknown. However, a significant body of experimental evidence points to an early role of MeCP2, including family videos of the perinatal age of young patients, ultrasound recordings of Mecp2-null mouse pups a few days old, and human hemizygous male patients who show the disease immediately after birth (48, 49).
Considering all of the above, the adult reversibility of the disease and recent data demonstrating that ablation of Mecp2 in adult mice results in features resembling those of the animals constitutively missing MeCP2 (50–52), we suggest that both the maturing and mature brain continuously rely on the methyl binding protein. Our future studies will address the relevance of Tyr-120 for neural stem cell proliferation, differentiation, and migration.
Acknowledgments
We thank Dr. Andrea Musacchio, Dr. Marco Muzi Falconi, and Dr. Anna de Antoni for technical assistance, scientific suggestions, and reagents.
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) Grant IG-10319 (to N. L.), by Telethon Grants GGP10032 (to N. L.) and GGP12094 (to F. D. C.), by Ministero della Salute (Ricerca finalizzata 2008–Bando Malattie Rare) (to N. L.), by a Fondation Jerome Lejeune grant (to F. D. C.), by the Italian Association proRETT Ricerca, and by ProRETT Ricerca, an Italian association of parents.

This article contains supplemental Movies 1–6.
A. Bergo, M. Strollo, M. Gai, I. Barbiero, G. Stefanelli, S. Sertic, C. Cobolli Gigli, F. Di Cunto, C. Kilstrup-Nielsen, and N. Landsberger, unpublished data.
- RTT
- Rett syndrome
- PTM
- posttranslational modification
- MEF
- mouse embryonic fibroblast
- λ-PPase
- λ-phosphatase
- WB
- Western blot.
REFERENCES
- 1. Jones P. L., Veenstra G. J., Wade P. A., Vermaak D., Kass S. U., Landsberger N., Strouboulis J., Wolffe A. P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191 [DOI] [PubMed] [Google Scholar]
- 2. Nan X., Ng H. H., Johnson C. A., Laherty C. D., Turner B. M., Eisenman R. N., Bird A. (1998) Transcriptional repression by the methyl-CpG binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389 [DOI] [PubMed] [Google Scholar]
- 3. Amir R. E., Van den Veyver I. B., Wan M., Tran C. Q., Francke U., Zoghbi H. Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188 [DOI] [PubMed] [Google Scholar]
- 4. Guy J., Gan J., Selfridge J., Cobb S., Bird A. (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bedogni F., Rossi R. L., Galli F., Cobolli Gigli C., Gandaglia A., Kilstrup-Nielsen C., Landsberger N. (2014) Rett syndrome and the urge of novel approaches to study MeCP2 functions and mechanisms of action. Neurosci. Biobehav. Rev. 46, 187–201 [DOI] [PubMed] [Google Scholar]
- 6. Georgel P. T., Horowitz-Scherer R. A., Adkins N., Woodcock C. L., Wade P. A., Hansen J. C. (2003) Chromatin compaction by human MeCP2: assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 278, 32181–32188 [DOI] [PubMed] [Google Scholar]
- 7. Skene P. J., Illingworth R. S., Webb S., Kerr A. R., James K. D., Turner D. J., Andrews R., Bird A. P. (2010) Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muotri A. R., Marchetto M. C., Coufal N. G., Oefner R., Yeo G., Nakashima K., Gage F. H. (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chahrour M., Jung S. Y., Shaw C., Zhou X., Wong S. T., Qin J., Zoghbi H. Y. (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Y., Wang H., Muffat J., Cheng A. W., Orlando D. A., Lovén J., Kwok S. M., Feldman D. A., Bateup H. S., Gao Q., Hockemeyer D., Mitalipova M., Lewis C. A., Vander Heiden M. G., Sur M., Young R. A., Jaenisch R. (2013) Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 13, 446–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mellén M., Ayata P., Dewell S., Kriaucionis S., Heintz N. (2012) MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Young J. I., Hong E. P., Castle J. C., Crespo-Barreto J., Bowman A. B., Rose M. F., Kang D., Richman R., Johnson J. M., Berget S., Zoghbi H. Y. (2005) Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U.S.A. 102, 17551–17558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ricciardi S., Boggio E. M., Grosso S., Lonetti G., Forlani G., Stefanelli G., Calcagno E., Morello N., Landsberger N., Biffo S., Pizzorusso T., Giustetto M., Broccoli V. (2011) Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 20, 1182–1196 [DOI] [PubMed] [Google Scholar]
- 14. Zhou Z., Hong E. J., Cohen S., Zhao W. N., Ho H. Y., Schmidt L., Chen W. G., Lin Y., Savner E., Griffith E. C., Hu L., Steen J. A., Weitz C. J., Greenberg M. E. (2006) Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tao J., Hu K., Chang Q., Wu H., Sherman N. E., Martinowich K., Klose R. J., Schanen C., Jaenisch R., Wang W., Sun Y. E. (2009) Phosphorylation of MeCP2 at serine 80 regulates its chromatin association and neurological function. Proc. Natl. Acad. Sci. U.S.A. 106, 4882–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bellini E., Pavesi G., Barbiero I., Bergo A., Chandola C., Nawaz M. S., Rusconi L., Stefanelli G., Strollo M., Valente M. M., Kilstrup-Nielsen C., Landsberger N. (2014) MeCP2 post-translational modifications: a mechanism to control its involvement in synaptic plasticity and homeostasis? Front. Cell. Neurosci. 8, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Inui K., Akagi M., Ono J., Tsukamoto H., Shimono K., Mano T., Imai K., Yamada M., Muramatsu T., Sakai N., Okada S. (2001) Mutational analysis of MECP2 in Japanese patients with atypical Rett syndrome. Brain Dev. 23, 212–215 [DOI] [PubMed] [Google Scholar]
- 18. Agarwal N., Becker A., Jost K. L., Haase S., Thakur B. K., Brero A., Hardt T., Kudo S., Leonhardt H., Cardoso M. C. (2011) MeCP2 Rett mutation affects large scale chromatin organization. Hum. Mol. Genet. 20, 4187–4195 [DOI] [PubMed] [Google Scholar]
- 19. Kudo S., Nomura Y., Segawa M., Fujita N., Nakao M., Schanen C., Tamura M. (2003) Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J. Med. Genet. 40, 487–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rusconi L., Salvatoni L., Giudici L., Bertani I., Kilstrup-Nielsen C., Broccoli V., Landsberger N. (2008) CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 283, 30101–30111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guy J., Hendrich B., Holmes M., Martin J. E., Bird A. (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326 [DOI] [PubMed] [Google Scholar]
- 22. Reber S. (2011) Isolation of centrosomes from cultured cells. Methods Mol. Biol. 777, 107–116 [DOI] [PubMed] [Google Scholar]
- 23. Damen D., Heumann R. (2013) MeCP2 phosphorylation in the brain: from transcription to behavior. Biol. Chem. 394, 1595–1605 [DOI] [PubMed] [Google Scholar]
- 24. Thambirajah A. A., Eubanks J. H., Ausió J. (2009) MeCP2 post-translational regulation through PEST domains: two novel hypotheses: potential relevance and implications for Rett syndrome. BioEssays 31, 561–569 [DOI] [PubMed] [Google Scholar]
- 25. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 [DOI] [PubMed] [Google Scholar]
- 27. Azimzadeh J., Hergert P., Delouvée A., Euteneuer U., Formstecher E., Khodjakov A., Bornens M. (2009) hPOC5 is a centrin-binding protein required for assembly of full-length centrioles. J. Cell Biol. 185, 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mazzorana M., Montoya G., Mortuza G. B. (2011) The centrosome: a target for cancer therapy. Current Cancer Drug. Targets 11, 600–612 [DOI] [PubMed] [Google Scholar]
- 29. Balmer D., Arredondo J., Samaco R. C., LaSalle J. M. (2002) MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum. Genet. 110, 545–552 [DOI] [PubMed] [Google Scholar]
- 30. Guy J., Cheval H., Selfridge J., Bird A. (2011) The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652 [DOI] [PubMed] [Google Scholar]
- 31. Nagai K., Miyake K., Kubota T. (2005) A transcriptional repressor MeCP2 causing Rett syndrome is expressed in embryonic non-neuronal cells and controls their growth. Brain Res. Dev. Brain Res. 157, 103–106 [DOI] [PubMed] [Google Scholar]
- 32. Babbio F., Castiglioni I., Cassina C., Gariboldi M. B., Pistore C., Magnani E., Badaracco G., Monti E., Bonapace I. M. (2012) Knock-down of methyl CpG-binding protein 2 (MeCP2) causes alterations in cell proliferation and nuclear lamins expression in mammalian cells. BMC Cell Biol. 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bracaglia G., Conca B., Bergo A., Rusconi L., Zhou Z., Greenberg M. E., Landsberger N., Soddu S., Kilstrup-Nielsen C. (2009) Methyl-CpG binding protein 2 is phosphorylated by homeodomain interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 10, 1327–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delaval B., Bright A., Lawson N. D., Doxsey S. (2011) The cilia protein IFT88 is required for spindle orientation in mitosis. Nat. Cell Biol. 13, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ballas N., Lioy D. T., Grunseich C., Mandel G. (2009) Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Derecki N. C., Cronk J. C., Lu Z., Xu E., Abbott S. B., Guyenet P. G., Kipnis J. (2012) Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alvarez-Saavedra M., Carrasco L., Sura-Trueba S., Demarchi Aiello V., Walz K., Neto J. X., Young J. I. (2010) Elevated expression of MeCP2 in cardiac and skeletal tissues is detrimental for normal development. Hum. Mol. Genet. 19, 2177–2190 [DOI] [PubMed] [Google Scholar]
- 38. Webb R., Wren J. D., Jeffries M., Kelly J. A., Kaufman K. M., Tang Y., Frank M. B., Merrill J., Kimberly R. P., Edberg J. C., Ramsey-Goldman R., Petri M., Reveille J. D., Alarcón G. S., Vilá L. M., Alarcón-Riquelme M. E., James J. A., Vyse T. J., Moser K. L., Gaffney P. M., Gilkeson G. S., Harley J. B., Sawalha A. H. (2009) Variants within MECP2, a key transcriptional regulator, are associated with increased susceptibility to lupus and differential gene expression in patients with systemic lupus erythematosus. Arthritis Rheum. 60, 1076–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klose R. J., Bird A. P. (2004) MeCP2 behaves as an elongated monomer that does not stably associates with the Sin3A chromatin remodeling complex. J. Biol. Chem. 279, 46490–46496 [DOI] [PubMed] [Google Scholar]
- 40. Ausió J., de Paz A. M., Esteller M. (2014) MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 20, 487–498 [DOI] [PubMed] [Google Scholar]
- 41. Adams V. H., McBryant S. J., Wade P. A., Woodcock C. L., Hansen J. C. (2007) Intrinsic disorder and autonomous domain function in the multi functional nuclear protein, MeCP2. J. Biol. Chem. 282, 15057–15064 [DOI] [PubMed] [Google Scholar]
- 42. Lyst M. J., Ekiert R., Ebert D. H., Merusi C., Nowak J., Selfridge J., Guy J., Kastan N. R., Robinson N. D., de Lima Alves F., Rappsilber J., Greenberg M. E., Bird A. (2013) Rett syndrome mutations abolish the interaction of MeCP2 with NCoR/SMRT co-repressor. Nat. Neurosci. 16, 898–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ebert D. H., Gabel H. W., Robinson N. D., Kastan N. R., Hu L. S., Cohen S., Navarro A. J., Lyst M. J., Ekiert R., Bird A. P., Greenberg M. E. (2013) Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499, 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delépine C., Nectoux J., Bahi-Buisson N., Chelly J., Bienvenu T. (2013) MeCP2 deficiency is associated with impaired microtubule stability. FEBS Letters 587, 245–253 [DOI] [PubMed] [Google Scholar]
- 45. Yaqinuddin A., Abbas F., Naqvi S. Z., Bashir M. U., Qazi R., Qureshi S. A. (2008) Silencing of MBD1 and MeCP2 in prostate-cancer-derived PC3 cells produces differential gene expression profiles and cellular phenotypes. Biosci. Rep. 28, 319–326 [DOI] [PubMed] [Google Scholar]
- 46. Roux J. C., Zala D., Panayotis N., Borges-Correia A., Saudou F., Villard L. (2012) Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway. Neurobiol. Dis. 45, 786–795 [DOI] [PubMed] [Google Scholar]
- 47. Li H., Zhong X., Chau K. F., Santistevan N. J., Guo W., Kong G., Li X., Kadakia M., Masliah J., Chi J., Jin P., Zhang J., Zhao X., Chang Q. (2014) Cell cycle-linked MeCP2 phosphorylation modulates adult neurogenesis involving the Notch signalling pathway. Nat. Commun. 5, 5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marschik P. B., Kaufmann W. E., Sigafoos J., Wolin T., Zhang D., Bartl-Pokorny K. D., Pini G., Zappella M., Tager-Flusberg H., Einspieler C., Johnston M. V. (2013) Changing the perspective on early development of Rett syndrome. Res. Dev. Disabil. 34, 1236–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Filippis B., Ricceri L., Laviola G. (2010) Early postnatal behavioral changes in the Mecp2–308 truncation mouse model of Rett syndrome. Genes Brain Behav. 9, 213–223 [DOI] [PubMed] [Google Scholar]
- 50. Cheval H., Guy J., Merusi C., De Sousa D., Selfridge J., Bird A. (2012) Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum. Mol. Genet. 17, 3806–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nguyen M. V., Du F., Felice C. A., Shan X., Nigam A., Mandel G., Robinson J. K., Ballas N. (2012) MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McGraw C. M, Samaco R. C., Zoghbi H. Y. (2011) Adult neural function requires MeCP2. Science 333, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]