

Background: Persistent STAT3 phosphorylation is detected in medulloblastoma and represents a viable target for therapeutic drug discovery.
Results: LY5, a novel STAT3 inhibitor, suppressed STAT3 phosphorylation, nuclear translocation, target gene expression, cell migration, angiogenesis, and induced apoptosis in medulloblastoma cells.
Conclusion: LY5 exhibited potent activity against STAT3 signaling in medulloblastoma cells.
Significance: LY5 is a promising drug candidate for medulloblastoma therapy.
Keywords: Apoptosis, Cancer Therapy, Interleukin 6 (IL-6), Medulloblastoma, STAT3
Abstract
Signal transducers and activators of transcription 3 (STAT3) signaling is persistently activated and could contribute to tumorigenesis of medulloblastoma. Numerous studies have demonstrated that inhibition of the persistent STAT3 signaling pathway results in decreased proliferation and increased apoptosis in human cancer cells, indicating that STAT3 is a viable molecular target for cancer therapy. In this study, we investigated a novel non-peptide, cell-permeable small molecule, named LY5, to target STAT3 in medulloblastoma cells. LY5 inhibited persistent STAT3 phosphorylation and induced apoptosis in human medulloblastoma cell lines expressing constitutive STAT3 phosphorylation. The inhibition of STAT3 signaling by LY5 was confirmed by down-regulating the expression of the downstream targets of STAT3, including cyclin D1, bcl-XL, survivin, and micro-RNA-21. LY5 also inhibited the induction of STAT3 phosphorylation by interleukin-6 (IL-6), insulin-like growth factor (IGF)-1, IGF-2, and leukemia inhibitory factor in medulloblastoma cells, but did not inhibit STAT1 and STAT5 phosphorylation stimulated by interferon-γ (IFN-γ) and EGF, respectively. In addition, LY5 blocked the STAT3 nuclear localization induced by IL-6, but did not block STAT1 and STAT5 nuclear translocation mediated by IFN-γ and EGF, respectively. A combination of LY5 with cisplatin or x-ray radiation also showed more potent effects than single treatment alone in the inhibition of cell viability in human medulloblastoma cells. Furthermore, LY5 demonstrated a potent inhibitory activity on cell migration and angiogenesis. Taken together, these findings indicate LY5 inhibits persistent and inducible STAT3 phosphorylation and suggest that LY5 is a promising therapeutic drug candidate for medulloblastoma by inhibiting persistent STAT3 signaling.
Introduction
STAT3, one important member of signal transducers and activators of transcription family, is constitutively activated in a wide variety of human malignancies, including medulloblastoma (1–3). STAT3 acts as signal messenger and transcription factor that participate in normal cellular responses to cytokines and growth factors (4, 5). However, persistent activation of STAT3 is associated with oncogenesis and tumor progression (6–8). The function domains of the STAT3 protein comprise the dimerization domain at the N terminus, SH22 domain, the DNA-binding domain, and the transcription activation domain at the C-terminal end. SH2 domain and the transcription activation domain at the C-terminal end are very important for activation of STAT3. When one of the gp130-acting cytokines and growth factors such as IL-6, IL-11, oncostatin M, leukemia inhibitory factor (LIF), and insulin-like growth factor (IGF) interacts with its receptor, STAT3 is activated through phosphorylation at its tyrosine 705 (Tyr-705) in the transcription activation domain (9–12). After phosphorylation, two STAT3 monomers form dimers through reciprocal phosphotyrosine-SH2 interactions, translocate from the cytoplasm to the nucleus, and bind to specific DNA sequence to regulate target genes transcription (13–15).
Once activated, STAT3 plays a critical role in oncogenesis, proliferation, survival, invasion, and inflammation of various human cancers and cancer cell lines. The persistently activated Stat3 up-regulates its downstream gene expression such as cyclinD1, c-myc, bcl-2, survivin, bcl-XL, and VEGF, which contribute to uncontrolled proliferation of cancer cells through promoting cell cycle progression, inhibiting apoptosis, stimulating tumor angiogenesis, and metastasis (16–19). STAT3 has a oncogenic potential to be involved in cell malignant transformation. Compelling evidence has confirmed that aberrant activated STAT3 participated in the process of oncogenesis in tumor cells by oncogenic tyrosine kinases (20–22). Further support comes from animal experiments that a constitutively activated STAT3 mutant alone can directly induce tumor formation in nude mice, which suggested that STAT3 activation plays a critical role in oncogenesis (6). Also, it has been demonstrated that up-regulated STAT3 phosphorylation also has important functions in inflammatory response and immune regulation (23, 24). Many drivers of STAT3 activation, including the anti-inflammatory IL-10, pro-inflammatory IL-6, and type I and II interferons (IFNs), ensure constitutive Stat3 activation, which may account for the fact that STAT3 can mediate both anti-inflammatory and pro-inflammatory responses (4, 25, 26). STAT3 is persistently activated in the tumor microenvironment, which not only down-regulates Th1 cytokines and other critical mediators for potent anti-tumor immune responses, but also activates many genes involved in immune suppression through a cross-talk between immune cells and tumor cells (27). In addition, STAT3 has been implicated as a facilitator of tumor angiogenesis and metastasis. It has been reported that persistently activated STAT3 up-regulates expression of VEGF, MMP-2, and MMP-9, which are both involved in tumor angiogenesis and metastasis (28–30).
Medulloblastoma is the most common malignant brain tumor of childhood. Medulloblastoma remain largely incurable cancers, with patients facing the poor 5-year survival rate (31). The current challenge is to identify and design effective molecular-targeted strategies that improve prognosis in the patient. Given the important role of constitutive STAT3 signaling in tumors, it provides a potential therapeutic target in treatment of this tumor. Mounting evidence has shown that disrupting STAT3 activation can inhibit the growth of tumor cells and induce apoptosis in tumor cells (32–35). Based on these findings, devising an inhibitor of STAT3 is becoming more and more attractive for development of cancer therapeutic drugs. In this study, using an in silico site-directed fragment-based drug design, we developed a novel small-molecule STAT3 inhibitor, LY5, that selectively disrupted STAT3-STAT3 dimer formation as shown by computer models with docking simulation (36). We have evaluated the inhibitory effect of LY5 on STAT3 activation and functions in human medulloblastoma cells. Studies shown here for LY5 not only selectively inhibited STAT3 phosphorylation, STAT3 nuclear translocation, and STAT3 target genes expression, but also induced apoptosis in medulloblastoma cells with persistent STAT3 phosphorylation, blocked cell migration, and suppressed angiogenesis. These results suggested that LY5 is a potent inhibitor against persistent STAT3 signaling in medulloblastoma.
EXPERIMENTAL PROCEDURES
Synthesis of LY5
LY5 was designed and synthesized as previously described (36). First, we designed a new STAT3 inhibitor. A new fragment-based drug design (FBDD) approach, in silico site-directed FBDD, was used in this study. To develop a new lead library, we linked the selected fragments from different fragment sublibraries that were built according to the binding mode of the known STAT3 dimerization inhibitors to the STAT3 SH2 domain (Protein Data Bank code 1BG1). The new compound was ultimately chosen for synthesis by repositioning the compounds in the lead library to the STAT3 SH2 domain. The Schrodinger software and computational docking program AutoDock4 (37) were applied. Second, we used chemistry synthesis of LY5. Naphthalenesulfonyl chloride reacted with ammonium hydroxide at room temperature for 3 h to get highly pure naphthalenesulfonamide (90.2%), which was subsequently dissolved in warm glacial acetic acid and mixed with chromium trioxide to synthesize the fragment of naphthalene-5,8-dione-1-sulfonamide. This fragment (237 mg), amine (1.2 mmol), and Cu(OAc)2·H2O (20 mg), was solubilized in a mixture of AcOH and H2O (1:10, v/v, 5.5 ml), refluxing for about 3 h. The product was purified by silica gel column chromatography eluting with CH2Cl2/EtOAc to harvest the compound 5,8-dioxo-6-(pyridin-3-ylamino)-5,8-dihydronaphthalene-1-sulfonamide, which was named LY5.
Cell Lines and Reagents
The medulloblastoma cell lines (UW426, UW288-1, and DAOY) were kindly provided by Dr. Corey Raffel and maintained in Dulbecco's modified Eagle's medium (DMEM, HyClone) supplemented with 10% FBS, 4.5 g/liter of l-glutamine, sodium pyruvate, and 1% penicillin/streptomycin. Normal human skeletal muscle myoblasts were purchased from Lonza Walkersville, Inc. (Walkersville, MD) and maintained in Ham's F-12 medium (Mediatech) supplemented with 5 μg/ml of insulin, 1 μg/ml of hydrocortisone, 10 μg/ml of epidermal growth factor, 100 μg/ml of cholera toxin, 5% fetal bovine serum (FBS). The human hepatocytes and normal human coronary artery smooth muscle cells were both bought from ScienCell cultured in hepatocyte medium (ScienCell) with 5% FBS plus hepatocyte growth supplement and in DMEM with 2% FBS plus smooth muscle cell growth supplement, respectively. Human umbilical vein endothelial cells (HUVEC) were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in endothelial cell growth medium M200 (Invitrogen) in high glucose-supplemented medium with 15% FBS, endothelial cell growth supplements (LSGS Medium, Cascade Biologics), and 2 mm glutamine. All cell lines were cultured in a humidified 37 °C incubator with 5% CO2. IL-6, LIF, EGF, and IFN-γ were purchased from Cell Signaling Technology. VEGF was purchased from R&D Systems Inc. Human recombinant IGF-I and IGF-2 were purchased from PeproTech Inc. The powder of LY5 was dissolved in sterile dimethyl sulfoxide to make a 20 mm stock solution and stored at −20 °C.
Western Blot Analysis
Cells were harvested after treatment with LY5 or dimethyl sulfoxide at 60–80% confluence for 24 h, then lysed in cold RIPA lysis buffer containing a protease inhibitor mixture and phosphatase inhibitor mixture. The lysates were subjected to 10 or 12% SDS-PAGE gel and transferred to a PVDF membrane. Membranes were incubated with a 1:1000 dilution of specific primary antibody and 1:10,000 HRP-conjugated secondary antibody. Primary antibodies including phospho-STAT3 (Tyr-705), STAT3, phosphor-STAT1 (Tyr-701), STAT1, phospho-STAT5 (Tyr-694), STAT5, cleaved caspase-3, GAPDH, and secondary antibody are all from Cell Signaling Technology. Membranes were analyzed using enhanced chemiluminescence Plus reagents and scanned with the Storm Scanner (Amersham Biosciences Inc.).
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
RNA was extracted from the cell using RNeasy Mini Kits (Qiagen) according to the manufacturer's instructions. Reverse transcription was done using an OmniScript reverse transcription kit (Qiagen). Polymerase chain reaction (PCR) amplification was performed under the following conditions: 5 min at 94 °C followed by 30 cycles of 30 s at 94 °C, 30 s at 53–55 °C, and 60 s at 72 °C with a final extension of 10 min at 72 °C. Primer sequences and source information of STAT3 downstream target genes were reported previously (38–41).
STATs Phosphorylation Induced by Cytokines or Growth Factors
DAOY cells were seeded to grow overnight. Then the cells were cultured in serum-free medium for 24 h and pretreated with 1, 2.5, or 5.0 μm LY5 for 4 h, followed by the stimulating with 50 ng/ml of IL-6, LIF, IGF-1, IGF-2, IFN-γ, or EGF for 30 min. The cells were harvested and analyzed by Western blot for P-STAT3, P-STAT1, or P-STAT5.
Immunofluorescence
UW288-1 cells were seeded on glass coverslips in a 6-well plate. The next day, the cells were cultured in serum-free medium for 24 h and pretreated with LY5 (5 μm) for 4 h, followed by induction with 50 ng/ml of IL-6, IFN-γ, or 100 ng/ml of EGF for 30 min. Cells were fixed with cold methanol for 15 min and blocked with 5% normal goat serum and 0.3% Triton X-100 in PBS for 1 h. The cells were incubated with primary antibodies of STAT3, P-STAT1, or P-STAT5 (Cell Signaling, 1:100) overnight at 4 °C. After incubation with anti-rabbit IgG (H+L), F(ab′)2 fragment Alexa Fluor® 555 secondary antibody (Cell Signaling, 1:200), the cells were mounted using Vectashield Hardset mounting medium with DAPI (Vector Laboratories). Photomicrographs were captured by Leica Microsystems.
Cell Migration Assay (in Vitro Wound-healing Assay)
HUVEC and UW288-1 cell migrations were detected using the wound-healing assay described by Thaloor et al. (42). Briefly, when cells grew into confluent monolayer in plate, we scratched the cells in the same width using yellow tip. After washing, HUVEC cells were incubated with medium containing VEGF (10 ng/ml) with or without LY5 (300 nm). UW288-1 cells were treated with LY5 (0.5 and 1 μm) or dimethyl sulfoxide. Cells were allowed to migrate into the scratched area for 22 or 48 h, and images were captured with a microscopic camera system. The percentage of wound healing was calculated by the equation: (percent wound healing) = average of ((gap area: 0 h) − (gap area: 48 h))/(gap area: 0 h) (43).
Confocal Microscopy
HUVEC cells were grown in Lab-Tek 4-well chamber slides (Nunc/Thermo Scientific) to ∼60% confluence, and treated with PBS, VEGF (10 ng/ml), dimethyl sulfoxide with VEGF, or 300 nm LY5 for 18 h with VEGF. The cultures were fixed in cold 4% paraformaldehyde for 20 min at 4 °C and permeabilized in 0.1% Triton X-100. The cells were probed using 10 μg/ml of mouse anti-human β-tubulin I primary antibody (Sigma, clone TUB 2.1) overnight at 4 °C, and then stained using 10 μg/ml of goat anti-mouse Alexa Fluor® 488-conjugated F(ab′)2 (Invitrogen) for 1 h at room temperature. F-actin in the cells was stained at room temperature for 1 h using 4 units/ml of Alexa Fluor 633-conjugated phalloidin (Invitrogen). The cells nuclei were then stained by incubating with 300 nm DAPI (Invitrogen) for 5 min at room temperature. After mounting a coverslip, 1024 × 1024 pixel images were taken using a Zeiss Axiovert 710 confocal microscope with a slice depth of 1 μm. Image processing was performed using the Zeiss Image Browser software package.
HUVEC Tube Formation
Cell culture plates (96-well) were bottom-coated with a thin layer of ECM gel (50 μl/well), which was left to polymerize at 37 °C for 60 min. HUVEC (2–3 × 104 cells) were stimulated with VEGF in 150 μl of medium and added to each well on the solidified ECM gel. Culture medium was added to each well in the presence or absence of LY5 (300 nm). The plates were incubated at 37 °C for 12–18 h and the endothelial tubes were quantified using a fluorescent microscope after staining with Calcein AM dye. Tube forming ability was quantified by counting the total number of cell clusters and branches under a ×4 objective and four different fields per well. The results are expressed as mean-fold change of branching compared with the control groups. Each experiment was performed at least three times.
In Vitro Three-dimensional Angiogenesis Assays
The three-dimensional in vitro angiogenesis assay was performed essentially as described by the manufacturer's instructions (Promo cell). Briefly, the assay plate was placed in a humidified incubator (37 °C, 5% CO2) and HUVEC (400 cells each well) implanted in collagen gels were prepared. The transport medium from the bottom of the well was aspirated and added to 200 μl of test medium including VEGF (20 ng/ml) with or without LY5 (300 nm). The image was taken after HUVECs were cultured in test medium for 24 h, and the average endothelial vascular sprout length was measured without staining using a phase-contrast microscope and an ocular micrometer. Sprout formation was assessed in two independent experiments.
Human Angiogenesis Array
Proteome profiler antibody array (R & D Systems, catalogue number ARY007) was used according to manufacturer's instructions to detect the relative levels of expression of 55 angiogenesis related proteins in HUVEC cells treated with or without LY5 (300 nm). After blocking the membranes, 300 μg of protein of lysed HUVECs in control and LY5-treated groups was added and incubated overnight at 4 °C. The membranes were washed and incubated with streptavidin-HRP for 30 min. Immunoreactive signals were visualized by using Super Signal Chemiluminescence substrate (Pierce). Array data on developed x-ray film was quantified by scanning the film using Bio-Rad Molecular Image Gel DocTM XR+ and data were analyzed using Image LabTM software.
STAT3 siRNA Transfection
Human STAT3 siRNA and negative control siRNA (Cell Signaling Technology, 100 nm) were transfected into UW288-1, UW426, or DAOY cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 48 h, the cells were harvested and lysed for protein or gene analysis as described above or processed to cell viability assay.
MTT Cell Viability Assay
Cells were seeded in 96-well plates at a density of 3000 cells/well and cultured for 24 h. For the cell viability experiment, cells were treated by LY5 at 0.5, 1, 5, and 10 μm in triplicate for 24 h. For the STAT3 siRNA experiment, cells were transfected with or without STAT3 siRNA for 48 h and treated with LY5 (1 and 2 μm) in triplicate for 24 h. For a combination effect experiment, cells were irradiated with 4 gray x-ray and cultured for 7 days, then treated by LY5 (2, 3 μm) in triplicate for 24 h. 25 μl of MTT (Sigma) was added to each sample and incubated for 4 h. Then 100 μl of N,N-dimethylformamide (Sigma) solubilization solution was added to each well. The absorbance was read at 595 nm.
Quantitative Reverse Transcriptase-PCR
Mature microRNA-21 (miR-21) gene expression was determined by quantitative reverse transcriptase (44). Briefly, total mRAN, including micro-RNAs, was isolated from cells using a miRNeasy Mini kit (Qiagen). The cDNA was generated using the miScript II reverse transcription kit (Qiagen). Real-time PCR amplification was performed using the miScript SYBR Green PCR Kit and miScript Primer Assays (primer sequence of miR-21: 5′-UAGCUUAUCAGACUGAUGUUGA) (Qiagen) according to the manufacturer's protocol with an Applied Biosystems 7900 HT Fast Real-time PCR System. U6 was used as an internal control for template normalization. The relative gene expression level between treatments was calculated using the following equation: relative gene expression = 2−(ΔCTsample−ΔCTcontrol).
Statistical Analysis
Significance of correlations was done using GraphPad Prism software. Unpaired t tests were used for analyses assuming Gaussian populations with a 95% confidence interval. Data are presented as mean ± S.E. Differences were analyzed with the Student's t test, and significance was set at p < 0.05.
RESULTS
LY5, a Novel Small Molecule That Targets STAT3
As the STAT3 SH2 domain is critical for the activation and biological function of STAT3, we designed a new small molecule inhibitor blocking its SH2 domain phosphotyrosine binding site to inhibit STAT3 activation. A new FBDD approach, in silico site-directed FBDD, was applied to identify the fragments from known STAT3 inhibitors that target to the STAT3 SH2 domain. On the basis of the binding mode, two fragments that were specific for each of the two binding sites: site Tyr(P)-705 and side pocket were selected out and linked to form a new compound, named LY5. This new compound was screened via computational docking mode and demonstrated most favorable docking energy and binding mode to STAT3 SH2 domain. Fig. 1A shows a docking model of LY5 binding to the STAT3 SH2 domain (Protein Data Bank code 1BG1) and LY5 fits well to the two key binding pockets, “site Tyr(P)-705” and “side pocket,” predicting that LY5 will be a selective inhibitor of STAT3. The chemical structure and synthesis of LY5 was shown in Fig. 1B. Strong binding energy of LY5 to STAT3 (−7.9 kcal/mol) also suggested that LY5 is a potent small molecule inhibitor that targets STAT3.
FIGURE 1.
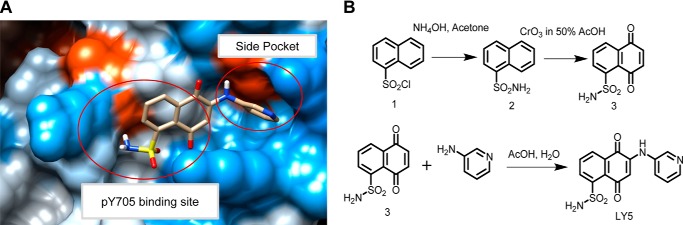
The computer model of LY5 binds to the STAT3 SH2 domain, chemical structure, and synthesis of LY5. A, docking model of LY5 binding to the STAT3 SH2 domain (Protein Data Bank code 1BG1), generated by AutoDock4 and viewed by Chimera. LY5 fits to the two key binding pockets, site Tyr(P)-705 (pY705) and side pocket. Carbon atoms of LY5 are colored gray. The molecular surface is colored according to the element property. B, synthesis of LY5 (includes chemical structure).
LY5 Diminishes STAT3 Phosphorylation and Induces Apotosis in Human Medulloblastoma Cells
LY5 was analyzed for its inhibitory effect on STAT3 phosphorylation in human medulloblastoma cell lines (DAOY, UW426, and UW288-1), which express persistent STAT3 phosphorylation. LY5 dramatically decreased the level of STAT3 phosphorylated at tyrosine residue 705 (Tyr-705) in all three medulloblastoma cell lines (Fig. 2A). Considering that activated STAT3 binds to the promoter regions to regulate the transcription of target genes that are involved in tumor cell proliferation and anti-apoptotic processes (16, 18), we measured the expression of downstream target genes of STAT3 by reverse transcriptase PCR to analyze the impact of LY5 on the inhibition of STAT3. As shown in Fig. 2B, downstream targeted genes of STAT3 such as cyclinD1, survivin, and bcl-XL in UW426, UW288-1, and DAOY medulloblastoma cell lines were down-regulated when treated with LY5. We also observed LY5 treatment suppressed the expression of miR-21 (Fig. 2C) in which gene expression was elevated in many kinds of cancer (45, 46). This result was consistent with the report that miR-21 was directly activated by STAT3 and the inhibition of STAT3 by siRNA strongly decreased miR-21 expression (47), which indicated the inhibitory effect of LY5 on STAT3. In addition, we found that treatment with LY5 resulted in apoptosis in human medulloblastoma cells as evidenced by the cleaved caspase-3 (Fig. 2, A and D). Cell viability was decreased by LY5 treatment (Fig. 2D), which confirmed the inhibitory effect of LY5 on tumor cell proliferation. However, LY5 did not induce apoptosis in normal human cell lines including human hepatocytes, human skeletal muscle myoblasts, and human coronary smooth muscle cells (Fig. 2D). These results indicated that LY5 selectively induced apoptosis in cancer cells expressing persistent STAT3 phosphorylation. Furthermore, the inhibitory effect of LY5 on STAT3 phosphorylation was evaluated in other types of cancer cell lines including sarcoma, breast, and pancreatic cancers. LY5 significantly suppressed the level of STAT3 phosphorylated in a dose-dependent manner in all three human cancer cell lines as shown in Fig. 2E. The effects of LY5 on P-STAT3, P-JAK2, P-P65 NF-κB, and P-Smad1/5 were shown in Fig. 2F. LY5 showed dramatic inhibition of STAT3 phosphorylation but did not inhibit or did not significantly inhibit P-JAK2, P-P65 NF-κB, and P-Smad1/5. Only certain reduction of P-P65 NF-κB in UW426 cells was observed but P-P65 NF-κB in UW288-1 cells was slightly increased by LY5.
FIGURE 2.

LY5 inhibits STAT3 phosphorylation, down-regulates expression of STAT3 target genes, and induces apoptosis in human medulloblastoma cells expressing persistent STAT3 phosphorylation. A, Western blot analysis of P-STAT3 and cleaved caspase-3 in medulloblastoma cells treated with LY5 at the indicated concentrations in DAOY, UW288-1, and UW426 cells. B, downstream target genes of STAT3, including cyclinD1, bcl-XL, and survivin in DAOY, UW288-1, and UW426 treated by LY5 were detected by RT-PCR as described under “Experimental Procedures.” C, miR-21 expression was measured in UW288-1 and UW426 cells treated by LY5 using quantitative RT-PCR as described under “Experimental Procedures.” The results are representative of 3 independent experiments. D, LY5 induced apoptosis in medulloblastoma cells, but did not induce apoptosis in human normal cell lines that do not express elevated levels of STAT3 phosphorylation. Following treatment with LY5, Western blot analysis showed the level of cleaved caspase-3 in UW288-1 and UW426 cell lines, as well as in normal cell lines including human coronary artery smooth muscle cells (HCSMC), human hepatocytes (HH), and human skeletal muscle myoblasts (HSMM). UW288-1 and UW426 cells were treated by LY5 at the indicated concentration for 24 h. MTT assay was processed to analyze cell viability. E, Western blot analysis of P-STAT3 in other types of cancer including pancreatic cancer (MIA-PACA-2), sarcoma (RH30), and breast cancer (MDA-MB-231) cells treated with LY5 at the indicated concentration. F, P-STAT3, P-JAK2, P-Smad1/5, and P-P65 NF-κΒ expression were detected in UW288-1 and UW426 cells treated with LY5 at the indicated concentration by Western blot analysis.
We next used a small interfering RNA (siRNA) approach to specifically knockdown STAT3 to assess the effects on medulloblastoma cell viability. STAT3 inhibition by STAT3 siRNA suppressed persistent STAT3 phosphorylation (Fig. 3A), expression of the STAT3 downstream targets (Fig. 3, B and C), and survival/cell viability (Fig. 3D), and induced apoptosis (Fig. 3A) in human medulloblastoma cells. These results indicate that medulloblastoma cells are addictive to a STAT3-dependent survival pathway and are sensitive to STAT3 inhibition, which strongly supports a rationale to target persistent STAT3 signaling with a potential for cancer therapy. The inhibition of STAT3 phosphorylation by the small molecular inhibitor LY5 is consistent with reduced phosphorylated STAT3 protein and STAT3 downstream target gene expression as well as induction of apoptosis by STAT3-specific siRNA in human medulloblastoma cells. Furthermore, Fig. 3D showed that cell viability was decreased in UW426 and UW288-1 when STAT3 was silenced by STAT3 siRNA, but LY5 treatment did not enhance the inhibition of cell viability, suggesting that LY5 is selective for STAT3.
FIGURE 3.

STAT3 siRNA decreases the expression level of P-STAT3, inhibits cell growth, and induces apoptosis in medulloblastoma cells. A, DAOY, UW288-1, and UW426 cells were transfected with STAT3 siRNA for 48 h as described under “Experimental Procedures.” P-STAT3 (Y705), P-STAT3 (S727), total STAT3, and Caspase-3 were assessed by Western blot analysis (No, no treatment; C. siRNA, control siRNA). B, downstream target genes of STAT3, including cyclinD1, bcl-XL, and survivin in UW288-1 and UW426 transfected with STAT3 siRNA were detected by RT-PCR. C, miR-21 expression was measured in UW288-1 and UW426 cells transfected with STAT3 siRNA using quantitative RT-PCR as described under “Experimental Procedures.” D, UW288-1 and UW426 cells were transfected with or without STAT3 siRNA for 48 h, followed by LY5 treatment at the indicated concentration for 24 h. MTT assay was processed to analyze cell viability. The results are representative of 3 independent experiments.
LY5 Inhibits STAT3 Phosphorylation and Blocks STAT3 Nuclear Translocation Induced by Growth Factors and Cytokines
It is known that when cytokines and growth factors specifically bind to their respective receptors, STAT3 is activated by the phosphorylation of its tyrosine residue in a SH2 domain through cytokine receptors and the Janus kinase (JAK) (48, 49). The activated STAT3 dimers translocate from the cytoplasm to the nucleus to exert its function as a transcription factor. To determine whether LY5 suppress STAT3 activation induced by cytokines or growth factors, human medulloblastoma cells (DAOY) were cultured in serum-free medium for 24 h and then pretreated with LY5 followed by stimulating with growth factors or cytokines including IL-6, IGF-1, IGF-2, LIF, EGF, as well as IFN-γ, then the levels of STAT3, STAT1, and STAT5 protein phosphorylation were examined. The STAT3 phosphorylation level was increased by stimulation of IL-6, IGF-1, IGF-2, or LIF, but this increase of STAT3 phosphorylation was inhibited by LY5 treatment in a dose-dependent manner. In contrast, LY5 had no inhibitory effect on increased STAT1 or STAT5 phosphorylation stimulated by IFN-γ or EGF, respectively, in DAOY cells (Fig. 4A).
FIGURE 4.

LY5 selectively inhibits STAT3 activation induced by cytokines and growth factors. A, DAOY medulloblastoma cells with lower expressing P-STAT3 were serum starved overnight, then treated or untreated with the indicated concentrations of LY5. After 4 h, the untreated and LY5-treated cells were stimulated by IL-6, IGF-1, IGF-2, LIF, EGF, or IFN-γ (50 ng/ml). The cells were harvested at 30 min and analyzed P-STAT3, P-STAT5, or P-STAT1 by Western blot. B, UW288 cells were plated on coverslides overnight and cultured in serum-free medium for 24 h. Then, cells were pretreated with LY5 (5 μm) for 4 h followed by IL-6 (50 ng/ml) stimulation for 30 min, and processed for STAT3 nuclear translocation detection by immunofluorescence as described under “Experimental Procedures.” STAT3 nuclear translocation positive cell percentage was quantified. C, UW288 cells were treated the same as described in B, except using stimulation with IFN-γ (50 ng/ml). STAT1 nuclear translocation was detected by immunofluorescence. D, UW288 cells were treated the same as described in B, except stimulation with EGF (100 ng/ml). STAT5 nuclear translocation was detected by immunofluorescence.
As mentioned above, LY5 was designed to inhibit STAT3 activation and dimerization, so we hypothesize that LY5 should be able to prevent STAT3 from translocating to the nucleus induced by IL-6. To test this hypothesis, UW288-1 cells were starved in serum-free medium for 24 h and pretreated with LY5 followed by IL-6 stimulation, after fixation, immunofluorescence was conducted as described under “Experimental Procedures.” As illustrated in Fig. 4B, IL-6 induced STAT3 translocation from the cytoplasm to the nucleus in UW288-1 cells, but we observed that most of STAT3 was retained in the cytoplasm when cells were treated with LY5, suggesting LY5 treatment blocked STAT3 nuclear translocation mediated by IL-6. However, LY5 treatment did not block STAT1 or STAT5 nuclear translocation induced by IFN-γ or EGF, respectively, in UW288-1 cells (Fig. 4, C and D).
LY5 Inhibits Cell Migration
We have shown that LY5 effectively inhibits STAT3 activation, suppresses its downstream target gene expression, and induces apoptosis in human medulloblastoma cells that harbor persistent STAT3 phosphorylation. Therefore, we next examined whether LY5 blocks STAT3-dependent cell migration, which may be an important process in tumor metastasis. For this purpose, a wound healing assay was conducted. As shown in Fig. 5A, HUVEC cells that were stimulated with VEGF migrated within 22 h to fill the scratched area, but LY5 treatment prevented this migration. Consistent with the above result, LY5 also significantly blocked medulloblastoma cells to migrate through the scratched area (Fig. 5B). In addition, considering F-actin and microtubule cytoskeletal elements that are regulated by STAT3 both involve the function of cell migration (50, 51), the ability of LY5 to inhibit F-actin fiber and microtubule formation was evaluated by confocal microscopy as described under “Experimental Procedures.” In the presence of LY5 treatment, Fig. 5C shows the cells had a condensed, rounded morphology, F-actin had compressed into fewer fibers or disappeared from the leading edge, and microtubules curled over without extending to the leading edge of the cells. However, F-actin in cells treated by VEGF without the presence of LY5 produced thin, identical fibers spanning the length of the cells, and microtubules formed a dense lattice that emanated from the center of the cells. This result suggests that inhibition of STAT3 by LY5 disrupts the F-actin and microtubule cytoskeletal elements.
FIGURE 5.

LY5 inhibits migration in HUVECs and medulloblastoma cells harboring high level of P-STAT3. A, wound healing assay for migration was carried out by scratching the cells with yellow tip when HUVECs grew into monolayer. Then, cells were incubated with the medium containing VEGF (10 ng/ml) with or without LY5 (300 nm) and allowed to migrate into the scratched area for 22 h. B, wound healing assay as described under “Experimental Procedures” was conducted for migration in UW288-1 medulloblastoma cells treated with the indicated concentrations of LY5. The arrow showed the gap of the scratched area and the percentage of migrating cells in wound healing assay was quantified. C, inhibition of STAT3 disrupts the F-actin and microtubule cytoskeletal elements in HUVECs. Cells cultured in 4-well chamber slides were treated with VEGF (10 ng/ml) in the presence or absence of LY5 (300 nm) for 18 h, then probed using anti-β-tubulin primary antibodies (green), and F-actin was stained using phalloidin (red). In the confocal photo, white arrows highlight F-actin localization at the leading edge, whereas white arrowheads indicate the curling of microtubules at the cell periphery. Slice depth = 1 μm. Scale bar, 20 μm. Inset, ×400 magnification.
Inhibition of STAT3 by LY5 Represses Angiogenesis in Vitro
Increasing evidence suggests that STAT3 is also an important facilitator of angiogenesis under both physiological and pathological situations (30, 52). The activation of STAT3 not only regulates VEGF production and function in a variety of human cancers, but also correlates with the expression of other critical angiogenic factors including angiopoietin, MMP-9, CXCL16, as well as IGF-binding protein (28, 53). To study the anti-angiogenic activity of LY5, human HUVEC were stimulated with VEGF in the absence or presence of LY5 and tube formation, a three-dimensional angiogenesis assay as well as angiogenesis array were examined. As shown in Fig. 6A, the number of branches was obviously decreased by LY5 treatment, which showed that LY5 inhibited HUVEC tube formation. The treatment also abolished VEGF-induced sprouting from pre-formed spheroids of HUVEC in a three-dimensional in vitro angiogenesis assay (Fig. 6B), suggesting that LY5 may play a crucial role in VEGF-induced angiogenic sprouting of blood vessels. Finally, to evaluate the inhibitory effect of LY5 on various important angiogenic factors including VEGF, we examined the levels of 55 angiogenesis-associated proteins using a human angiogenesis array. Compared with the control group, the LY5-treated group showed a dramatic decrease of VEGF, MMP-9, Angiopoietin, tissue factor, and many other critical regulators of angiogenesis (Fig. 6C). Taken together, data from the above experiments suggests that targeting STAT3 by LY5 inhibits angiogenesis in vitro.
FIGURE 6.

Inhibition of STAT3 by LY5 represses angiogenesis in vitro. A, LY5 inhibits HUVEC tube formation. HUVECs were grown in M200 with VEGF (10 ng/ml) for 18–20 h in the absence or presence of LY5 (300 nm). The number of branches was determined by CalcinM staining and tube formation was quantified by counting the total number of branches and cell clusters as described under “Experimental Procedures.” B, LY5 treatment decreased the length of endothelial vascular sprouts. Representative phase-contrast photographs of endothelial vascular sprouts were shown in a three-dimensional angiogenesis assay. Length of endothelial vascular sprouts in a three-dimensional angiogenic assay (bottom panel) is expressed as mean of vascular sprout length ± S.E. for three independent experiments. *, p < 0.05. C, the expression of various critical angiogenic factors in human endothelial cells was determined using a Proteome profiler antibody array as described under “Experimental Procedures.” The effect of LY5 treatment is quantified in the histogram.
The Effects of LY5 Combined with Cisplatin and X-ray Irradiation
Cisplatin is one of the first line chemotherapeutic agents used in medulloblastoma. However, cancer cells often develop resistance to cisplatin, which limits clinical therapeutic effectiveness. It is found that cisplatin-resistant cancer cells frequently express elevated levels of P-STAT3/STAT3 and is one of the mechanisms of drug resistance (54, 55), because the constitutive STAT3 signaling pathway can mediate the survival signals and confer resistance to apoptosis induced by chemotherapeutic agents (56–59). We therefore rationed that the STAT3-selective inhibitor LY5 should be able to enhance cisplatin-mediated inhibition of cell viability. Indeed, we observed that a combination of LY5 and cisplatin generates a statistically significant decrease of cell viability than single agents alone in UW288-1 and UW426 human medulloblastoma cell lines (Fig. 7A). Furthermore, radiotherapy has been commonly used for treating medulloblastoma (60–62). To evaluate the combinational effect of LY5 and irradiation, cell viability was measured for UW288-1 or UW426 cells, which was irradiated with 4 gray x-ray and cultured for 7 days, then treated by LY5. Our result showed that combinational treatments of LY5 and x-ray radiation produced a statistically better inhibitory effect than a single treatment alone (Fig. 7B). These results suggested that the small molecule inhibitor LY5 enhanced the killing efficiency of nontoxic doses of cisplatin and x-ray, which provided promising improvement for cancer therapy.
FIGURE 7.

The effects of LY5 combined with cisplatin and x-ray irradiation. A, UW288-1 and UW426 cells were seeded in 96-well plate at a density of 3000 cells/well and cultured for 24 h. Cells were treated by LY5 and cisplatin (CDDP) at the indicated concentrations in triplicate for 24 h and processed for MTT assay to analyze cell viability. B, UW288-1 and UW426 cells were seeded the same as described for A. Cells were irradiated with 4 gray x-ray and cultured for 7 days. LY5 treatment was performed at the indicated concentrations in triplicate for 24 h to detect a combinational effect of LY5 and irradiation using MTT assay. The results are representative of 3 independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
DISCUSSION
Constitutive activation of STAT3 signaling is crucial to tumorigenesis, proliferation, survival, and invasion of various human cancers and cancer cell lines, promoting it as a very attractive drug development target for tumor treatment (63–65). The first phosphotyrosyl peptide was reported as a STAT3 inhibitor in 2001 to inhibit STAT3 activity in vitro and in vivo by competing with the native phosphopetide of STAT3 protein (15). This peptide and subsequently developed peptidomimetics inhibitors elucidate the role of STAT3 and the impact of the Stat3 inhibitor. Because of the challenge of the metabolic instability and poor cell permeability, the use of peptide and peptidomimetics inhibitors was limited. Therefore, additional research works focus on developing non-peptide small molecule inhibitors of STAT3, which were mostly designed to directly inhibit STAT3 activity through disrupting STAT3-STAT3 dimerization. Among all these reagents, STA-21 was discovered by a structure-based virtual screening as one small molecule compound that blocks STAT3 dimerization, DNA binding, and transcriptional activity in human breast cancer cells (3). Another example of the non-peptide small molecule STAT3 inhibitor (66), S3I-201 was identified as a STAT3-STAT3 dimerization disruptor to inhibit STAT3 activity, and induce apoptosis of tumor cells harboring aberrantly active STAT3, and suppress tumor growth in human breast cancer xenograft models. A couple of its analogs with improved potency, including S3I-201.1066, BP-1-102, and S3I-1757, were proved to inhibit malignant transformation, tumor cell growth, migration, and invasion (67, 68). To date, however, few STAT3 inhibitors are available as cancer therapy drugs used in clinical trials (69). More research efforts to discover more druggable small molecule STAT3 inhibitors with high potency and bioavailability for tumor therapy are still needed. In this study, we have demonstrated a novel developed small molecule STAT3 inhibitor, LY5, inhibited STAT3 phosphorylation, decreased STAT3 downstream gene expression, and induced apoptosis in human medulloblastoma cell lines expressing persistent STAT3 phosphorylation. LY5 also selectively decreased inducible STAT3 phosphorylation, blocked STAT3 nuclear localization upon IL-6 stimulation, inhibited cell migration, and suppressed angiogenesis in vitro.
Using the new in silico site-directed computational fragment-based drug design approach, we identified LY5 as a non-peptide cell-permeable inhibitor of STAT3 dimerization to block STAT3 activation with low IC50 values (0.5–1.4 μm) and strong binding affinity to the STAT3 SH2 domain confirmed by the fluorescence polarization assay (36). The IC50 values of the inhibition of cell viability by LY5 measured in UW288-1, UW426, and DAOY are 0.364, 0.318, and 0.488 μm, respectively. The ability of LY5 to significantly inhibit STAT3 phosphorylation at the lower drug concentration in medulloblastoma expressing constitutive STAT3 suggested its high potency. Several known STAT3 downstream targets, such as cyclinD1, survivin, and bcl-XL, were decreased by LY5 treatment as demonstrated by RT-PCR analysis and angiogenesis array, which also support the idea of LY5 as a potent STAT3 inhibitor. In addition, the result that miR-21 gene expression was down-regulated by LY5 gave another confirmation of the inhibitory effect of LY5 on STAT3. The fact that LY5 inhibited STAT3 phosphorylation stimulated by cytokines and growth factors including IL-6, IGF-1, IGF-2, and LIF, but had no effect on STAT1 or STAT5 phosphorylation induced by IFN-γ or EGF, suggested that LY5 selectively suppressed STAT3 activation. All these results that indicate the potency and selectivity of LY5 for STAT3 are consistent with the prediction derived from its original design of a novel STAT3 inhibitor scaffold with most favorable docking energies and binding pose to STAT3 SH2 domain screened via computational docking.
LY5 showed its anti-tumor cell function by inducing apoptosis in medulloblastoma cells due to STAT3 inhibition. The fact that LY5 blocked tumor cell migration and angiogenesis in vitro through inhibition of STAT3 gave more supports for this suggestion. In addition, the result that LY5 only induced apoptosis in tumor cell, but no influence on normal human cells showed LY5 is potent in inhibiting cancer cells but lower toxicity to normal human cells. We also observed that LY5 is a potent inhibitor of persistent STAT3 phosphorylation in cancer cells from other types of cancer including sarcoma, breast, and pancreatic cancers as shown in Fig. 2E. Furthermore, our results showed that treatment of LY5 combined with cisplatin or x-ray radiation produced a statistically better inhibitory effect than single treatment alone, which suggested LY5 enhanced the killing efficiency of nontoxic doses of cisplatin and x-ray. On the basis of these findings, LY5 could be extended to an application to investigate its inhibitory effect on other tumor types of cancer cells with persistent STAT3 signaling or animal tumor in vivo.
In summary, we developed a novel non-peptide small molecule STAT3 inhibitor, LY5, which selectively inhibited persistent STAT3 activation and induced apoptosis in medulloblastoma cells. This study demonstrated LY5 is a promising therapeutic drug candidate for human medulloblastoma through inhibiting STAT3 signaling.
Acknowledgments
We thank members of the laboratory and our collaborators for their ideas and comments about this work.
Note Added in Proof
The images representing GAPDH and STAT3 blots for RH30 cells in Fig. 2E, GAPDH blots for LY5+LIF in Fig. 4A, and the top three subfigures of DMSO-treated cells in Fig. 4D were mistakenly switched in the original version of Figs. 2 and 4, respectively, that were published on October 13, 2014 as a Paper in Press. These mistakes have been corrected.
This work was supported by Alex's Lemonade Stand Foundation and the Technology Development Fund from The Research Institute at Nationwide Children's Hospital.
- SH2
- Src homology domain 2
- STAT
- signal transducer and activator of transcription
- IL-6
- interleukin 6
- LY5
- 5,8-dioxo-6-(pyridin-3-ylamino)-5,8-dihydronaphthalene-1-sulfonamide
- HUVEC
- human umbilical vein endothelial cell
- MTT
- 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide
- LIF
- leukemia inhibitory factor
- IGF
- insulin-like growth factor
- MMP
- matrix metalloprotease
- FBDD
- fragment-based drug design
- miR
- microRNA.
REFERENCES
- 1. Barton B. E., Karras J. G., Murphy T. F., Barton A., Huang H. F. (2004) Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol. Cancer Ther. 3, 11–20 [PubMed] [Google Scholar]
- 2. Turkson J., Jove R. (2000) STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19, 6613–6626 [DOI] [PubMed] [Google Scholar]
- 3. Song H., Wang R., Wang S., Lin J. (2005) A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 102, 4700–4705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Darnell J. E., Jr., Kerr I. M., Stark G. R. (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421 [DOI] [PubMed] [Google Scholar]
- 5. Zhong Z., Wen Z., Darnell J. E., Jr. (1994) Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98 [DOI] [PubMed] [Google Scholar]
- 6. Bromberg J. F., Wrzeszczynska M. H., Devgan G., Zhao Y., Pestell R. G., Albanese C., Darnell J. E., Jr. (1999) Stat3 as an oncogene. Cell 98, 295–303 [DOI] [PubMed] [Google Scholar]
- 7. Schlessinger K., Levy D. E. (2005) Malignant transformation but not normal cell growth depends on signal transducer and activator of transcription 3. Cancer Res. 65, 5828–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calò V., Migliavacca M., Bazan V., Macaluso M., Buscemi M., Gebbia N., Russo A. (2003) STAT proteins: from normal control of cellular events to tumorigenesis. J. Cell Physiol. 197, 157–168 [DOI] [PubMed] [Google Scholar]
- 9. Clark J., Edwards S., Feber A., Flohr P., John M., Giddings I., Crossland S., Stratton M. R., Wooster R., Campbell C., Cooper C. S. (2003) Genome-wide screening for complete genetic loss in prostate cancer by comparative hybridization onto cDNA microarrays. Oncogene 22, 1247–1252 [DOI] [PubMed] [Google Scholar]
- 10. Zong C. S., Zeng L., Jiang Y., Sadowski H. B., Wang L. H. (1998) Stat3 plays an important role in oncogenic Ros- and insulin-like growth factor I receptor-induced anchorage-independent growth. J. Biol. Chem. 273, 28065–28072 [DOI] [PubMed] [Google Scholar]
- 11. Kuropatwinski K. K., De Imus C., Gearing D., Baumann H., Mosley B. (1997) Influence of subunit combinations on signaling by receptors for oncostatin M, leukemia inhibitory factor, and interleukin-6. J. Biol. Chem. 272, 15135–15144 [DOI] [PubMed] [Google Scholar]
- 12. Cressman D. E., Greenbaum L. E., DeAngelis R. A., Ciliberto G., Furth E. E., Poli V., Taub R. (1996) Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 274, 1379–1383 [DOI] [PubMed] [Google Scholar]
- 13. Darnell J. E., Jr. (1997) STATs and gene regulation. Science 277, 1630–1635 [DOI] [PubMed] [Google Scholar]
- 14. Bromberg J., Darnell J. E., Jr. (2000) The role of STATs in transcriptional control and their impact on cellular function. Oncogene 19, 2468–2473 [DOI] [PubMed] [Google Scholar]
- 15. Turkson J., Ryan D., Kim J. S., Zhang Y., Chen Z., Haura E., Laudano A., Sebti S., Hamilton A. D., Jove R. (2001) Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 276, 45443–45455 [DOI] [PubMed] [Google Scholar]
- 16. Aoki Y., Feldman G. M., Tosato G. (2003) Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 101, 1535–1542 [DOI] [PubMed] [Google Scholar]
- 17. Xu Q., Briggs J., Park S., Niu G., Kortylewski M., Zhang S., Gritsko T., Turkson J., Kay H., Semenza G. L., Cheng J. Q., Jove R., Yu H. (2005) Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552–5560 [DOI] [PubMed] [Google Scholar]
- 18. Amin H. M., McDonnell T. J., Ma Y., Lin Q., Fujio Y., Kunisada K., Leventaki V., Das P., Rassidakis G. Z., Cutler C., Medeiros L. J., Lai R. (2004) Selective inhibition of STAT3 induces apoptosis and G1 cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene 23, 5426–5434 [DOI] [PubMed] [Google Scholar]
- 19. Bollrath J., Phesse T. J., von Burstin V. A., Putoczki T., Bennecke M., Bateman T., Nebelsiek T., Lundgren-May T., Canli O., Schwitalla S., Matthews V., Schmid R. M., Kirchner T., Arkan M. C., Ernst M., Greten F. R. (2009) gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15, 91–102 [DOI] [PubMed] [Google Scholar]
- 20. Turkson J., Bowman T., Garcia R., Caldenhoven E., De Groot R. P., Jove R. (1998) Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 18, 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bowman T., Garcia R., Turkson J., Jove R. (2000) STATs in oncogenesis. Oncogene 19, 2474–2488 [DOI] [PubMed] [Google Scholar]
- 22. Yu C. L., Meyer D. J., Campbell G. S., Larner A. C., Carter-Su C., Schwartz J., Jove R. (1995) Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269, 81–83 [DOI] [PubMed] [Google Scholar]
- 23. Wang T., Niu G., Kortylewski M., Burdelya L., Shain K., Zhang S., Bhattacharya R., Gabrilovich D., Heller R., Coppola D., Dalton W., Jove R., Pardoll D., Yu H. (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10, 48–54 [DOI] [PubMed] [Google Scholar]
- 24. Yu H., Pardoll D., Jove R. (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fu X. Y. (2006) STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 16, 214–219 [DOI] [PubMed] [Google Scholar]
- 26. Hodge D. R, Hurt E. M., Farrar W. L. (2005) The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 41, 2502–2512 [DOI] [PubMed] [Google Scholar]
- 27. Yu H., Kortylewski M., Pardoll D. (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51 [DOI] [PubMed] [Google Scholar]
- 28. Wei D., Le X., Zheng L., Wang L., Frey J. A., Gao A. C., Peng Z., Huang S., Xiong H. Q., Abbruzzese J. L., Xie K. (2003) Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 22, 319–329 [DOI] [PubMed] [Google Scholar]
- 29. Dechow T. N., Pedranzini L., Leitch A., Leslie K., Gerald W. L., Linkov I., Bromberg J. F. (2004) Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. U.S.A. 101, 10602–10607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niu G., Wright K. L., Huang M., Song L., Haura E., Turkson J., Zhang S., Wang T., Sinibaldi D., Coppola D., Heller R., Ellis L. M., Karras J., Bromberg J., Pardoll D., Jove R., Yu H. (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21, 2000–2008 [DOI] [PubMed] [Google Scholar]
- 31. Jones D. T., Jäger N., Kool M., Zichner T., Hutter B., Sultan M., Cho Y. J., Pugh T. J., Hovestadt V., Stütz A. M., Rausch T., Warnatz H. J., Ryzhova M., Bender S., Sturm D., Pleier S., Cin H., Pfaff E., Sieber L., Wittmann A., Remke M., Witt H., Hutter S., Tzaridis T., Weischenfeldt J., Raeder B., Avci M., Amstislavskiy V., Zapatka M., Weber U. D., Wang Q., Lasitschka B., Bartholomae C. C., Schmidt M., von Kalle C., Ast V., Lawerenz C., Eils J., Kabbe R., Benes V., van Sluis P., Koster J., Volckmann R., Shih D., Betts M. J., Russell R. B., Coco S., Tonini G. P., Schüller U., Hans V., Graf N., Kim Y. J., Monoranu C., Roggendorf W., Unterberg A., Herold-Mende C., Milde T., Kulozik A. E., von Deimling A., Witt O., Maass E., Rossler J., Ebinger M., Schuhmann M. U., Fruhwald M. C., Hasselblatt M., Jabado N., Rutkowski S., von Bueren A. O., Williamson D., Clifford S. C., McCabe M. G., Collins V. P., Wolf S., Wiemann S., Lehrach H., Brors B., Scheurlen W., Felsberg J., Reifenberger G., Northcott P. A., Taylor M. D., Meyerson M., Pomeroy S. L., Yaspo M. L., Korbel J. O., Korshunov A., Eils R., Pfister S. M., Lichter P. (2012) Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ling X., Arlinghaus R. B. (2005) Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 65, 2532–2536 [DOI] [PubMed] [Google Scholar]
- 33. Yang F., Jove V., Xin H., Hedvat M., Van Meter T. E., Yu H. (2010) Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways. Mol. Cancer Res. 8, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burke W. M., Jin X., Lin H. J., Huang M., Liu R., Reynolds R. K., Lin J. (2001) Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 20, 7925–7934 [DOI] [PubMed] [Google Scholar]
- 35. Yu H., Jove R. (2004) The STATs of cancer: new molecular targets come of age. Nat. Rev. Cancer 4, 97–105 [DOI] [PubMed] [Google Scholar]
- 36. Yu W., Xiao H., Lin J., Li C. (2013) Discovery of novel STAT3 small molecule inhibitors via in silico site-directed fragment-based drug design. J. Med. Chem. 56, 4402–4412 [DOI] [PubMed] [Google Scholar]
- 37. Huey R., Morris G. M., Olson A. J., Goodsell D. S. (2007) A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 28, 1145–1152 [DOI] [PubMed] [Google Scholar]
- 38. Tian X., Li J., Ma Z. M., Zhao C., Wan D. F., Wen Y. M. (2009) Role of hepatitis B surface antigen in the development of hepatocellular carcinoma: regulation of lymphoid enhancer-binding factor 1. J. Exp. Clin. Cancer Res. 28, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paydas S., Tanriverdi K., Yavuz S., Disel U., Sahin B., Burgut R. (2003) Survivin and aven: two distinct antiapoptotic signals in acute leukemias. Ann. Oncol. 14, 1045–1050 [DOI] [PubMed] [Google Scholar]
- 40. Shimonovitz S., Hurwitz A., Dushnik M., Anteby E., Geva-Eldar T., Yagel S. (1994) Developmental regulation of the expression of 72 and 92 kDa type IV collagenases in human trophoblasts: a possible mechanism for control of trophoblast invasion. Am. J. Obstet. Gynecol. 171, 832–838 [DOI] [PubMed] [Google Scholar]
- 41. Yamaguchi H., Inokuchi K., Tarusawa M., Dan K. (2002) Mutation of bcl-x gene in non-Hodgkin's lymphoma. Am. J. Hematol. 69, 74–76 [DOI] [PubMed] [Google Scholar]
- 42. Thaloor D., Singh A. K., Sidhu G. S., Prasad P. V., Kleinman H. K., Maheshwari R. K. (1998) Inhibition of angiogenic differentiation of human umbilical vein endothelial cells by curcumin. Cell Growth Differ. 9, 305–312 [PubMed] [Google Scholar]
- 43. Shiota M., Bishop J. L., Nip K. M., Zardan A., Takeuchi A., Cordonnier T., Beraldi E., Bazov J., Fazli L., Chi K., Gleave M., Zoubeidi A. (2013) Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 73, 3109–3119 [DOI] [PubMed] [Google Scholar]
- 44. Dong S., Cheng Y., Yang J., Li J., Liu X., Wang X., Wang D., Krall T. J., Delphin E. S., Zhang C. (2009) MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J. Biol. Chem. 284, 29514–29525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grunder E., D'Ambrosio R., Fiaschetti G., Abela L., Arcaro A., Zuzak T., Ohgaki H., Lv S. Q., Shalaby T., Grotzer M. (2011) MicroRNA-21 suppression impedes medulloblastoma cell migration. Eur. J. Cancer 47, 2479–2490 [DOI] [PubMed] [Google Scholar]
- 46. Connolly E., Melegari M., Landgraf P., Tchaikovskaya T., Tennant B. C., Slagle B. L., Rogler L. E., Zavolan M., Tuschl T., Rogler C. E. (2008) Elevated expression of the miR-17–92 polycistron and miR-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am. J. Pathol. 173, 856–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iliopoulos D., Jaeger S. A., Hirsch H. A., Bulyk M. L., Struhl K. (2010) STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 39, 493–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu Y., Li P. K., Li C., Lin J. (2010) Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J. Biol. Chem. 285, 27429–27439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cheng J. G., Chen J. R., Hernandez L., Alvord W. G., Stewart C. L. (2001) Dual control of LIF expression and LIF receptor function regulate Stat3 activation at the onset of uterine receptivity and embryo implantation. Proc. Natl. Acad. Sci. U.S.A. 98, 8680–8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vogel S., Wottawa M., Farhat K., Zieseniss A., Schnelle M., Le-Huu S., von Ahlen M., Malz C., Camenisch G., Katschinski D. M. (2010) Prolyl hydroxylase domain (PHD) 2 affects cell migration and F-actin formation via RhoA/rho-associated kinase-dependent cofilin phosphorylation. J. Biol. Chem. 285, 33756–33763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Badr G., Mohany M., Abu-Tarboush F. (2011) Thymoquinone decreases F-actin polymerization and the proliferation of human multiple myeloma cells by suppressing STAT3 phosphorylation and Bcl2/Bcl-XL expression. Lipids Health Dis. 10, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Osugi T., Oshima Y., Fujio Y., Funamoto M., Yamashita A., Negoro S., Kunisada K., Izumi M., Nakaoka Y., Hirota H., Okabe M., Yamauchi-Takihara K., Kawase I., Kishimoto T. (2002) Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J. Biol. Chem. 277, 6676–6681 [DOI] [PubMed] [Google Scholar]
- 53. Wang L., Luo J., He S. (2007) Induction of MMP-9 release from human dermal fibroblasts by thrombin: involvement of JAK/STAT3 signaling pathway in MMP-9 release. BMC Cell Biol. 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang S., Chen M., Shen Y., Shen W., Guo H., Gao Q., Zou X. (2012) Inhibition of activated Stat3 reverses drug resistance to chemotherapeutic agents in gastric cancer cells. Cancer Lett. 315, 198–205 [DOI] [PubMed] [Google Scholar]
- 55. Kato K., Nomoto M., Izumi H., Ise T., Nakano S., Niho Y., Kohno K. (2000) Structure and functional analysis of the human STAT3 gene promoter: alteration of chromatin structure as a possible mechanism for the up-regulation in cisplatin-resistant cells. Biochim. Biophys. Acta 1493, 91–100 [DOI] [PubMed] [Google Scholar]
- 56. Real P. J., Sierra A., De Juan A., Segovia J. C., Lopez-Vega J. M., Fernandez-Luna J. L. (2002) Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 21, 7611–7618 [DOI] [PubMed] [Google Scholar]
- 57. Shen Y., Devgan G., Darnell J. E., Jr., Bromberg J. F. (2001) Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc. Natl. Acad. Sci. U.S.A. 98, 1543–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rebbaa A., Chou P. M., Mirkin B. L. (2001) Factors secreted by human neuroblastoma mediated doxorubicin resistance by activating STAT3 and inhibiting apoptosis. Mol. Med. 7, 393–400 [PMC free article] [PubMed] [Google Scholar]
- 59. Alas S., Bonavida B. (2003) Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin's lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin. Cancer Res. 9, 316–326 [PubMed] [Google Scholar]
- 60. Packer R. J., Cogen P., Vezina G., Rorke L. B. (1999) Medulloblastoma: clinical and biologic aspects. Neuro-oncology 1, 232–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Packer R. J., Gajjar A., Vezina G., Rorke-Adams L., Burger P. C., Robertson P. L., Bayer L., LaFond D., Donahue B. R., Marymont M. H., Muraszko K., Langston J., Sposto R. (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J. Clin. Oncol. 24, 4202–4208 [DOI] [PubMed] [Google Scholar]
- 62. Rossi A., Caracciolo V., Russo G., Reiss K., Giordano A. (2008) Medulloblastoma: from molecular pathology to therapy. Clin. Cancer Res. 14, 971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Buettner R., Mora L. B., Jove R. (2002) Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 8, 945–954 [PubMed] [Google Scholar]
- 64. Inghirami G., Chiarle R., Simmons W. J., Piva R., Schlessinger K., Levy D. E. (2005) New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell Cycle 4, 1131–1133 [DOI] [PubMed] [Google Scholar]
- 65. Chiarle R., Simmons W. J., Cai H., Dhall G., Zamo A., Raz R., Karras J. G., Levy D. E., Inghirami G. (2005) Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 11, 623–629 [DOI] [PubMed] [Google Scholar]
- 66. Siddiquee K., Zhang S., Guida W. C., Blaskovich M. A., Greedy B., Lawrence H. R., Yip M. L., Jove R., McLaughlin M. M., Lawrence N. J., Sebti S. M., Turkson J. (2007) Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. U.S.A. 104, 7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang X., Yue P., Fletcher S., Zhao W., Gunning P. T., Turkson J. (2010) A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem. Pharmacol. 79, 1398–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang X., Yue P., Page B. D., Li T., Zhao W., Namanja A. T., Paladino D., Zhao J., Chen Y., Gunning P. T., Turkson J. (2012) Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. U.S.A. 109, 9623–9628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miklossy G., Hilliard T. S., Turkson J. (2013) Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 12, 611–629 [DOI] [PMC free article] [PubMed] [Google Scholar]