

Background: PrsA is a foldase for secreted proteins and pathogenicity factors in Gram-positive bacteria.
Results: Crystallographic, enzymatic, and NMR spectroscopic analysis provide insights to PrsA function.
Conclusion: Substrate peptides interact around a unique crevice generated by PrsA dimerization via its chaperone-like domain.
Significance: The comprehensive characterization of PrsA promotes its utilization as foldase and drug target.
Keywords: Biophysics, Cell Wall, Chaperone, Gram-positive Bacteria, Prolyl Isomerase, Protein Folding, Protein Secretion, X-ray Crystallography
Abstract
Secretion of proteins into the membrane-cell wall space is essential for cell wall biosynthesis and pathogenicity in Gram-positive bacteria. Folding and maturation of many secreted proteins depend on a single extracellular foldase, the PrsA protein. PrsA is a 30-kDa protein, lipid anchored to the outer leaflet of the cell membrane. The crystal structure of Bacillus subtilis PrsA reveals a central catalytic parvulin-type prolyl isomerase domain, which is inserted into a larger composite NC domain formed by the N- and C-terminal regions. This domain architecture resembles, despite a lack of sequence conservation, both trigger factor, a ribosome-binding bacterial chaperone, and SurA, a periplasmic chaperone in Gram-negative bacteria. Two main structural differences are observed in that the N-terminal arm of PrsA is substantially shortened relative to the trigger factor and SurA and in that PrsA is found to dimerize in a unique fashion via its NC domain. Dimerization leads to a large, bowl-shaped crevice, which might be involved in vivo in protecting substrate proteins from aggregation. NMR experiments reveal a direct, dynamic interaction of both the parvulin and the NC domain with secretion propeptides, which have been implicated in substrate targeting to PrsA.
Introduction
Protein folding is assisted by molecular chaperones and by foldases. Peptidyl-prolyl cis-trans isomerases (PPIases)3 constitute a class of foldases that occur in all types of cells and cell compartments (1). They catalyze the isomerization of peptide bonds preceding proline residues (2), which is often a rate-limiting step during protein folding (3). Three families of PPIases are known: the cyclophilins (4), the FK506-binding proteins (FKBP) (5), and the parvulins (6). Parvulins are ubiquitous globular proteins or protein domains of about 100 residues characterized by the parvulin fold (7), a four-stranded antiparallel β-sheet, surrounded by four α-helices (βα3βαβ2). Human Pin1 is the most prominent member of the parvulin family and is involved in phosphorylation-dependent signal transduction pathways, in transcriptional regulation, and cell cycle control (8). In prokaryotes, parvulins assist in the maturation of intracellular proteins, e.g. nitrogenase reductase (9) and various virulence factors (10, 11). A network of periplasmic PPIases, including SurA, FkpA, CypB, and Par10, supports the folding of periplasmic and outer membrane proteins in Gram-negative bacteria (12, 13). In Gram-positive bacteria, the parvulin-type PPIase PrsA is the only general factor mediating folding of secreted proteins, which are essential for bacterial pathogenicity and cell wall biosynthesis.
PrsA is a ubiquitous 30-kDa lipoprotein localized to the space between the plasma membrane and cell wall. It is tethered to the outer leaflet of the cell membrane by a lipid anchor, which is attached to its N-terminal cysteine residue (14, 15). PrsA is essential under normal growth conditions in Bacillus subtilis (16) and PrsA-depleted cells are affected by decreased cell wall integrity (17, 18), osmotic shock susceptibility (19), and increased sensitivity to antibiotics (20, 21). Overexpression of PrsA improves the recombinant overproduction of biotechnologically important proteins (22, 23). PrsA plays an important role as a folding factor of secreted proteins (14) including enzymes involved in cell wall biogenesis (18), toxins (25), and virulence factors (21, 26). Due to its general role for the maturation of pathogenicity factors, PrsA is a potential target for novel antimicrobial drugs.
At the sequence level, PrsA consists of three regions: a large N-terminal part with unknown function, followed by a parvulin-type PPIase domain (27) and a small C-terminal region (15). Despite its importance for protein secretion and pathogenicity in Gram-positive bacteria, the structural basis for PrsA function has so far remained unknown. Here, we determine the crystal structure of PrsA from B. subtilis and characterize its functional properties. Our data reveal a di-domain architecture, in which the catalytically active parvulin domain is inserted into a composite N- and C-terminal chaperone-like domain. Dimerization of a soluble PrsA variant at high concentrations and presumably of membrane-tethered PrsA is mediated by the chaperone-like domain and creates a bowl-shaped crevice, which may be required for in vivo chaperone activity of PrsA. NMR spectroscopic experiments provide initial insights to substrate interactions of PrsA.
EXPERIMENTAL PROCEDURES
Expression and Purification of PrsA
For the expression of full-length PrsA (residues 21–292) and C-terminal truncated version (residues 21–280) the gene fragments were amplified by PCR from B. subtilis (strain 168) and cloned into the expression plasmid pNIC28-Bsa4 (28), where they are N-terminally linked to the His6-tag followed by a tobacco etch virus protease cleavage site. The proteins were overexpressed in Escherichia coli BL21(DE3). After lysis of the cells in 50 mm Hepes/NaOH, 500 mm NaCl, pH 7.4, 40 mm imidazole with a sonicator and centrifugation, both proteins were found in soluble form. The proteins were purified by immobilized metal-affinity chromatography on a nickel-nitrilotriacetic acid column, digested overnight by tobacco etch virus protease, followed by an orthogonal nickel-nitrilotriacetic acid column step and then subjected to size exclusion chromatography in 20 mm Hepes/NaOH, pH 7.4, on a Superdex S75 column (GE Healthcare). The protein-containing fractions were pooled and concentrated in Amicon Ultra units (Millipore). [U-15N]PrsA was obtained by growing E. coli BL21(DE3) cells in M9 minimal medium supplemented with 1 g/liter of (15NH4)Cl (Cambridge Isotope Labs) at 37 °C until an A600 = 0.8 was reached. Expression was induced by addition of 1 mm isopropyl β-d-thiogalactopyranoside. Selenomethionine-derivatized PrsA was produced in M9 minimal medium. Cells were grown at 37 °C. At an A600 of 0.6 100 mg/liter of lysine, 100 mg/liter of phenylalanine, 100 mg/liter of threonine, 50 mg/liter of isoleucine, 50 mg/liter of leucine, 50 mg/liter of valine, and 100 mg/liter of dl-selenomethionine were added, and after 30 min of incubation, protein expression was induced by addition of 1 mm isopropyl β-d-thiogalactopyranoside. Cells were harvested 6 h after induction and purified in the same manner as the unlabeled protein. Yields were about 15–30 mg/liter. Reduced and carboxymethylated ribonuclease T1 (RCM-T1) was expressed and purified as described (29).
Prolyl Isomerase and Chaperone Activity Assays
The prolyl isomerase activities were measured by a protease-free fluorescence assay, as described (30). The kinetics of the decrease in cis-content was measured by the change in fluorescence at 416 nm (5 nm bandwidth) after excitation at 316 nm (3 nm bandwidth) in 100 mm potassium phosphate, pH 7.4, at 15 °C. Under these conditions, the cis to trans isomerization of the prolyl bond was a monoexponential process, and its rate constant was determined by using GraFit 5.0 (Erithacus Software, Staines, UK). The folding experiments of RCM-T1 were performed as described (29). The citrate synthase aggregation assay was measured as described (31). Citrate synthase was unfolded in 50 mm Tris-HCl, pH 8.0, 20 mm dithioerythritol, 6 m GdmCl for 1 h and then diluted 200-fold to a final concentration of 0.15 μm (monomer) in 50 mm Tris-HCl, pH 8.0, 0.1 mm dithioerythritol, 30 mm GdmCl, and various concentrations of prolyl isomerase at 25 °C. Spontaneous aggregation was monitored by measuring the increase in light scattering at 360 nm.
Thermal-induced Unfolding Transitions
The heat-induced unfolding transitions were measured by CD at protein concentrations of 0.5–4.0 μm in 100 mm potassium phosphate, pH 7.4. Samples were heated at a rate of 60 K h−1. The transitions were monitored by the increase of the CD signal at 222 nm with 1-nm bandwidth and 10-mm path length with a JASCO J710A spectropolarimeter (JASCO, Tokyo, Japan). The experimental data were analyzed on the basis of a three-state approximation with a fixed heat capacity change ΔCp of 6000 J mol−1 K−1 for the first transition and a ΔCp of 6000 J mol−1 K−1 for the second transition (32, 33).
NMR Spectroscopy
NMR experiments of PrsA were performed in NMR buffer containing 25 mm MES, 150 mm NaCl, pH 6.5. The measurements were recorded at 298 K on a Bruker AscendII 700 MHz spectrometer equipped with a cryogenically cooled triple-resonance probe. For the titration with AmyE-(ETANKSNELTA)-peptide two-dimensional [15N,1H]-TROSY-HSQC (34) spectra of 0.5 mm [U-15N]PrsA at different peptide concentrations titrated from a 50 mm stock solution were collected. NMR data were processed using PROSA (35) and analyzed with CARA (36). The chemical shift changes of the amide moiety were normalized according to Equation 1.
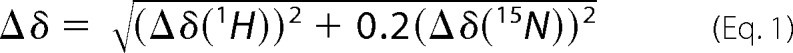 |
The chemical shift changes observed upon increasing AmyE-peptide concentrations were analyzed by non-linear regression to Equation 2,
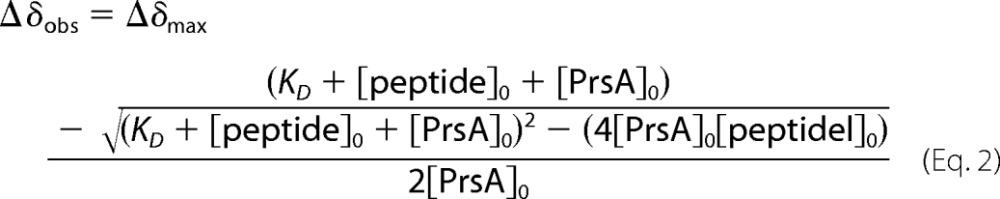 |
by using standard software.
SEC-MALS
For size exclusion chromatography coupled with multiangle light scattering (SEC-MALS) measurements, 20- or 100-μl samples of 1–4 mg/ml of protein were applied to a GE Healthcare Superdex 200 5/150 GL SEC column equilibrated overnight in 20 mm Hepes, pH 7.4, at 5 °C, using an Agilent 1100 series HPLC system. Light scattering and differential refractive index measurements were made using Wyatt miniDawn TriStar detector and a Wyatt Optilab rRex detector, respectively. The inter-detector delay volumes, band broadening, and the light scattering detector normalization, were calibrated according to the manufacturer's protocol using a 2 mg/ml of BSA solution (Thermo Pierce) run in the same buffer. The absolute refractive index of the buffer was measured using the refractive index detector. The data were collected and processed using Wyatt Astra 5 software. The molar mass was calculated from a global fit of the light scattering signals from three detectors at different angles, and the differential refractive index signal, using algorithms in the Astra 5 software.
Protein Crystallization and Structure Determination
Initial full-length PrsA crystals diffracted only to 6-Å resolution. After truncation of the 12 C-terminal residues (SNSTSSSSSNSK) PrsAΔC was crystallized under identical conditions as the full-length protein, but crystals diffracted to higher resolution. Best diffracting crystals were grown at 30 °C in 17% PEG2000MME, 0.1 m Hepes-NaOH, pH 7.5, within 4 days. The crystals were dehydrated and cryo-preserved by successively increasing the PEG2000MME to 50%. Diffraction data were collected at Swiss Light source X06DA beamline and processed using XDS (37). The crystals belong to space group C2221 with cell dimensions a = 71.5 Å, b = 88.4 Å, and c = 233.4 Å, α = β = γ = 90° and contain two molecules per asymmetric unit. The structure of PrsA was determined by selenomethionine single-wavelength anomalous dispersion phasing and refined to 2.63 Å. All expected 2 × 4 selenium sites were identified in the selenium-methionine dataset by Phenix-HySS (38). Initial automated model building was carried out with Phenix (39) and Buccaneer (40). Manual model building and structure refinement were performed with Coot (41) and PHENIX (42). The final model includes residues 3–259 of mature PrsA.
RESULTS
PrsA Is a Stable Two-domain Protein That Dimerizes at High Concentrations
In Gram-positive bacteria, PrsA-like lipoproteins have an N-terminal cysteine that is enzymatically modified upon signal sequence cleavage with a diacylglycerol residue to promote membrane association (43). We produced a recombinant soluble variant of the PrsA protein that comprises residues 21–292 excluding the signal peptide and amino-terminal residue Cys20. The circular dichroism spectrum shows a maximum near 190 nm and minima near 208 and 222 nm, as expected for a protein with a high content of helical secondary structure (Fig. 1A). Thermal unfolding of PrsA was monitored via the increase of the helical CD signal at 222 nm. The resulting curve can be decomposed into two transitions with midpoints (Tm1) at 46.5 °C and (Tm2) at 62.8 °C and unfolding enthalpies (ΔHD) of 247 and 211 kJ mol−1, respectively (Fig. 1B). To distinguish whether these transitions involve an oligomerization process, we set out to characterize the oligomeric state of soluble PrsA as a function of the protein concentration. The two thermal unfolding transitions are independent of the protein concentration in the range between 0.5 and 4 μm (Fig. 1C). Furthermore, at a concentration of about 25 μm, PrsA elutes in a single peak with monomer molecular weight from SEC-MALS (Fig. 2A). These data thus indicate that PrsA is monomeric in the concentration range up to at least 25 μm and that the two concentration-independent transitions observed during thermal unfolding (Fig. 1B) represent conformational unfolding reactions of individual domains of the monomeric PrsA.
FIGURE 1.
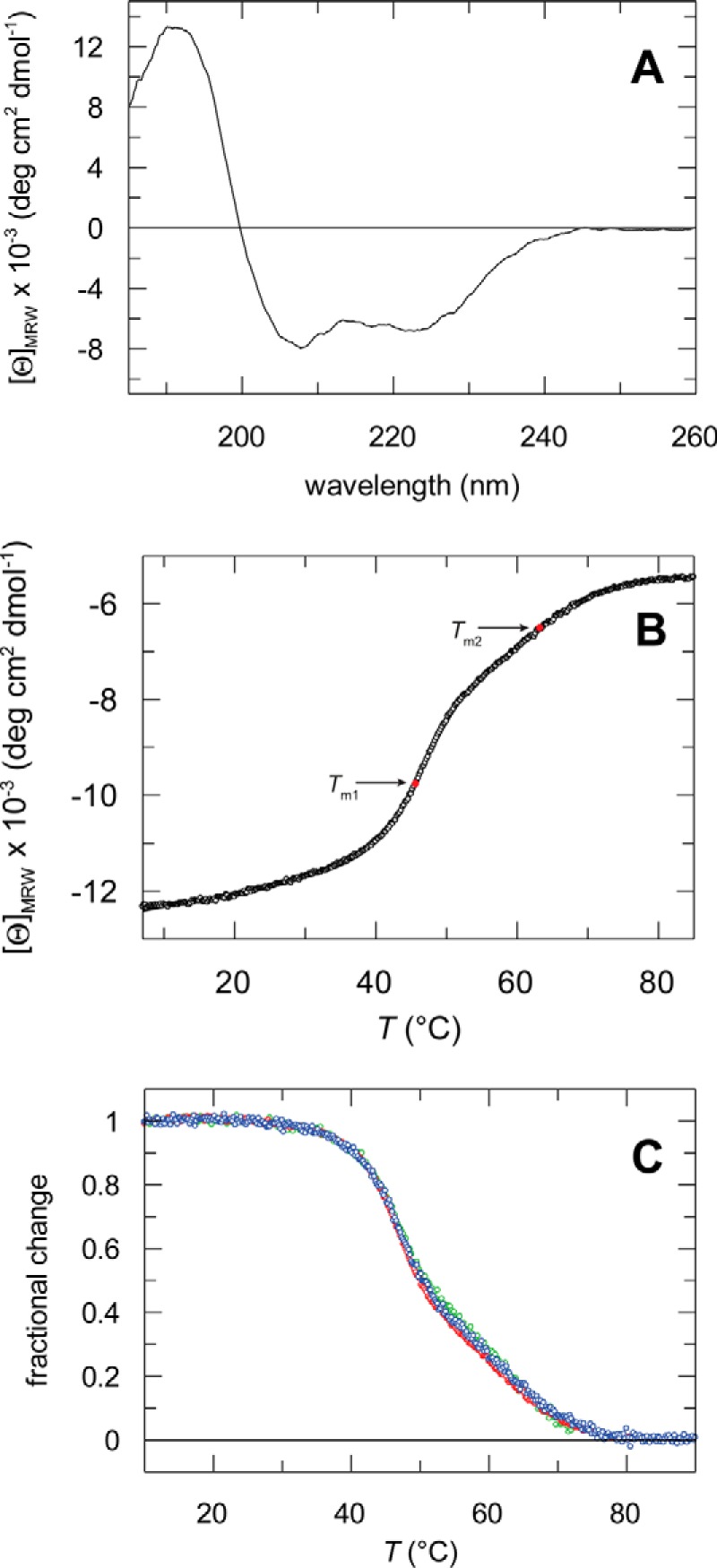
Circular dichroism spectra and thermal stability of PrsA. A, far-UV CD spectrum of PrsA at 15 °C. The spectrum was measured with 2 μm protein in 10 mm potassium phosphate, pH 7.0, at a path length of 1 mm and a bandwidth of 1 nm. B, thermal stability of PrsA. Thermal transition measured by CD at 222 nm for the wild-type protein. The change of the CD signal is shown as a function of temperature. The three-state analysis (continuous line) resulted in Tm1 = 46.5 ± 0.4 °C, ΔHD1 = 247 ± 15 kJ mol−1 and Tm2 = 62.8 ± 0.4 °C, ΔHD2 = 211 ± 15 kJ mol−1. Tm1 and Tm2 are indicated as red dots. The transition was measured with 2 μm protein in 100 mm potassium phosphate, pH 7.0, at a path length of 10 mm. C, thermal stability of PrsA at different protein concentrations. The fractional change after a three-state analysis is shown as a function of the temperature. The transitions were measured with 0.5 μm (red, Tm1 = 46.7 ± 0.2 °C, ΔHD1 = 259 ± 12 kJ mol−1, Tm2 = 62.4 ± 0.2 °C, ΔHD2 = 201 ± 13 kJ mol−1), 2 (green) and 4 μm (blue) (Tm1 = 46.2 ± 0.3 °C, ΔHD1 = 233 ± 14 kJ mol−1, Tm2 = 67.0 ± 0.3 °C, ΔHD2 = 218 ± 18 kJ mol−1) protein in 100 mm potassium phosphate (pH 7.0), at a path length of 10 mm.
FIGURE 2.
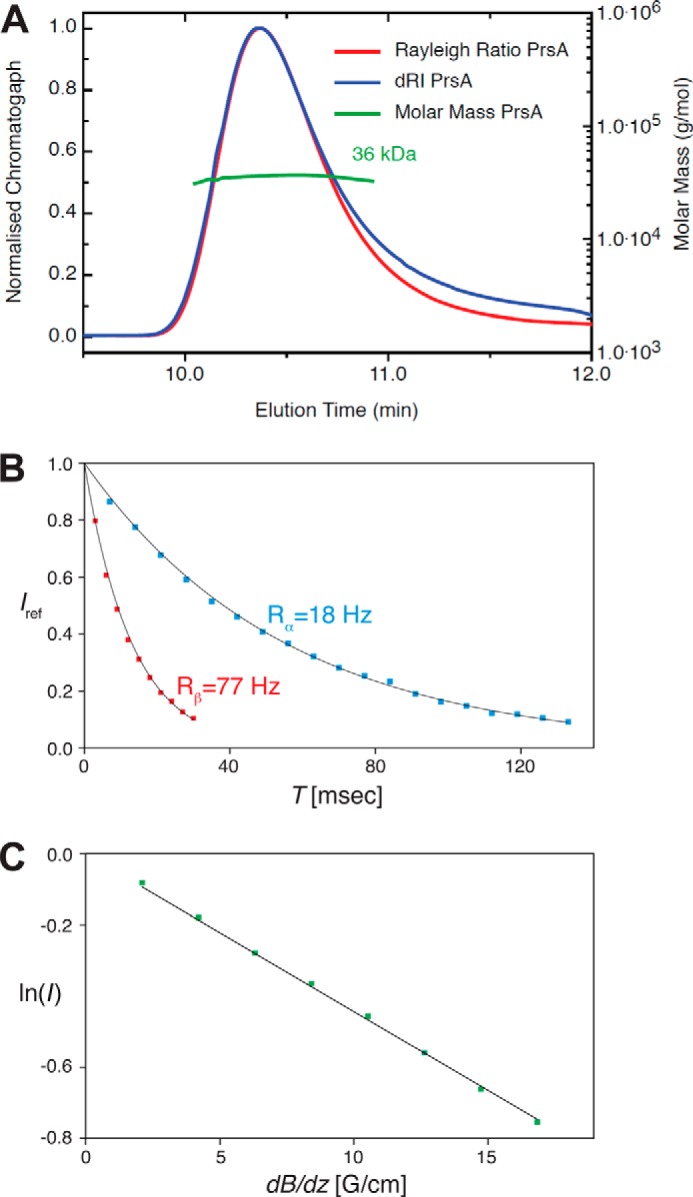
Molecular weight and biophysical molecular parameter determination of PrsA. A, size exclusion chromatography coupled to SEC-MALS of PrsA. The molecular mass was calculated throughout the eluting peaks and is indicated in green. The calculated mass of 36.0 kDa is in good agreement with the 33.4 kDa for the N-terminal His and tobacco etch virus protease cleavage site-tagged (MHHHHHHSSGVDLGTENLYFQ*SM (*, tobacco etch virus protease cleavage site) PrsA protein (residues 21–292). B, [15N,1H]-TRACT experiment (80) for the determination of the effective rotational correlation time constant τc. The one-dimensional proton signal intensity, Iref, of a sample of 750 μm PrsA at 25 °C was integrated between 10 and 8.5 ppm and plotted versus the relaxation period T. The transverse relaxation rates of the TROSY component (blue; black: least squares fit) and the anti-TROSY component (red; black: least squares fit) were determined to be Rα = 18 Hz and Rβ = 77 Hz, resulting in τc = 23 ns, that is typical for a globular protein of about 60 kDa size (81). C, measurement of the molecular diffusion constant in aqueous solution by the 15N-filtered diffusion BPP-LED NMR experiment (82) for 750 μm PrsA (green). The logarithm of the signal intensity, integrated between 10 and 8.5 ppm, is plotted versus the gradient strength of the pulsed field gradients. The black line represents the linear fit to the measured data. This measurement yielded the self-diffusion constants, D0 = 6.11 × 10−11 m2 s−1, that corresponds under the assumption of a spherical molecule and by using the Stokes-Einstein equation to an estimated molecular mass of 58 kDa.
Although SEC-MALS is an ideal tool to study oligomeric protein states at intermediate and low concentrations, it is not suited for measurements at high protein concentrations. However, increased concentrations best represent the in vivo situation of a membrane-anchored protein, where high local concentrations and molecular pre-orientation increases the likelihood of dimer formation by up to 106-fold (44). Thus, PrsA oligomerization was also analyzed at high protein concentrations by lateral diffusion and relaxation analyses of 15N-labeled protein by NMR spectroscopy. The TRACT and BPP-LED NMR experiments demonstrate that 750 μm PrsA is dimeric (Fig. 2, B and C), pointing to an inherent propensity of PrsA to dimerize based on direct interactions of its soluble regions. Lipid anchoring will further promote self-association, indicating that a dimer is the most likely physiological state of PrsA, in agreement with earlier in vivo data (18, 21).
Catalysis of Prolyl Isomerization by PrsA in Peptides and Proteins
To characterize the activity of PrsA as a prolyl isomerase and map its substrate specificity, we used a fluorimetric protease-free assay and a set of proline-containing tetrapeptides, which carry an aminobenzoyl (Abz) group at the amino terminus and a para-nitroanilide (pNA) group at the carboxyl terminus (30, 45). In the peptides of the general formula Abz-Ala-Xaa-Pro-Phe-pNA, the Xaa position was occupied by charged (Glu, Lys), aliphatic (Ala, Leu), or aromatic (Phe) residues. PrsA catalyzes peptide isomerization very well when hydrophobic residues such as Leu precede the proline (Abz-Ala-Leu-Pro-Phe-pNA), and 16 nm PrsA sufficed to double the isomerization rate (Fig. 3A). From the increase of the isomerization rate as a function of the PrsA concentration (Fig. 3C), the catalytic efficiencies (kcat/Km) of PrsA toward peptides with six different Xaa-Pro sequences were derived (Table 1). The highest catalytic efficiency was observed for Leu-Pro. For Phe-Pro the efficiency was 6-fold, for Ala-Pro, 60-fold lower. Peptides with charged residues such as Glu or Lys preceding the proline resembled the peptide with the Phe-Pro sequence in their kcat/Km values (Table 1). Our kcat/Km values are about 10–20-fold higher than those obtained previously for PrsA with a protease-coupled activity assay using N terminally succinylated Suc-Ala-Xaa-Pro-pNA peptides (Table 1) (15). The protease-coupled peptide assay can be used only for enzymes that are resistant to proteolysis, and therefore is unsuitable for most multidomain prolyl isomerases. Possibly, PrsA is sensitive to proteolysis and inactivated during the assay as found for trigger factor (46) and SlyD (47). With its kcat/Km value of 3 × 103 mm−1 s−1 for the Leu-Pro peptide, PrsA shows an activity that is 1000- and 10-fold higher than the activities of human parvulin 14 and E. coli SurA, respectively, and only 10-fold lower than the activity of E. coli parvulin 10, which is the most active parvulin known to date (48). In its substrate specificity with a preference for hydrophobic residues preceding proline, PrsA resembles other bacterial parvulin homologues (49, 50).
FIGURE 3.

Characterization of enzymatic activity of PrsA. A, kinetics of cis-trans isomerization of 3 μm Abz-Ala-Leu-Pro-Phe-pNa followed by fluorescence at 416 nm, without enzyme (black), with 12 (blue), 16 (green), and 20 nm (red) PrsA. B, refolding kinetics of RCM-T1 in the presence of increasing concentrations of PrsA, 0 (black), 300 (blue), 500 (green), and 1000 nm (red). Catalytic efficiencies of PrsA for (C) the cis-trans isomerization of Abz-Ala-Leu-Pro-Phe-pNA and (D) the refolding of RCM-T1. The measured rate constants kapp are shown as a function of the prolyl isomerase concentration. The kcat/Km values derived from the slopes are given in Table 1.
TABLE 1.
Catalytic efficiencies of B. subtilis PrsA for prolyl isomerization in peptide and protein substrates
| BsPrsAa (kcat/Km) | BsPrsAb (kcat/Km) | SaPrsA(140–245)c (kcat/Km) | |
|---|---|---|---|
| mm−1 s−1 | |||
| Peptide isomerization | |||
| Abz-Ala-Ala-Pro-Phe-pNA | 52 | 6 | 17 |
| Abz-Ala-Glu-Pro-Phe-pNA | 450 | 7 | 33 |
| Abz-Ala-Leu-Pro-Phe-pNA | 3 × 103 | NDd | ND |
| Abz-Ala-Lys-Pro-Phe-pNA | 300 | 15 | 5 |
| Abz-Ala-Phe-Pro-Phe-pNA | 500 | ND | ND |
| Abz-Ala-Asn-Pro-Phe-pNA | ND | 0.5 | 3 |
| Protein folding | |||
| RNase T1 | 8.7e | 25f | ND |
a Catalytic activities towards peptides of full-length PrsA from B. subtilis from protease-free experiments as described in the legend to Fig. 3. The confidence limits of the kcat/Km values were between 5 and 10%.
b Catalytic activities towards peptides of full-length PrsA from B. subtilis derived with a protease-coupled assay. Data taken from Refs. 15 and 64.
c Catalytic activities of the isolated parvulin domain of S. aureus SaPrsA(140–245). Data taken from Refs. 15 and 64.
d ND, not determined.
e Catalytic activity was determined using the refolding of RCM-T1 in 2 m NaCl, 100 mm Tris-HCl, pH 7.8, and 1 mm EDTA.
f Catalytic activity was determined using the refolding of urea denaturated RNase T1 in 0.28 m urea, 100 mm Tris-HCl, pH 7.8, and 1 mm EDTA.
The efficiency of PrsA as a catalyst of proline-limited protein folding (the protein folding activity) was determined by using RCM-T1 as a substrate protein. This variant cannot form disulfide bonds because the cysteine residues are modified. RCM-T1 is permanently unfolded in the absence of salt, but refolds spontaneously in 2 m NaCl, where electrostatic repulsion between negatively charged groups is decreased. In unfolded proteins, most of the proline residues adopt the trans conformation. Proline 39 is in the cis conformation in folded RCM-T1, but in the unfolded state 85% of the molecules contain trans-proline 39. As a consequence, the refolding of 85% of all molecules is limited in rate by trans → cis isomerization at Pro39, which shows a time constant of 530 s (at 15 °C, pH 8.0). The major part of the fluorescence change during RCM-T1 refolding reflects this isomerization and makes RCM-T1 an excellent substrate for assaying the folding activity PPIases (51–54). Relatively high PrsA concentrations were necessary to accelerate the refolding of RCM-T1 (Fig. 3B). The apparent rate constant of catalyzed folding increased in a linear fashion with the PrsA concentration suggesting that the affinity of PrsA for refolding RCM-T1 is rather low (Fig. 3D). A kcat/Km value of 8.7 mm−1 s−1 is derived from Fig. 3D. This value is similar to previously determined values (Table 1) (15) and also similar to the kcat/Km values obtained for the RCM-T1 refolding catalyzed by parvulin 10 or SurA from E. coli (48, 55). In its properties as a catalyst of protein folding, PrsA thus resembles other parvulins.
Crystal Structure of PrsA
PrsA(21–292) was crystallized under various conditions, but diffraction was always limited to 6-Å resolution. At their C termini, many PrsA orthologs contain serine-rich stretches of 5–15 residues. In PrsA, this sequence region was shown to be dispensable for function, in vivo and in vitro (15). The targeted deletion of the respective 12 C-terminal residues (SNSTSSSSSNSK) from PrsA improved diffraction to about 3.5-Å resolution under similar conditions as for the full-length protein. Crystal dehydration by successive addition of long chain polyethylene glycols (up to 50% PEG 2000 MME) led to a further improvement. Finally, the x-ray crystal structure of PrsA(21–280) was solved by single-wavelength anomalous dispersion for crystals of selenium-methionine-derivatized PrsA at 3.2-Å resolution. The structure of native PrsA was refined to R/Rfree of 22/25% at 2.63-Å resolution. Data collection and refinement statistics are given in Table 2.
TABLE 2.
Statistics on diffraction data and structure refinement of PrsA
| Data set | PrsA |
|---|---|
| Wavelength | 1.36246 |
| Space group | C 2 2 21 |
| Unit cell | 69.29 87.68 234.1 90 90 90 |
| Resolution (Å) | 117-2.63 (2.79-2.63)a |
| Total reflections | 144433 |
| Unique reflections | 21428 |
| Multiplicity | 6.7 (5.4) |
| Completeness (%) | 98.8 (90.2) |
| Mean I/σ(I) | 19.7 (1.6) |
| Wilson B-factor | 90.7 |
| R-merge | 0.054 (1.097) |
| CC1/2 | 0.998 (0.65) |
| Rwork | 0.2160 (0.4199) |
| Rfree | 0.2470 (0.4284) |
| Number of atoms | 8281 |
| Macromolecules | 4079 |
| Water | 37 |
| Protein residues | 513 |
| R.m.s. deviation (bonds) | 0.009 |
| R.m.s. deviation (angles) | 0.95 |
| Ramachandran favored (%) | 97 |
| Ramachandran outliers (%) | 0.2 |
| Clash score | 1.7 |
| Average B-factor | 109.1 |
| Macromolecules | 109.3 |
| Ligands | |
| Solvent | 89.9 |
a Values in parentheses are for highest resolution shell.
PrsA consists of two domains in a discontinuous arrangement (Fig. 4A): the central parvulin domain (Leu115-Glu207) (Fig. 4B) is connected via short linkers to a composite domain formed by the N-terminal region Ser4-Gly114 together with the C-terminal region Arg208-Ser260. This domain, which we call the NC domain, is almost exclusively α-helical, except for two short strands in the N-terminal region, which form an antiparallel β-sheet.
FIGURE 4.

Crystal structure of PrsA. A, schematic representation of the PrsA primary structure. B, PrsA monomer. The N-terminal domain, PPIase, and C-terminal domain are colored in blue, green, and red, respectively. C, superimposition of the parvulin domain of PrsA (gray) to the closest homologue of human Pin1 (Protein Data Bank code 1pin.pdb, green). Secondary structure elements as well as the substrate interacting loop (S1-H1) of Pin1 are indicated. D, comparison of the two PrsA molecules A and B in the asymmetric unit. PrsA molecule A is colored as in B. PrsA molecule B is shown in gray, superimposed on molecule A based on the parvulin domain. The tilt of secondary elements is indicated. E, dimeric structure of PrsA. PrsA molecules A and B are colored as in D. The estimated position of membrane and membrane anchor are indicated in schematic representation.
The PrsA parvulin domain consists of a four-stranded antiparallel β-sheet surrounded by four α-helices (βα3βαβ2 topology) and superimposes well with other parvulin-type PPIase domains (13, 27, 56–64). Structurally, it is most closely related to human Pin1 (56), although PrsA is of prokaryotic origin and exhibits a different substrate specificity. The strongest differences locate to the S1-H1 loop (Fig. 4C). In Pin1, this loop contains 17 residues including a cluster of positively charged residues, which determine the specificity of Pin1 for phosphorylated Ser/Thr-Pro substrates. In PrsA, this loop is shortened to only a 2-residue turn. The proline-binding pocket of PrsA is lined predominantly by hydrophobic residues. It also includes the highly conserved residues His123, Asp155, Thr195, and His200, which form an extended hydrogen bonding network (65) similar to the active sites of other parvulins (Fig. 5).
FIGURE 5.

Active site structure of PrsA (A) in comparison to Pin1 (B), SurA (C), and Par10 (D). Residues that contribute to the active site of the parvulin enzyme family are shown in stick representation and labeled. E, comparison of the sequences that define the active site of PPIase domains.
PrsA crystallizes with two monomers in the crystallographic asymmetric unit, which superimpose with an overall root mean square deviation (r.m.s. deviation) of 1.9 Å. The isolated domains are structurally very similar with r.m.s. deviation values of 0.7 Å for the PPIase and 1.2 Å for the NC domains; the larger overall r.m.s. deviation results from a slight hinge bending motion between the domains (Fig. 4D). The dimer interface involves 47 mainly N-terminal residues, it extends over an area of 1830 Å2 and is classified as stable (66). In the PrsA dimer, the N-terminal ends are in close proximity, in agreement with dimer formation of the full-length protein anchored via its lipid-linked N-terminal cysteine to the membrane anchors in B. subtilis (Fig. 4E).
The NC Domain Is Structurally Related to the Chaperone Domains of SurA and Trigger Factor
In addition to their catalytic domains, foldases often contain domains for transient and dynamic interactions with their substrates. The N-terminal region of PrsA starts with a pair of short antiparallel β-strands (Val9-Thr13 and Asp16-Thr19) followed by five α-helices before entering the parvulin domain. The C-terminal region is arranged into two long α-helices that tightly interact with the N-terminal helices. Together with the parvulin domain, the N- and C-terminal helices form an extended crevice (Fig. 4E). Interestingly, despite a lack of sequence homology, the NC domain is structurally related to the chaperone domains of trigger factor, which assists the folding of newly synthesized proteins at the ribosome (67, 68) and of SurA (61), a foldase for outer membrane proteins in Gram-negative bacteria. A related fold is also found in the less well characterized leptospiral protein LIC12922. Fig. 6 highlights the common structural elements of these four proteins. The 110 residues of PrsA (Met27-Leu115; Asp229–250) that build the central region of the NC domain superimpose well with SurA (Asp62-Ala161; Phe406-Ser427), trigger factor (Ser280-Lys361), and LIC12922 (Ser89-Val184; Glu322-Leu351) with r.m.s. deviation values of 3.3, 3.6, and 3.1 Å (62), respectively. The “arms” and “body” structure described initially for trigger factor (69) is present in all four proteins (Fig. 6A), but arm1 is significantly shortened in PrsA.
FIGURE 6.

The NC domain of PrsA shares a common fold with chaperone domains. A, schematic representation of the PrsA NC domain (cyan) with the body and arms modules, in comparison with the chaperone domains of SurA (red) and trigger factor (yellow), both from E. coli and LIC12922 from Leptospira interrogans (green). Structurally non-conserved regions are represented in gray, whereas conserved ones are highlighted. B, orientation of the PPIase (parvulin) domains (orange), relative to the NC domain. The color code is the same as in A. SurA contains two parvulin domains (ppiase1 and ppiase2). Trigger factor shows a FKBP-type PPIase and a ribosome binding domain (dark gray).
The linkers to the parvulin domain are relatively short (5 amino acids) in PrsA, and therefore the prolyl isomerase site is located in close proximity to the NC domain, such that helix α2 of the NC domain is in close proximity (5 Å) to helix α3′ of the parvulin domain (Fig. 6B). The orientation of the two domains is similar in LIC12922, although the parvulin domain is tilted relative to its position in PrsA by about 25 degrees, but differs from the organization of the well characterized trigger factor and SurA chaperones. In trigger factor, an FKBP-type PPIase is inserted into the chaperone domain, and the linkers between the two domains are much longer. SurA consists of a chaperone domain and two parvulin-type PPIase domains. In SurA, parvulin domain 1 is inactive and tightly associated with the chaperone (NC) domain, whereas the functionally active parvulin domain 2 is tethered to the NC domain also via a long, and presumably flexible linker (61) similar to the FKBP-type PPIase in trigger factor (Fig. 6B).
Monomeric PrsA in Solution Does Not Prevent Protein Aggregation
Many periplasmic folding factors are known to have dual functions as PPIases and chaperones (70). As the NC domain of PrsA shows structural similarity to chaperones we examined whether PrsA interacts with an unfolded protein. We performed citrate synthase (CS) aggregation and inactivation assays. GdmCl-unfolded CS aggregates spontaneously after dilution with refolding buffer, and this is accompanied by a strong increase in light scattering, unless aggregation is suppressed by a chaperone. PrsA was unable to inhibit the aggregation of CS (Fig. 7), whereas 3.0 μm SlyD from E. coli completely abolished aggregation of CS (71). Similar results have been previously obtained in a rhodanese aggregation assay (15). Importantly, under the assay conditions, non-membrane tethered PrsA is monomeric and does not self-associate into its presumably native dimeric form. As a distinct chaperone activity has been observed for PrsA in vivo, these results strongly suggest that dimerization, in line with our structural data, may be a pre-requisite for chaperone function of PrsA, although we cannot rule out that PrsA may only be active toward a limited set of substrates different from CS.
FIGURE 7.
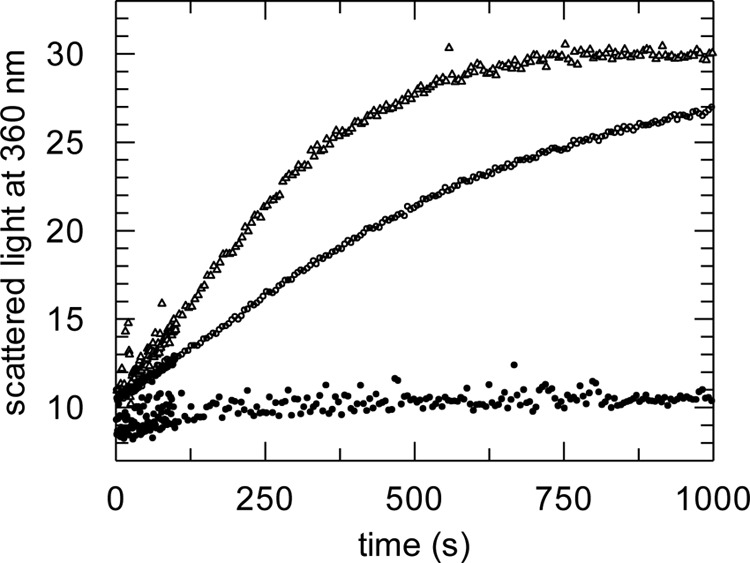
Monomeric PrsA in solution has no generic chaperone function. Citrate synthase chaperone assay. Influence of PrsA on the aggregation of chemically denatured citrate synthase at 25 °C. Denatured citrate synthase was diluted to a final concentration of 0.15 μm (monomer) in 100 mm Tris-HCl, pH 8.0, 1 mm EDTA, 30 mm GdmCl, 50 mm NaCl, and 0.1 mm dithioerythritol. Light scattering at 360 nm was monitored in the absence of enzyme (○) and in the presence of 3.0 μm SlyD* (●) and presence of 3.0 μm PrsA (Δ).
The Binding of PrsA to Presequence Involves Residues from the Parvulin and NC Domain
In Gram-positive organisms, exported proteins commonly contain a propeptide sequence, which follows the export signal sequence and increases export efficiency (23). The AmyE propeptide from the α-amylase of B. subtilis is 11 amino acids long (ETANKSNELTA) and contains no proline residues. When attached to other proteins, it improves their export (23, 72). If PrsA is overexpressed in addition to the AmyE attachment, the protein export is further increased (23), suggesting functional interplay and possibly a direct interaction of PrsA and the AmyE propetide.
To examine how PrsA interacts with the AmyE propeptide, we recorded two-dimensional [15N,1H]-TROSY spectra of 15N-labeled PrsA in the presence of the AmyE-peptide at increasing concentrations. The [15N,1H]-TROSY cross-peaks of [1H,15N]PrsA were well resolved and enabled a detailed analysis of the chemical shift changes upon addition of the AmyE propeptide (Fig. 8A). The resonances of the isolated parvulin domain of PrsA (residues 116–206 (27)) closely match the corresponding resonances of full-length PrsA. This indicates that most amide moieties of the parvulin domain in isolation and in full-length PrsA are in similar chemical environments and allows a direct transfer of sequence-specific resonance assignments for the analysis of chemical shift changes induced by binding of the AmyE propeptide to full-length PrsA.
FIGURE 8.

Experimental mapping of PrsA residues involved in AmyE-propeptide binding. A, left panel: [15N,1H]-TROSY spectra of 0.5 mm [U-15N]PrsA in 25 mm MES, pH 6.5, with increasing AmyE-peptide concentrations, as indicated. The protein to ligand ratio is given in parentheses. Right panels: enlargement of two regions with the assignment derived from the NMR structure of the PPIase domain alone ((27) BMRB 6601) as well as peaks belonging to the NC domain, that were analyzed. All spectra were measured at 25 °C with eight scans and 1024 × 200 complex points. B, NMR peak shift analysis of AmyE-peptide to PrsA titration. Combined chemical shift changes of the amide moiety upon titration with a 10-fold excess of AmyE-peptide (blue) versus the residue number. The broken line indicates a significance level of 0.05 ppm. * denotes unassigned residues as well as Pro192. C, backbone chemical shift perturbations for residues located in the parvulin domain of 0.5 mm PrsA upon titrating increasing AmyE-peptide. D, backbone chemical shift perturbations for residues located in the NC domain of 0.5 mm PrsA upon titrating increasing AmyE-peptide. The solid lines in B and C represent nonlinear least square best fits of the normalized changes of the 1H and 15N chemical shifts to all of the titration data simultaneously, using a bimolecular equilibrium binding model. E, surface representation of the PrsA dimer (monomer A in dark gray and monomer B in light gray). PrsA residues of the parvulin domain involved in AmyE propeptide binding identified by NMR are shown in red. The dimensions of the PrsA cavity are indicated.
Addition of 5.0 mm AmyE propeptide to 0.5 mm PrsA (final ligand to protein ratio 10:1) led to chemical shifts larger than 0.05 ppm for the amide moieties of 18 residues in the parvulin and 37 residues in the NC domain (Fig. 8B). In all cases, the resonances changed their positions continuously during the titration (Fig. 8, C and D), indicating that exchange kinetics between the bound and unbound forms are occurring in the fast exchange limit of the NMR chemical shift time scale (>1000 s−1). The residues in the PPIase domain with the largest chemical shift changes are located around the active site (Fig. 8E). From the concentration dependence of the chemical shift change we derived dissociation constants for the interaction between the propeptide and individual residues in the PPIase or NC domain (Tables 3 and 4; Fig. 8, C and D). With the caveat that substrate saturation was not reached for all residues in this experiment, the data are well suited to establish an overall estimate of the binding constant of the substrate peptide to the PPIase (in the range of 1.5 to 4.5 mm) and NC domain (in the range of 1.2 to 4.9 mm). These dissociation constants are identical within the experimental precision. Overall, the NMR experiments thus demonstrate that PrsA directly interacts with the AmyE propeptide and that both the NC and the parvulin domain contribute to the interaction, even though the propeptide does not contain proline residues.
TABLE 3.
Interaction of the AmyE-peptide with the parvulin domain of PrsA
Dissociation constants for the interaction of PrsA with the AmyE peptide for selected PrsA residues of the parvulin domain, as determined by NMR spectroscopy titrations.
| Residue | AmyE KD |
|---|---|
| mm | |
| Thr131 | 3.9 ± 0.5 |
| Ala132 | 3.7 ± 1.3 |
| Lys194 | 4.8 ± 0.7 |
| Ile201 | 3.1 ± 0.7 |
| Ile202 | 1.2 ± 0.2 |
| Glu206 | 1.7 ± 0.5 |
| Median | 3.4 ± 1.6 |
TABLE 4.
Interaction of the AmyE-peptide with the NC domain of PrsA
Dissociation constants for the interaction of PrsA with AmyE peptide for selected PrsA residues of the NC domain, as determined by NMR spectroscopy titrations.
| Residue | 1H | 15N | PrsA KD |
|---|---|---|---|
| ppm | mm | ||
| A | 8.52 | 123.4 | 4.5 ± 1.2 |
| B | 7.52 | 118.5 | 2.9 ± 0.7 |
| C | 7.50 | 118.0 | 1.5 ± 0.4 |
| D | 8.00 | 117.8 | 2.0 ± 0.7 |
| E | 7.44 | 112.3 | 2.9 ± 0.5 |
| Median | 2.8 ± 1.1 | ||
DISCUSSION
In Gram-positive bacteria, most secreted proteins are translocated in an unfolded state into the space between the cell membrane and the cell wall (73, 74). This compartment is characterized by high concentrations of cations bound to teichoic acid, a high negative charge density, and a low pH (75), which together form a challenging environment for protein folding. PrsA-like proteins are key folding factors to support post-membrane protein maturation and secretion in Gram-positive bacteria (76). The crystal structure of PrsA demonstrates that PrsA folds into two subdomains, the chaperone-like helical NC domain and the PPIase domain, which is inserted into the NC domain. PrsA shares this domain architecture with folding factors from Gram-negative organisms such as trigger factor or SurA (61), despite a lack of sequence homology in the respective N- and C-terminal regions. In PrsA, PPIase activity is independent of the presence of the NC domain, as demonstrated by the fact that the isolated, monomeric parvulin domain resembles the catalytic properties of the full-length protein (15). It should also be noted that other chaperones such as prefoldin, Skp, or the mitochondrial small Tim proteins also use helical protrusions (“tentacles”) to interact with folding proteins (70, 77, 78).
Despite the structural homology of the NC domain to the chaperone domains of other folding factors, non-membrane tethered PrsA is not active in common chaperone assays (Fig. 7). So is the NC domain a true chaperone relevant for PrsA function? In vivo experiments have clearly demonstrated that the isolated NC domain alone is sufficient for promoting the secretion of various substrates and even rescues a PrsA deletion knock-out (15, 21); still all domains of a PrsA homologue in Listeria monocytogenes (PrsA2) are required for full virulence (21). Together, these results indicate that the PPIase activity of PrsA is relevant for a subset of substrates, but that the most essential functions of PrsA are mediated by the NC domain.
The most likely reason for the absence of chaperone activity of PrsA in solution assays is a requirement on dimerization, although other reasons, such as mismatch between test substrates and PrsA specificity cannot be completely ruled out at this point. We demonstrate that PrsA lacking its membrane anchor is monomeric, but has an inherent tendency for dimer formation at high protein concentrations. It crystallizes in a dimeric arrangement, which is fully compatible with membrane tethering and employs exclusively the NC domain for dimerization. In vivo, lipid anchoring dramatically favors dimer formation, and in fact, PrsA2 of L. monocytogenes and PrsA of B. subtilis have been reported to form dimers at the cell membrane (18, 21).
In the dimeric assembly, PrsA forms a bowl-like crevice (Fig. 4E) between the NC domains of two monomers. The inside surface of the crevice is enriched in hydrophobic residues (Fig. 9A), whereas the outside contains many charged amino acids (Fig. 9B). Analogous hydrophobicity profiles are observed for the NC domains of related bona fide chaperones, e.g. trigger factor (69), suggesting that the inside of the crevice could provide a chaperone function in vivo. Enlarging the interaction surface for unfolded peptides in the dimer might be critical to permit productive substrate interaction by avidity of multiple, low affinity interactions. With dimension of about 20 × 30 × 35 Å (Fig. 9, C and D), the bowl-shaped crevice in the PrsA dimer could completely accommodate polypeptides of up to 20 kDa, but the observed flexibility of the NC domains (Fig. 4D) and the attached parvulin domain may even permit adaptation to larger substrates. The only other chaperone with PPIase/NC domain architecture, for which dimerization and the formation of an enlarged crevice has been observed, is Peb4 from Campylobacter jejuni (13). However, its NC region is more distantly related to the SurA/trigger factor and dimerization under unusual domain swapping is required for completion of the NC-domain fold. Many Gram-positive bacteria contain only a single PrsA-like protein. Others, such as Bacillus cereus, contain several PrsA homologs (76). For L. monocytogenes and Bacillus anthracis it was shown that PrsA homologs have non-overlapping functions and cannot substitute each other (25, 26). Interestingly, the parvulin domains, but not the NC chaperone domains of PrsA homologues are highly conserved (Fig. 10). The high conservation in the PPIase domain is necessary to preserve the enzymatic function; the variations in the NC domain may reflect differing substrate specificities.
FIGURE 9.

Distribution of hydrophobic and charged amino acids on PrsA and size of the PrsA crevice. A, surface representation of PrsA color coded ranging from hydrophobic (green) to hydrophilic (gray) according to the normalized consensus hydrophobicity scale of the exposed residues (83). B, surface representation of PrsA color coded ranging from negative charged (red) to positive charged (blue). C, the crevice of the PrsA dimer can accommodate domain-sized polypeptides. Monomer structure of PrsA: the parvulin domain and the NC domain are colored blue and yellow, respectively. The PPIase active site is indicated as an ellipse. The arm1-arm2 distance (20 Å) as well as the distance from the parvulin domain to the opposite site of the NC domain (35 Å) within one PrsA molecule are given. D, representation of PrsA dimer with approximate dimensions of the cavity indicated; PrsA molecule A, is colored as in C. For PrsA molecule B, the parvulin domain and the NC domain are colored red and gray, respectively. The intermolecular distance of arm1, arm2, and parvulin regions are 28, 40, and 29 Å, respectively.
FIGURE 10.

Sequence conservation in the PrsA family. A, consurf representation (84) of sequence conservation within the PrsA family; residues that are highly conserved are shown in red, sequences with lower identity are shown in yellow, green, and blue. B, multiple sequence alignment to analyze the sequence and secondary structure conservation of PrsA. Highly conserved residues are red (>70% conservation) or white in red boxes (100% conservation). The secondary structure of B. subtilis PrsA is shown on top of the protein sequence. Sequences of representative PrsA proteins were retrieved from the UniProt database (85) and aligned using MULTALIN (86). The final figure was generated using the ESPript server (24). Species abbreviations and UniProt accession numbers are BsPrsA, B. subtilis (P24327); BaPrsA1, B. anthracis (Q81U45); LmPrsA1 L. monocytogenes (Q71ZM6); LmPrsA2, L. monocytogenes (Q71XE6); SpPrsA, Streptococcus pneumoniae (B2IPD4); SgPrsA, Streptococcus gordonii (A8AYJ0); LlPrsA, Lactococcus lactis (P0C2B5); SaPrsA, Staphylococcus aureus (A6QI23).
PrsA substrates exert functions in cell wall metabolism (penicillin-binding proteins), swimming motility (flagellin), oligopeptide transport (OppA), and membrane bioenergetics (quinol oxidase), act as virulence factors or as enzymes such as α-amylase or subtilisin (76, 79). The molecular mass of substrates range from 20 to 80 kDa, and thus includes proteins, which are clearly too large to be accommodated in the PrsA crevice. The lateral openings of the crevice might serve to provide the possibility for interactions of multiple PrsA molecules with one substrate polypeptide, as found for trigger factor, where e.g. three molecules bind to a 45-kDa protein (87). We show that PrsA directly interacts with substrate propeptides and analyze the binding of PrsA to a prototypic 11-residue peptide. The NMR experiments indicate that PrsA uses both the NC chaperone and the parvulin domain for transient and dynamic substrate interactions.
To our knowledge, PrsA is the first authentic member of the SurA/trigger factor class of foldases, which has been structurally characterized as a symmetric dimer and may require dimer formation for exerting chaperone activity. Our current structural and functional characterization of PrsA provides a basis for future in-depth studies on the role of self-association, e.g. by targeted trapping of dimeric states for solution studies. The structural information will help to identify PrsA regions involved in polypeptide recognition, and to understand its role in the maturation of larger proteins. This paves the way for new strategies to optimize protein export in biotechnological applications and may yield opportunities for developing small molecule inhibitors of extracellular protein folding as antibacterial agents against Gram-positive pathogens.
Acknowledgments
We thank the staff at the Swiss Light Source (Villigen, Switzerland) for assistance during data collection. We thank Tim Sharpe and the Biophysics Facility of the Biozentrum, University of Basel, for assistance with SEC-MALS experiments and members of our groups for many fruitful discussions.
This work was supported by Swiss National Science Foundation Grants PP00P3_152989 and R'EQUIP 145023.
The atomic coordinates and structure factors (code 4WO7) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PPIase
- peptidyl-prolyl cis-trans isomerase
- ΔHD
- van't Hoff enthalpy of denaturation at Tm
- SEC-MALS
- size exclusion chromatography coupled to multiangle light scattering
- Abz
- aminobenzoyl
- pNA
- para-nitroanilide
- RCM-T1
- reduced and carboxymethylated RNase T1
- FKBP
- FK506-binding protein
- CS
- citrate synthase
- GdmCl
- guanidinium chloride.
REFERENCES
- 1. Fanghänel J., Fischer G. (2004) Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front. Biosci. 9, 3453–3478 [DOI] [PubMed] [Google Scholar]
- 2. Schmid F. X. (2001) Prolyl isomerases. Adv. Protein Chem. 59, 243–282 [DOI] [PubMed] [Google Scholar]
- 3. Schmid F. X., Mayr L. M., Mücke M., Schönbrunner E. R. (1993) Prolyl isomerases: role in protein folding. Adv. Protein Chem. 44, 25–66 [DOI] [PubMed] [Google Scholar]
- 4. Lang K., Schmid F. X., Fischer G. (1987) Catalysis of protein folding by prolyl isomerase. Nature 329, 268–270 [DOI] [PubMed] [Google Scholar]
- 5. Siekierka J. J., Hung S. H., Poe M., Lin C. S., Sigal N. H. (1989) A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 341, 755–757 [DOI] [PubMed] [Google Scholar]
- 6. Rahfeld J. U., Rücknagel K. P., Schelbert B., Ludwig B., Hacker J., Mann K., Fischer G. (1994) Confirmation of the existence of a third family among peptidyl-prolyl cis/trans isomerases. Amino acid sequence and recombinant production of parvulin. FEBS Lett. 352, 180–184 [DOI] [PubMed] [Google Scholar]
- 7. Yaffe M. B., Schutkowski M., Shen M., Zhou X. Z., Stukenberg P. T., Rahfeld J. U., Xu J., Kuang J., Kirschner M. W., Fischer G., Cantley L. C., Lu K. P. (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278, 1957–1960 [DOI] [PubMed] [Google Scholar]
- 8. Lu K. P., Zhou X. Z. (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916 [DOI] [PubMed] [Google Scholar]
- 9. Harris G. S., White T. C., Flory J. E., Orme-Johnson W. H. (1990) Genes required for formation of the apoMoFe protein of Klebsiella pneumoniae nitrogenase in Escherichia coli. J. Biol. Chem. 265, 15909–15919 [PubMed] [Google Scholar]
- 10. Emelyanov V. V., Loukianov E. V. (2004) A 29.5 kDa heat-modifiable major outer membrane protein of Rickettsia prowazekii, putative virulence factor, is a peptidyl-prolyl cis/trans isomerase. IUBMB Life 56, 215–219 [DOI] [PubMed] [Google Scholar]
- 11. Hodak H., Wohlkönig A., Smet-Nocca C., Drobecq H., Wieruszeski J. M., Sénéchal M., Landrieu I., Locht C., Jamin M., Jacob-Dubuisson F. (2008) The peptidyl-prolyl isomerase and chaperone Par27 of Bordetella pertussis as the prototype for a new group of parvulins. J. Mol. Biol. 376, 414–426 [DOI] [PubMed] [Google Scholar]
- 12. Behrens-Kneip S. (2010) The role of SurA factor in outer membrane protein transport and virulence. Int. J. Med. Microbiol. 300, 421–428 [DOI] [PubMed] [Google Scholar]
- 13. Kale A., Phansopa C., Suwannachart C., Craven C. J., Rafferty J. B., Kelly D. J. (2011) The virulence factor PEB4 (Cj0596) and the periplasmic protein Cj1289 are two structurally related SurA-like chaperones in the human pathogen Campylobacter jejuni. J. Biol. Chem. 286, 21254–21265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacobs M., Andersen J. B., Kontinen V., Sarvas M. (1993) Bacillus subtilis PrsA is required in vivo as an extracytoplasmic chaperone for secretion of active enzymes synthesized either with or without pro-sequences. Mol. Microbiol. 8, 957–966 [DOI] [PubMed] [Google Scholar]
- 15. Vitikainen M., Lappalainen I., Seppala R., Antelmann H., Boer H., Taira S., Savilahti H., Hecker M., Vihinen M., Sarvas M., Kontinen V. P. (2004) Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J. Biol. Chem. 279, 19302–19314 [DOI] [PubMed] [Google Scholar]
- 16. Vitikainen M., Pummi T., Airaksinen U., Wahlström E., Wu H., Sarvas M., Kontinen V. P. (2001) Quantitation of the capacity of the secretion apparatus and requirement for PrsA in growth and secretion of α-amylase in Bacillus subtilis. J. Bacteriol. 183, 1881–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo L., Wu T., Hu W., He X., Sharma S., Webster P., Gimzewski J. K., Zhou X., Lux R., Shi W. (2013) Phenotypic characterization of the foldase homologue PrsA in Streptococcus mutans. Mol. Oral Microbiol. 28, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hyyryläinen H. L., Marciniak B. C., Dahncke K., Pietiäinen M., Courtin P., Vitikainen M., Seppala R., Otto A., Becher D., Chapot-Chartier M. P., Kuipers O. P., Kontinen V. P. (2010) Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol. Microbiol. 77, 108–127 [DOI] [PubMed] [Google Scholar]
- 19. Drouault S., Anba J., Bonneau S., Bolotin A., Ehrlich S. D., Renault P. (2002) The peptidyl-prolyl isomerase motif is lacking in PmpA, the PrsA-like protein involved in the secretion machinery of Lactococcus lactis. Appl. Environ. Microbiol. 68, 3932–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jousselin A., Renzoni A., Andrey D. O., Monod A., Lew D. P., Kelley W. L. (2012) The posttranslocational chaperone lipoprotein PrsA is involved in both glycopeptide and oxacillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 3629–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alonzo F., 3rd, Xayarath B., Whisstock J. C., Freitag N. E. (2011) Functional analysis of the Listeria monocytogenes secretion chaperone PrsA2 and its multiple contributions to bacterial virulence. Mol. Microbiol. 80, 1530–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu S. C., Ye R., Wu X. C., Ng S. C., Wong S. L. (1998) Enhanced secretory production of a single-chain antibody fragment from Bacillus subtilis by coproduction of molecular chaperones. J. Bacteriol. 180, 2830–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kakeshita H., Kageyama Y., Endo K., Tohata M., Ara K., Ozaki K., Nakamura K. (2011) Secretion of biologically-active human interferon-β by Bacillus subtilis. Biotechnol. Lett. 33, 1847–1852 [DOI] [PubMed] [Google Scholar]
- 24. Gouet P., Robert X., Courcelle E. (2003) ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31, 3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Williams R. C., Rees M. L., Jacobs M. F., Prágai Z., Thwaite J. E., Baillie L. W., Emmerson P. T., Harwood C. R. (2003) Production of Bacillus anthracis protective antigen is dependent on the extracellular chaperone, PrsA. J. Biol. Chem. 278, 18056–18062 [DOI] [PubMed] [Google Scholar]
- 26. Alonzo F., 3rd, Freitag N. E. (2010) Listeria monocytogenes PrsA2 is required for virulence factor secretion and bacterial viability within the host cell cytosol. Infect. Immun. 78, 4944–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tossavainen H., Permi P., Purhonen S. L., Sarvas M., Kilpeläinen I., Seppala R. (2006) NMR solution structure and characterization of substrate binding site of the PPIase domain of PrsA protein from Bacillus subtilis. FEBS Lett. 580, 1822–1826 [DOI] [PubMed] [Google Scholar]
- 28. Savitsky P., Bray J., Cooper C. D., Marsden B. D., Mahajan P., Burgess-Brown N. A., Gileadi O. (2010) High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 172, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmid F. X. (1993) Prolyl isomerase: enzymatic catalysis of slow protein-folding reactions. Annu. Rev. Biophys. Biomol. Struct. 22, 123–142 [DOI] [PubMed] [Google Scholar]
- 30. Zoldák G., Aumüller T., Lücke C., Hritz J., Oostenbrink C., Fischer G., Schmid F. X. (2009) A library of fluorescent peptides for exploring the substrate specificities of prolyl isomerases. Biochemistry 48, 10423–10436 [DOI] [PubMed] [Google Scholar]
- 31. Buchner J., Schmidt M., Fuchs M., Jaenicke R., Rudolph R., Schmid F. X., Kiefhaber T. (1991) GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry 30, 1586–1591 [DOI] [PubMed] [Google Scholar]
- 32. Myers J. K., Pace C. N., Scholtz J. M. (1995) Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4, 2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kather I., Jakob R., Dobbek H., Schmid F. X. (2008) Changing the determinants of protein stability from covalent to non-covalent interactions by in vitro evolution: a structural and energetic analysis. J. Mol. Biol. 381, 1040–1054 [DOI] [PubMed] [Google Scholar]
- 34. Pervushin K., Riek R., Wider G., Wüthrich K. (1997) Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guntert P., Dötsch V., Wider G., Wüthrich K. (1992) Processing of multi-dimensional NMR data with the new software PROSA. J. Biomol. NMR 2, 619–629 [Google Scholar]
- 36. Keller R. L. J. (2004) The computer aided resonance assignment tutorial. Cantina Verlag, Goldau [Google Scholar]
- 37. Kabsch W. (2010) Xds. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zwart P. H., Afonine P. V., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., McKee E., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Storoni L. C., Terwilliger T. C., Adams P. D. (2008) Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435 [DOI] [PubMed] [Google Scholar]
- 39. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cowtan K. (2006) The Buccaneer software for automated model building: 1. tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 41. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 42. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 43. Leskelä S., Wahlström E., Kontinen V. P., Sarvas M. (1999) Lipid modification of prelipoproteins is dispensable for growth but essential for efficient protein secretion in Bacillus subtilis: characterization of the Lgt gene. Mol. Microbiol. 31, 1075–1085 [DOI] [PubMed] [Google Scholar]
- 44. Grasberger B., Minton A. P., DeLisi C., Metzger H. (1986) Interaction between proteins localized in membranes. Proc. Natl. Acad. Sci. U.S.A. 83, 6258–6262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jakob R. P., Zoldák G., Aumüller T., Schmid F. X. (2009) Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc. Natl. Acad. Sci. U.S.A. 106, 20282–20287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scholz C., Stoller G., Zarnt T., Fischer G., Schmid F. X. (1997) Cooperation of enzymatic and chaperone functions of trigger factor in the catalysis of protein folding. EMBO J. 16, 54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hottenrott S., Schumann T., Plückthun A., Fischer G., Rahfeld J. U. (1997) The Escherichia coli SlyD is a metal ion-regulated peptidyl-prolyl cis/trans-isomerase. J. Biol. Chem. 272, 15697–15701 [DOI] [PubMed] [Google Scholar]
- 48. Scholz C., Rahfeld J., Fischer G., Schmid F. X. (1997) Catalysis of protein folding by parvulin. J. Mol. Biol. 273, 752–762 [DOI] [PubMed] [Google Scholar]
- 49. Uchida T., Fujimori F., Tradler T., Fischer G., Rahfeld J. U. (1999) Identification and characterization of a 14 kDa human protein as a novel parvulin-like peptidyl prolyl cis/trans isomerase. FEBS Lett. 446, 278–282 [DOI] [PubMed] [Google Scholar]
- 50. Schmidpeter P. A., Jahreis G., Geitner A. J., Schmid F. X. (2011) Prolyl isomerases show low sequence specificity toward the residue following the proline. Biochemistry 50, 4796–4803 [DOI] [PubMed] [Google Scholar]
- 51. Kiefhaber T., Grunert H. P., Hahn U., Schmid F. X. (1990) Replacement of a cis proline simplifies the mechanism of ribonuclease T1 folding. Biochemistry 29, 6475–6480 [DOI] [PubMed] [Google Scholar]
- 52. Mayr L. M., Odefey C., Schutkowski M., Schmid F. X. (1996) Kinetic analysis of the unfolding and refolding of ribonuclease T1 by a stopped-flow double-mixing technique. Biochemistry 35, 5550–5561 [DOI] [PubMed] [Google Scholar]
- 53. Mücke M., Schmid F. X. (1992) Enzymatic catalysis of prolyl isomerization in an unfolding protein. Biochemistry 31, 7848–7854 [DOI] [PubMed] [Google Scholar]
- 54. Mücke M., Schmid F. X. (1994) Folding mechanism of ribonuclease T1 in the absence of the disulfide bonds. Biochemistry 33, 14608–14619 [DOI] [PubMed] [Google Scholar]
- 55. Behrens S., Maier R., de Cock H., Schmid F. X., Gross C. A. (2001) The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J. 20, 285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ranganathan R., Lu K. P., Hunter T., Noel J. P. (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89, 875–886 [DOI] [PubMed] [Google Scholar]
- 57. Sekerina E., Rahfeld J. U., Müller J., Fanghänel J., Rascher C., Fischer G., Bayer P. (2000) NMR solution structure of hPar14 reveals similarity to the peptidyl prolyl cis/trans isomerase domain of the mitotic regulator hPin1 but indicates a different functionality of the protein. J. Mol. Biol. 301, 1003–1017 [DOI] [PubMed] [Google Scholar]
- 58. Li Z., Li H., Devasahayam G., Gemmill T., Chaturvedi V., Hanes S. D., Van Roey P. (2005) The structure of the Candida albicans Ess1 prolyl isomerase reveals a well-ordered linker that restricts domain mobility. Biochemistry 44, 6180–6189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Landrieu I., Wieruszeski J. M., Wintjens R., Inzé D., Lippens G. (2002) Solution structure of the single-domain prolyl cis/trans isomerase PIN1At from Arabidopsis thaliana. J. Mol. Biol. 320, 321–332 [DOI] [PubMed] [Google Scholar]
- 60. Kühlewein A., Voll G., Hernandez Alvarez B., Kessler H., Fischer G., Rahfeld J. U., Gemmecker G. (2004) Solution structure of Escherichia coli Par10: the prototypic member of the Parvulin family of peptidyl-prolyl cis/trans isomerases. Protein Sci. 13, 2378–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bitto E., McKay D. B. (2002) Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10, 1489–1498 [DOI] [PubMed] [Google Scholar]
- 62. Giuseppe P. O., Von Atzingen M., Nascimento A. L., Zanchin N. I., Guimarães B. G. (2011) The crystal structure of the leptospiral hypothetical protein LIC12922 reveals homology with the periplasmic chaperone SurA. J. Struct. Biol. 173, 312–322 [DOI] [PubMed] [Google Scholar]
- 63. Weininger U., Jakob R. P., Kovermann M., Balbach J., Schmid F. X. (2010) The prolyl isomerase domain of PpiD from Escherichia coli shows a parvulin fold but is devoid of catalytic activity. Protein Sci. 19, 6–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heikkinen O., Seppala R., Tossavainen H., Heikkinen S., Koskela H., Permi P., Kilpeläinen I. (2009) Solution structure of the parvulin-type PPIase domain of Staphylococcus aureus PrsA: implications for the catalytic mechanism of parvulins. BMC Struct. Biol. 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mueller J. W., Link N. M., Matena A., Hoppstock L., Rüppel A., Bayer P., Blankenfeldt W. (2011) Crystallographic proof for an extended hydrogen-bonding network in small prolyl isomerases. J. Am. Chem. Soc. 133, 20096–20099 [DOI] [PubMed] [Google Scholar]
- 66. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 67. Lill R., Crooke E., Guthrie B., Wickner W. (1988) The trigger factor cycle includes ribosomes, presecretory proteins, and the plasma-membrane. Cell 54, 1013–1018 [DOI] [PubMed] [Google Scholar]
- 68. Stoller G., Rücknagel K. P., Nierhaus K. H., Schmid F. X., Fischer G., Rahfeld J. U. (1995) A ribosome-associated peptidyl-prolyl cis/trans isomerase identified as the trigger factor. EMBO J. 14, 4939–4948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ferbitz L., Maier T., Patzelt H., Bukau B., Deuerling E., Ban N. (2004) Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature 431, 590–596 [DOI] [PubMed] [Google Scholar]
- 70. Goemans C., Denoncin K., Collet J. F. (2014) Folding mechanisms of periplasmic proteins. Biochim. Biophys. Acta 1843, 1517–1528 [DOI] [PubMed] [Google Scholar]
- 71. Kovermann M., Schmid F. X., Balbach J. (2013) Molecular function of the prolyl cis/trans isomerase and metallochaperone SlyD. Biol. Chem. 394, 965–975 [DOI] [PubMed] [Google Scholar]
- 72. Kakeshita H., Kageyama Y., Ara K., Ozaki K., Nakamura K. (2011) Propeptide of Bacillus subtilis amylase enhances extracellular production of human interferon-α in Bacillus subtilis. Appl. Microbiol. Biotechnol. 89, 1509–1517 [DOI] [PubMed] [Google Scholar]
- 73. Freudl R. (2013) Leaving home ain't easy: protein export systems in Gram-positive bacteria. Res. Microbiol. 164, 664–674 [DOI] [PubMed] [Google Scholar]
- 74. Matias V. R., Beveridge T. J. (2005) Cryo-electron microscopy reveals native polymeric cell wall structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol. Microbiol. 56, 240–251 [DOI] [PubMed] [Google Scholar]
- 75. Sarvas M., Harwood C. R., Bron S., van Dijl J. M. (2004) Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim. Biophys. Acta 1694, 311–327 [DOI] [PubMed] [Google Scholar]
- 76. Cahoon L. A., Freitag N. E. (2014) Listeria monocytogenes virulence factor secretion: don't leave the cell without a chaperone. Front. Cell Infect. Microbiol. 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Burmann B. M., Wang C., Hiller S. (2013) Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat. Struct. Mol. Biol. 20, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 78. Ceh-Pavia E., Spiller M. P., Lu H. (2013) Folding and biogenesis of mitochondrial small Tim proteins. Int. J. Mol. Sci. 14, 16685–16705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Forster B. M., Marquis H. (2012) Protein transport across the cell wall of monoderm Gram-positive bacteria. Mol. Microbiol. 84, 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee D., Hilty C., Wider G., Wüthrich K. (2006) Effective rotational correlation times of proteins from NMR relaxation interference. J. Magn. Reson. 178, 72–76 [DOI] [PubMed] [Google Scholar]
- 81. Fernández C., Adeishvili K., Wüthrich K. (2001) Transverse relaxation-optimized NMR spectroscopy with the outer membrane protein OmpX in dihexanoyl phosphatidylcholine micelles. Proc. Natl. Acad. Sci. U.S.A. 98, 2358–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chou J. J., Baber J. L., Bax A. (2004) Characterization of phospholipid mixed micelles by translational diffusion. J. Biomol. NMR 29, 299–308 [DOI] [PubMed] [Google Scholar]
- 83. Eisenberg D., Schwarz E., Komaromy M., Wall R. (1984) Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179, 125–142 [DOI] [PubMed] [Google Scholar]
- 84. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jain E., Bairoch A., Duvaud S., Phan I., Redaschi N., Suzek B. E., Martin M. J., McGarvey P., Gasteiger E. (2009) Infrastructure for the life sciences: design and implementation of the UniProt website. BMC bioinformatics 10, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Corpet F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Saio T., Guan X., Rossi P., Economou A., Kalodimos C. G. (2014) Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 10.1126/science.1250494 [DOI] [PMC free article] [PubMed] [Google Scholar]