

Background: Exosomes are a novel mechanism of intercellular transmission of infectious prions.
Results: Chemical and RNAi inhibition of the neutral sphingomyelinase (nSMase) pathway impairs exosome formation and prion packaging.
Conclusion: The nSMase pathway regulates exosome formation and packaging of infectious prions.
Significance: This reveals a novel role for the nSMase pathway in exosomal prion packaging and identifies a direct pathway, which mediates prion transmission.
Keywords: Ceramide, Exosome, Extracellular Vesicles, Prion, Prion Disease, Exosomes, nSMase2, Neutral Sphingomyelinase
Abstract
Prion diseases are a group of transmissible, fatal neurodegenerative disorders associated with the misfolding of the host-encoded prion protein, PrPC, into a disease-associated form, PrPSc. The transmissible prion agent is principally formed of PrPSc itself and is associated with extracellular vesicles known as exosomes. Exosomes are released from cells both in vitro and in vivo, and have been proposed as a mechanism by which prions spread intercellularly. The biogenesis of exosomes occurs within the endosomal system, through formation of intraluminal vesicles (ILVs), which are subsequently released from cells as exosomes. ILV formation is known to be regulated by the endosomal sorting complexes required for transport (ESCRT) machinery, although an alternative neutral sphingomyelinase (nSMase) pathway has been suggested to also regulate this process. Here, we investigate a role for the nSMase pathway in exosome biogenesis and packaging of PrP into these vesicles. Inhibition of the nSMase pathway using GW4869 revealed a role for the nSMase pathway in both exosome formation and PrP packaging. In agreement, targeted knockdown of nSMase1 and nSMase2 in mouse neurons using lentivirus-mediated RNAi also decreases exosome release, demonstrating the nSMase pathway regulates the biogenesis and release of exosomes. We also demonstrate that PrPC packaging is dependent on nSMase2, whereas the packaging of disease-associated PrPSc into exosomes occurs independently of nSMase2. These findings provide further insight into prion transmission and identify a pathway which directly assists exosome-mediated transmission of prions.
Introduction
Prion diseases, such as scrapie in sheep, bovine spongiform encephalopathy in cattle and Creutzfeldt-Jakob disease in humans are a family of invariably fatal neurodegenerative diseases characterized by a progressive loss of neurons and spongiform vacuolation of the brain. According to the protein only hypothesis, the underlying cause of prion disease is misfolding of the normal soluble cellular prion protein, PrPC, into the disease-associated isoform, PrPSc, which has an increased propensity to aggregate (1). Prion diseases are unique among neurodegenerative diseases as they can be acquired through exposure to the infectious agent, giving the disease a transmissible etiology.
The exact mechanism by which prion disease is transmitted intercellularly is yet to be identified, although three mechanisms have been proposed, including: direct cell-cell contact; tunnelling nanotubes, which are fine membrane protrusions connecting the cytoplasm of one cell with a neighboring cell; and extracellular vesicles such as microvesicles and exosomes (2–5). Exosomes are small membrane-bound vesicles released by cells, ∼30–100 nm in diameter, as determined using TEM, and contain proteins, mRNAs and miRNAs (6–10). Whereas microvesicles are generated via budding at the plasma membrane, exosomes are distinct and are generated within the endosomal system (11). As early endosomes mature into late endosomes, the limiting membrane of late endosomes invaginate to form intraluminal vesicles (ILVs),3 generating the multivesicular body (MVB) (6). The MVB either fuses with lysosomes, resulting in degradation of the ILVs, a well-known process for down-regulating transmembrane receptors, or it fuses with the plasma membrane, resulting in release of the ILVs as exosomes. Consistent with the endosomal origin, exosomes have a unique protein and lipid composition, and contain proteins such as Tsg101, a component of the endosomal sorting complex required for transport (ESCRT) machinery, and are enriched in raft-associated lipids such as cholesterol, sphingolipids, ceramide, and glycerophospholipids (6, 7, 12).
A link between exosomes and prion disease was first established in rabbit kidney epithelial cells expressing ovine PrP (ROV). The ROV cells release exosomes which contain PrPSc and are capable of transmitting infection to mice (4). In support of this, exosomes released from a prion-infected mouse neuronal cell line are also capable of transmitting infection to naïve cells and to mice (10, 13). Exosomes are also able to transmit infectivity between heterologous cell types, suggesting that exosomes could assist prion transmission from the periphery to the central nervous system (13). In addition to prion disease, exosomes have also been implicated in other neurodegenerative diseases such as Alzheimer and Parkinson disease, and Amyotrophic lateral sclerosis, where the disease-associated proteins have also been found in exosomes (14–18). Furthermore, HIV-1 has also been proposed to hijack the exosomal pathway to produce new viral particles (19).
Given the proposed role of exosomes in facilitating the spread of disease-related proteins involved in neurodegenerative disorders, elucidating pathways which regulate exosome protein packaging could be valuable for understanding disease pathogenesis (20). The most common mechanism of protein targeting into ILVs is mono-ubiquitination and recruitment of the ESCRT machinery (21, 22). However, ubiquitin-independent mechanisms have also been described, including lipid-raft-association, higher-ordered oligomerization, association with tetraspanin-enriched domains, and the generation of ceramide via the neutral sphingomyelinase (nSMase) pathway (23–26). These alternative pathways are dependent on a reduction in the diffusion rate of cargo proteins, either through association with other proteins or with microdomains within the limiting membrane of endosomes, leading to retention of the proteins and subsequent incorporation into budding ILVs. The nSMase pathway was first demonstrated to assist ILV formation and packaging of the proteolipid protein in mouse oligodendroglial cells, and have since then being found to also regulate packaging of miRNA into ILVs (27). Furthermore, nSMase-dependent exosome biogenesis has also been linked to Alzheimer's disease as the exosomes were proposed to promote clearance of Aβ by microglia (28).
Four mammalian nSMase isoforms have been identified: nSMase1, nSMase2, nSMase3, and the recently identified mitochondrial-associated nSMase (MA-nSMase) (29–32). Both nSMase1 and nSMase2 have been reported to have nSMase activity in vitro, although only nSMase2 has nSMase activity in vivo. With exosomes implicated as vehicles for intercellular transmission of prions, we aimed to investigate a role for the nSMase pathway in exosome biogenesis and packaging of PrP into exosomes by inhibiting the nSMase pathway using a chemical inhibitor, GW4869, and using lentivirus-mediated RNAi to silence nSMase1 and nSMase2.
EXPERIMENTAL PROCEDURES
Reagents
Unless otherwise specified, all reagents are from Sigma-Aldrich. Antibodies used and their working dilutions are as follows: α-PrP antibody ICSM-18 (1:10,000; D-GEN, London, UK); 03R19 (1:25,000; in-house) (33); L3 (1:5,000; in-house); α-GM130 (1:1,000), α-Bcl-2 (1:1,000), α-flotillin-1 (1:3,000), and α-nucleoporin (1:1,000) from BD Biosciences (Sydney, NSW, Australia); α-Tsg101 (M-19; 1:3,000; Santa Cruz Biotechnology, Santa Cruz, CA); α-nSMase1 (1:1,000; Abcam, Waterloo, NSW, Australia); α-tubulin (1:25,000); α-TfR (1:1,000; Invitrogen, Melbourne, VIC, Australia). α-mouse HRP (1:25,000), and α-rabbit HRP (1:25,000) from GE Healthcare (Sydney, NSW, Australia); and α-goat HRP (1:100,000).
Maintenance and Infection of Cultured Cells Lines with Prions, and the Cell Blot Assay
GT1–7 cells were cultured in Opti-MEM (Invitrogen, Melbourne, VIC, Australia), and infected with a mouse-adapted strain of human prions (M1000), as previously described (10). At 6 passages postinfection, stable infection of cells was assessed using a cell blot assay. Cells were seeded onto sterile plastic cover slips (Thermanox; Nunc-Thermo, Scoresby, VIC, Australia) in 6-well plates (Nunc-Thermo) with 250,000 cells/well and grown to confluence before assay was carried out as previously described (10).
Generation of Lentiviral Supernatants and Stable Transduction of GT1–7 Cells
A panel of 3 (A-C) and 4 (A-D) commercial lentiviruses for murine nSMase1 and nSMase2, respectively, GIPZ Non-silencing lentiviral control and TRC lentiviral pLKO.1 empty vector control were purchased as glycerol stocks from Open Biosystems (Millennium Science, Surrey Hills, VIC, Australia). The construct ID and mature antisense sequences are: nSMase1A (V2LMM_27471; TAAGCTCTTAAGCTCTGTC); nSMase1B (V2LMM_29867; TCAGACTGTAGACATGCTG); nSMase1C (V2LMM_38704; ACAAAGTAGCCATGAGGGC); nSMase2A (TRCN0000099415; AAATCGTGACTGAAAGAATGG); nSMase2B (TRCN0000099416; ATACAGCAGCTTGCTGTTAGC); nSMase2C (TRCN0000099418; ATGTAGATCTTGATCTGAGGC); nSMase2D (TRCN0000099419; ATAGAGACCGTTTGTGTCCAG). Plasmid DNA was isolated using the PureYield™ Plasmid Midiprep System (Promega, Fitchburg, WI) according to manufacturer's instructions. The 2nd generation lentiviral packaging plasmids, psPAX2 (Addgene plasmid 12260; Addgene) and pMD2.G (Addgene plasmid 12259; Addgene) were used to generate lentiviral particles in HEK293FT cells. 2.5 μg pMD2.G, 6.5 μg of psPAX2 and 2 μg of lentiviral construct were combined and used to transfect a 75 cm2 flask (Nunc-Thermo) of HEK293FT cells using Lipofectamine 2000 (Invitrogen), according to manufacturer's instructions. 24 h after transfection, the medium is replaced with fresh DMEM (Invitrogen) supplemented with 10% (v/v) heat inactivated FCS, 1 mm sodium pyruvate (Invitrogen), 1× MEM Non-essential Amino Acids, 100 units of penicillin, and 100 μg/ml streptomycin as penicillin-streptomycin 100× and 1× GlutaMAX. Lentiviral supernatants were harvested at 48 h post-transfection and centrifuged for 5 min, 1,400 × g, then aliquoted and stored at −80 °C. GT1–7 cells were seeded into 6-well plates (Nunc-Thermo) at 400,000 cells/well and allowed to recover for 24 h before being overlaid with 1 ml of lentiviral supernatant diluted in 1 ml complete Opti-MEM culture medium. 24 h after transduction, the lentiviral supernatant is replaced with fresh complete culture medium and 0.2 μg/ml puromycin was introduced at 48 h post-transduction. The degree of KD achieved was assessed at 3 passages post-transduction.
RNA Isolation and Quantitative Real Time RT-PCR
Total RNA was extracted from cells using the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's protocol. 2 μg of RNA was converted to cDNA using High Capacity cDNA Kit (Invitrogen) according to manufacturer's instructions. Real Time RT-PCR samples were prepared using TaqMan® Gene Expression Master Mix (Invitrogen) and murine specific TaqMan® Gene Expression Assays (Invitrogen) for Prnp (Mm00448389_m1), nSMase1 (Mm01188195_g1) and nSMase2 (Mm00491359_m1), with endogenous controls for murine Tbp (Mm00446971_m1) and Hprt1 (Mm00446968_m1). Samples were prepared on the QIAgility liquid-handling robot (Qiagen) and run on a StepOnePlus qRT-PCR instrument (Invitrogen) using manufacturer's recommended cycling conditions. Data were analyzed by the comparative ΔΔCt method as previously described (34).
Exosome Isolation
For exosome collection, 175 cm2 flasks (Nunc-Thermo) were seeded with 7 × 106 cells and allowed to recover for 24 h. For stably transduced cells, the culture medium is replaced with exosome-free medium (complete medium containing exosome-depleted FCS) and cultured for 2 days, without puromycin. For GW4869 treatment, the culture medium is replaced with exosome-free medium containing 4 μm GW4869 and cultured for 2 days. Exosomes were isolated from the culture supernatant using differential ultracentrifugation, as previously described (10). Exosome pellets were resuspended using 70 μl of filtered PBS.
Transmission Electron Microscopy
10-μl aliquots of exosomes were fixed with 10 μl of 2% (v/v) EM grade glutaraldehyde and incubated for 30 min, room temperature. A 6-μl aliquot of fixed exosome solution was then absorbed onto glow-discharged 300 mesh carbon coated formvar-copper TEM grids (ProSciTech, SA, Australia) and analyzed using TEM, as previously described (10).
Acetylcholinesterase (AChE) Assay
AChE activity assay was carried out as previously described (35–37). Briefly, freshly collected exosomes in 70 μl of PBS were diluted 1:10 with PBS, and 50 μl of the diluted samples were aliquoted into a clear 96-well colorimetric plate in triplicate. The reaction was initiated by adding 50 μl of AChE assay solution (1.25 mm acetylthiocholine, 0.1 mm 5,5-dithio-bis(2-nitrobenzoic acid), PBS pH 7.4). The plate was wrapped in foil and incubated for 10 min, 37 °C. Absorbance at 450 nm was measured every 30 s for 10 min, room temperature, using a Varioskan® multi-mode plate reader. The rate of change (per min) was determined over the 10 min using the SkanIt Software 2.4.3 RE (Thermo Scientific, Australia), adjusted to be rate of change per ml, and then scaled by 1000 for data handling.
qNano
Scanning ion occlusion sensing analysis using the qNano instrument (Izon, Christchurch, New Zealand) was carried out as previously described, with minor alterations (38). Cell culture supernatant was spiked with 40× qNano supplement (3.2 m KCl, 100 mm EDTA pH 8.0, 0.1% Tween 20) to reach a final concentration of 1×. The supernatant was cleared using a 0.22 μm filter (Merck Millipore, Darmstadt, Germany). The samples were vortexed for 30 s immediately prior to measurement.
Protease Digestion, SDS-PAGE, and Immunoblotting
Cells were lysed on ice for 20 min using ice-cold lysis buffer (150 mm NaCl, 50 mm Tris pH 7.4, 1% (v/v) Triton X-100, 1% (w/v) sodium deoxycholate containing complete protease inhibitor mixture (Roche, Castle Hill, NSW, Australia)). Exosomes were lysed on ice for 20 min using 30 μl of lysis buffer. Total protein concentrations were determined using the bicinchoninic acid (BCA) protein assay (Pierce, Thermo Scientific), according to manufacturer's protocol. 100 μg of protein were treated with proteinase K (PK; 25 μg/ml), as previously described (10). 40 μg of protein were incubated on ice for non-PK-treated samples. Ice-cold methanol (4× volume of lysate) was added to the samples, vortexed briefly, and then stored overnight at −20 °C. Protein precipitates were pelleted by centrifugation for 15 min, 16,100 × g, room temperature, and the pellets were air-dried for 30 min. Pellets were resuspended with 20 μl 1× SDS-PAGE sample buffer, vortexed, heated for 8 min, 100 °C, then centrifuged for 2 min, 10,000 × g, prior to SDS-PAGE and immunoblotting analysis, as previously described (10).
Toxicity Assay
GT1–7 cells were seeded into μClear black 96-well plates with clear bottom (Greiner Bio-One, Frickenhausen, Germany) at 12,000 cells/well and allowed to attach overnight. Cells were treated with DMSO-only, 0.5 μm, 1 μm, 2 μm and 4 μm GW4869 for 48 h, and toxicity was determined using the LIVE/DEAD® Viability/Cytotoxicity Kit (Invitrogen), according to manufacturer's instructions. 15 min prior to the assay, 1% (v/v) Triton X-100 (Ajax Finechem; Thermo Scientific), diluted in complete Opti-MEM, was added to positive control cells to induce cell death. Data were analyzed according to the manufacturer's protocol and represented as % live cells compared with DMSO-only control.
Lipidomic Analysis
GT1–7 cells, were seeded with 7 × 106 cells into 175 cm2 flasks (Nunc-Thermo) and allowed to recover for 24 h. Cells were treated with DMSO-only or 4 μm GW4869 and cultured for 2 days in exosome-free medium. Cells were washed in warm medium and PBS. Cells were then detached in PBS, pelleted by centrifuagtion, snap frozen in liquid nitrogen and stored at −80 °C before lipidomic extractions. Lipids were extracted using monophasic extraction with chloroform:methanol:water (1:2:0.6,v/v/v). After removal of insoluble material by centrifugation (15,000 × g, 15 min), extracts were dried under nitrogen and dried cellular extracts were resuspended in 100 μl of butanol/methanol (1:1, v/v) containing 5 μm ammonium formate. Cellular lipids were separated using an Agilent LC 1200 and lipids were analyzed by electrospray ionisation-mass spectrometry (ESI-MS) using an Agilent Triple Quad 6460 (Mulgrave, Australia), as previously described (39). The molecular species of the lipid classes were identified using precursor ion scanning from 100 to 1000 m/z in positive ion mode, ceramide (m/z 264.6). The peak area of each lipid species was normalized to abundance per cell.
Image Processing and Statistical Analysis
Densitometric analysis was carried out using ImageJ (40). Statistical analyses were performed using GraphPad Prism 6 (GraphPad, San Diego, CA). Data is analyzed as follows: qRT-PCR and toxicity assay data are shown as mean of triplicate and quadruplicate experiments, respectively, ± S.E., and statistical significance tests were performed with two sample t tests; AChE, qNano, and densitometric analyses data are shown as mean change in comparison to control of triplicate experiments ± S.E., and statistical significance tests were performed with one sample t tests to compare each value against a standardized control of 100%. Advice was sought from The University of Melbourne's Statistical Consulting Centre, and it was advised that where there is a difference observed for samples, but the p value is not < 0.05, it is still acceptable to note that there is a change. Statistical significance is defined as *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
RESULTS
Isolating Exosomes from Prion-infected GT1–7 Cells
The GT1–7 mouse hypothalamic neuronal cell line was used as a model of prion disease as it expresses endogenous levels of PrPC and is one of few cell lines capable of sustaining persistent prion infection (41, 42). Non- and M1000 prion-infected GT1–7 cells were generated and the infection was confirmed through detection of PrPSc by the cell blot assay (Fig. 1A). Exosomes isolated from non- and prion-infected GT1–7 cells were characterized by probing for the presence of exosomal protein markers, Tsg101 and flotillin-1, and the absence of cytosolic contaminants markers such as GM130 (Golgi), Bcl-2 (mitochondria) and nucleoporin (nucleus) (Fig. 1B). In addition, both PrPC and PrPSc are detected within exosomes (Fig. 1C). Morphological analysis using TEM reveals a homogenous population of round membrane-bound vesicles, ∼85 nm in diameter (Fig. 1D). However, reports using techniques such as cryo-EM and nanoparticle tracking analysis to visualize the hydrated structure of exosomes indicate that exosomes are slightly larger, with an approximate diameter of 80–160 nm (10, 43). In agreement with this, qNano analysis of the conditioned medium from non- and prion-infected cells reveal that the exosomes are ∼116 nm in diameter (Fig. 1E).
FIGURE 1.

PrPC and PrPSc associate with vesicles from GT1–7 cells that display features characteristic of exosomes. A, cell blot assay confirming presence of PrPSc and transmission of prion infection to GT1–7 cells. B, immunoblot characterization of exosomes (Exo) with a panel of protein markers: GM130 (Golgi); Bcl-2 (mitochondrial); Nucleoporin (nuclear); Flotillin-1 (exosomal); and Tsg101 (exosomal). C, immunoblotting of PK-treated and untreated lysates from non- (NI) and prion-infected (INF) cells and exosomes for PrP, showing that both PrPC and PrPSc can be found within exosomes. D, exosomes were further characterized using TEM, confirming that the isolated vesicles have biochemical and biophysical properties characteristic of exosomes. E, size distribution analysis of exosomes from culture supernatants of non- and prion-infected cells using qNano, revealing a diameter of ∼116 nm for hydrated exosomes. Values represent mean or mode size ± S.E., n = 3.
Inhibition of the nSMase Pathway with GW4869 Impedes Exosome Biogenesis and PrP Packaging
GW4869, a nSMase inhibitor, was used to investigate the role of the nSMase pathway in exosome biogenesis and PrP packaging (26). GW4869 was tested for toxicity to GT1–7 cells and analysis of the percentage live cells relative to vehicle-only control reveal no toxic effects for all concentrations tested (Fig. 2A). GW4869 was thus used at a concentration of 4 μm, which is comparable to the 5 μm used in the study which reported a link between the nSMase pathway and packaging of the proteolipid protein into exosomes (26). GC-MS analysis of ceramide levels in control and GW4869-treated cells reveal a reduction for several ceramide species, confirming the inhibitory activity of the compound on the nSMase pathway (Fig. 2B).
FIGURE 2.

Treatment with GW4869 impairs exosome biogenesis. A, increasing concentrations of GW4869 were tested for toxicity to GT1–7 cells, and no toxicity is observed at all concentrations tested. Note that the highest concentration tested is limited by the solubility of GW4869 and toxicity of DMSO alone to GT1–7 cells. B, GC-MS analysis of ceramide species, confirming the decrease in ceramide levels in response to GW4869 treatment. C, qNano analysis of the size distribution of exosomes in the culture supernatant of DMSO- and GW4869-treated non-infected cells and (D) prion-infected cells. E, qNano quantitation of exosomes in the culture supernatant following DMSO or GW4869 treatment, showing decreasing exosome release with GW4869 treatment. The decrease in non-infected exosomes is close to, but did not achieve statistical significance (p < 0.061). F, AChE quantitation of isolated exosomes, confirming a decrease in exosome release upon GW4869 treatment. Data presented as mean ± S.E., n = 3, *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The effect of GW4869 on exosome biogenesis was assessed initially using the qNano instrument to quantitate exosomes in culture supernatant. qNano uses scanning ion occlusion sensing technology to determine particle size and concentration (44). While the size distribution of non- and prion-infected exosomes do not change with treatment (Fig. 2, C and D), the amount of exosomes present is decreased upon GW4869 treatment (Fig. 2E). To further validate this finding, exosomes isolated from culture supernatant were quantitated using the acetyl cholinesterase (AChE) activity assay, which provides a quantitative measure for exosomes via detection of acetyl choline present on the surface of exosomes (36, 37). In agreement, exosome release is also decreased in both non- and prion-infected cells upon GW4869 treatment (Fig. 2F). This indicates that the nSMase pathway also regulates exosome biogenesis in GT1–7 cells, similar to oligodendroglial cells (26).
The effect of GW4869 on the levels of cellular and exosomal PrPC and PrPSc were also examined. Analyses of vehicle- and GW4869-treated cells show that treatment with the GW4869 decreases both cellular PrPTot (total PrP prior to PK treatment) and PrPSc levels in prion-infected cells (Fig. 3, A and B). In exosomes, a significant decrease is observed for PrPC from non-infected cells, while PrPTot and PrPSc is decreased in exosomes from infected cells (Fig. 3, A and B). Treatment of both non- and prion-infected cells with GW4869 also reduced the levels of exosome markers Flotillin-1 and Tsg101 (Fig. 3, C and D), in agreement with a reduction in exosome release. Together, these findings indicate that packaging of PrP into exosomes is regulated by the nSMase pathway.
FIGURE 3.

Treatment with GW4869 impairs exosomal packaging of PrPC and PrPSc. A, immunoblot analyses of PrP in control and GW4869-treated non- and prion-infected GT1–7 cells and exosomes. (B) Densitometric analyses show that PrPC, PrPTot and PrPSc levels are decreased in exosomes released from GW4869-treated cells. Cellular PrPSc is also decreased in GW4869-treated prion-infected cells, along with a concomitant, but not statistically significant, decrease of PrPTot. C, immunoblot analyses of Flotillin-1 and Tsg101 in control and GW4869-treated non- and prion-infected GT1–7 exosomes. D, densitometric analyses show that Flotillin-1 and Tsg101 levels are decreased in exosomes released from GW4869-treated cells. Data presented as mean ± S.E., n = 3, *, p < 0.05; **, p < 0.01.
Depletion of nSMase1 and nSMase2 Impairs Exosome Biogenesis
To further investigate the nSMase pathway, two isoforms, nSMase1 and nSMase2, were targeted for RNAi. As GT1–7 are neuronal cells, lentivirus-mediated RNAi was used to generate stably knocked down cells in order to maximize RNAi efficiency. A panel of 3 lentiviruses were used for nSMase1 (nSMase1A-C) and 4 were used for nSMase2 (nSMase2A-D). RNAi was quantitated using qRT-PCR for nSMase1 (Fig. 4A) and nSMase2 (Fig. 4B). The levels of nSMase1 KD were also quantitated via immunoblotting, the same could not to be carried out for nSMase2 due to the lack of a suitable antibody (Fig. 4C). Lentiviruses producing successful KD were used for ensuing analyses.
FIGURE 4.

RNAi quantification using qRT-PCR and immunoblotting. A, qRT-PCR quantification of nSMase1 KD showing successful KD in nSMase1B and nSMase1C. B, qRT-PCR quantification of nSMase2 KD showing successful KD with all lentiviruses. Due to the higher level of KD induced, nSMase2A and nSMase2B were chosen for subsequent analyses. C, immunoblot quantification of nSMase1 KD, showing a decrease for nSMase1 protein levels in nSMase1B and nSMase1C cells. Data presented as mean ± S.E., n = 3, *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Using the AChE assay, exosomes from nSMase1 and nSMase2 KD cells were quantified following isolation from the culture supernatant. In agreement with GW4869 treatment, exosome release from non- and prion-infected nSMase1B and nSMase1C KD cells are decreased, indicating that nSMase1 regulates exosome biogenesis (Fig. 5A). Similarly, exosome release is also decreased from non- and prion-infected nSMase2A and nSMase2B KD cells, albeit the decrease is less in the latter (Fig. 5B). Together, these results confirm a role for the nSMase pathway in modulating exosome biogenesis in GT1–7 cells.
FIGURE 5.

Depletion of nSMase1 and nSMase2 reduces exosome release. A, AChE quantification of exosomes from nSMase1B and nSMase1C, revealing a decrease in exosome release upon nSMase1 KD. The decrease for non-infected nSMase1B exosomes is close to, but did not achieve statistical significance (p < 0.094). B, AChE quantification of exosomes from nSMase2A and nSMase2B, also revealing a decrease in exosome release upon nSMase2 KD, although the decrease is slightly less from prion-infected than non-infected cells. The decrease for non-infected nSMase2B and prion-infected nSMase2A exosomes is close to, but did not achieve statistical significance (p < 0.078 and p < 0.061, respectively). Data presented as mean ± S.E., n = 3, *, p < 0.05.
The nSMase Pathway Regulates PrP Packaging into Exosomes
The effect of nSMase1 and nSMase2 KD on the levels of cellular and exosomal PrP were also analyzed. For both nSMase1B and nSMase1C KD, no significant changes are detected for the levels of cellular PrPC, PrPTot, and PrPSc (Fig. 6, A and B). Similarly, no significant changes are detected for exosomal PrPC, PrPTot, and PrPSc. However, an apparent decrease is observed for PrPTot and PrPSc in prion-infected nSMase1C exosomes, suggesting that the KD slightly hinders packaging of PrP into exosomes. In agreement, the levels of exosome markers Flotillin-1 and Tsg101 also show an apparent decrease in prion-infected nSMase1C exosomes (Fig. 6, C and D). Together, these results indicate that nSMase1 could play a minor role in exosome protein packaging in prion-infected cells, which correlates with the reported activity of nSMase1 only under stressed cellular conditions (45, 46).
FIGURE 6.

Depletion of nSMase1 does not significantly impair exosomal PrP packaging. A, immunoblot analyses of PrP in nSMase1B and nSMase1C cells and exosomes. B, densitometric analyses show that cellular PrPC, PrPTot, and PrPSc levels remain unchanged in nSMase1B and nSMase1C. While exosomal PrPC levels remain unchanged, exosomal PrPTot and PrPSc levels decrease slightly in prion-infected nSMase1C exosomes, although this is not statistically significant (p < 0.142 and p < 0.214, respectively). C, immunoblot analyses of Flotillin-1 and Tsg101 in nSMase1B and nSMase1C exosomes. D, densitometric analyses show that while exosomal Flotillin-1 levels remain unchanged in non-infected nSMase1B and nSMase1C exosomes, a small decrease is observed in prion-infected nSMase1C exosomes, although this is not statistically significant (p < 0.115). A similar decrease is also observed for Tsg101 in prion-infected nSMase1C exosomes (p < 0.140). Data presented as mean ± S.E., n = 3.
Analyses of nSMase2A and nSMase2B KD also indicated no changes in cellular levels of PrPC, PrPTot, and PrPSc (Fig. 7, A and B). However, a decrease is observed for PrPC in nSMase2B exosomes. While a slight decrease in PrPC is also observed for nSMase2A exosomes, the decrease did not achieve statistical significance. Most significantly, no changes are observed for PrPTot or PrPSc in prion-infected exosomes, suggesting that the nSMase pathway is altered in prion-infected cells such that nSMase2 no longer mediates exosomal packaging of PrPSc. Analysis of exosome markers Flotillin-1 and Tsg101 levels in non- and prion-infected exosomes also supported nSMase2B KD reducing exosome biogenesis in non-infected cells, with no changes observed for nSMase2B KD in prion-infected cells (Fig. 7, C and D). While the effect of nSMase2 KD on PrP levels in non-infected cells and exosomes is in agreement with GW4869 treatment, the effects of nSMase2 KD on PrP levels in prion-infected cells differs from that of GW4869 treatment. This discrepancy suggests that there could be compensation between the nSMase isoforms. Together, these findings suggest that the nSMase pathway regulates exosome protein packaging in non-infected GT1–7 cells, with the process predominantly mediated by the nSMase2 isoform. However, upon prion infection, the pathway is altered such that nSMase2 no longer governs sorting of the infectious prion protein PrPSc.
FIGURE 7.

Depletion of nSMase2 impairs PrPC packaging into exosomes. A, immunoblot analyses of PrP in nSMase2A and nSMase2B cells and exosomes. B, densitometric analyses show that cellular PrPC, PrPTot, and PrPSc levels remain unchanged. In contrast, PrPC is decreased in non-infected nSMase2B exosomes. However, both PrPTot and PrPSc levels do not change in prion-infected exosomes. C, immunoblot analyses of Flotillin-1 and Tsg101 in nSMase2A and nSMase2B exosomes. D, densitometric analyses show that both Flotillin-1 and Tsg101 levels decrease in non-infected nSMase2B exosomes, but remain unchanged in prion-infected exosomes. Data presented as mean ± S.E., n = 3, *, p < 0.05.
DISCUSSION
Multiple studies have now shown that the prion protein is secreted from cells in exosomes, highlighting a novel role for these extracellular vesicles in intercellular dissemination of prions (4, 10, 13, 47). Despite the growing number of reports establishing an association of PrP with exosomes, the cellular pathways which regulate packaging of PrP has eluded identification. We aimed to address this by investigating the potential involvement of the nSMase pathway in exosome biogenesis and protein packaging. Using chemical inhibition and RNAi, we have shown that the nSMase pathway regulates exosome biogenesis in mouse neuronal cells. In addition, the nSMase pathway also modulates PrP packaging into exosomes. While nSMase2 appears to be the predominant isoform responsible for PrPC packaging, an alternative isoform could be responsible for packaging PrPSc into exosomes.
In agreement with Trajkovic et al., treatment with GW4869 impairs exosome biogenesis and protein packaging, and this is further supported by the findings obtained using RNAi (26). Silencing of nSMase1 and nSMase2 decreases exosome release, indicating that exosome biogenesis in GT1–7 cells is regulated by the nSMase pathway. In addition, it also reveals that nSMase1 and nSMase2 share redundancy with respect to regulating exosome biogenesis. Comparison of the effect of nSMase1 and nSMase2 KD on exosome release from non- and prion-infected cells reveal an intriguing relationship. nSMase2 KD causes a larger decrease of exosome release from non-infected cells than nSMase1 KD. Conversely, nSMase1 KD causes a larger decrease of exosome release from infected cells than nSMase2 KD. This suggests that while the two isoforms share functional redundancy in terms of exosome formation, the relative contribution of the two isoforms toward this process varies depending on prion infection.
A similar scenario is observed with packaging of proteins into exosomes. While exosomal protein packaging in non-infected cells is not dependent on nSMase1, in infected cells, loss of nSMase1 decreases the amount of PrPTot and PrPSc in exosomes, albeit the decrease does not achieve statistical significance. Nonetheless, this suggests that prion infection alters the nSMase pathway such that nSMase1 plays a small role in protein packaging in prion-infected cells. The change with nSMase1 suggests that this isoform displays more nSMase activity in prion-infected cells than in non-infected cells. This is in accordance with the reported nSMase activity for nSMase1 under conditions of stress (45, 46). Likewise, a change in nSMase2-regulated packaging of proteins is also observed upon infection, lending support to the proposal that the nSMase pathway is altered in prion-infected cells. Both nSMase1 and nSMase2 therefore mediate exosome biogenesis and the relative contribution of these isoforms differ depending on the prion-infected state of the cell.
While neither nSMase1 nor nSMase2 KD produces appreciable decreases of PrPTot or PrPSc in exosomes, treatment with GW4869 significantly impairs sorting of these proteins into exosomes in infected cells. This difference also suggests that there could be compensation occurring between nSMase2 and an alternative nSMase isoform, one which is also susceptible to inhibition by GW4869. GW4869 is proposed to inhibit the nSMase pathway by targeting nSMase2 and inhibiting activation of the isoform by anionic phospholipids (48). Co-incidentally, MA-nSMase has also been reported to be susceptible to activation by anionic phospholipids (32). Thus, GW4869 could also negatively regulate MA-nSMase activity, although how MA-nSMase could regulate exosome formation and protein packaging, given that it is located in the mitochondria, is enigmatic. Perhaps, upon prion infection, nSMase2 and MA-nSMase are redistributed subcellularly, resulting in a switch of exosome biogenesis regulation (Fig. 8). Indeed, mitochondrial structural abnormalities have been observed in prion-infected hamsters and mice (49–52). Components of the mitochondria were detected in lysosomes, and as the endosomal system also sorts proteins destined for lysosomal degradation, it is possible that MA-nSMase could traverse the endosomal system in prion-infected cells en route to the lysosome. In doing so, it could also regulate exosome formation and protein packaging, and thus compensate for disruptions in nSMase2 activity. As tools for studying MA-nSMase become available, the localization of this isoform in prion-infected cells and contribution toward exosome biogenesis and protein packaging will require investigation.
FIGURE 8.
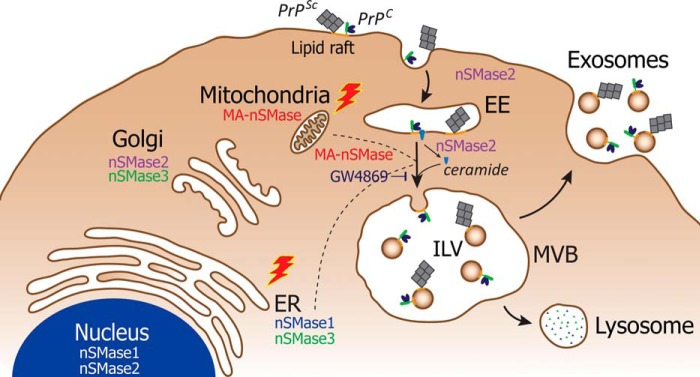
Schematic representation of nSMase-dependent exosome biogenesis and PrP packaging. Exosome biogenesis and PrP packaging are mediated by the nSMase pathway. In non-infected cells, nSMase2 mediates packaging of PrPC into exosomes. However, this regulation is abolished upon prion infection (red lightning bolt). Treatment of infected cells with GW4869 impedes sorting of PrPSc into exosomes, indicating that compensatory mechanisms exist between the nSMase isoforms. Possible mechanisms include the cellular redistribution of MA-nSMase or nSMase1 from the mitochondria or endoplasmic reticulum, respectively, to the endosomal system in prion-infected cells (dotted line).
The roles of nSMase1 and nSMase2 in exosomal protein packaging is different and is also likely to be affected by their subcellular localization. Whereas nSMase1 is located in the ER and the nuclear matrix, nSMase2 is reported to be located in the Golgi, but can also be shuttled between the Golgi and plasma membrane via anterograde and retrograde transport pathways (53–55). Once exported to the plasma membrane, nSMase2 activity is enhanced by anionic phospholipids in the membrane (56). Internalization and retrograde transport of nSMase2, however, negatively regulates the activity of the enzyme (54). As nSMase2 transits the endosomal compartments, the isoform is able to hydrolyze sphingomyelin, leading to production of ceramide and initiation of exosome formation and protein packaging. Since the activity of nSMase2 is regulated by intracellular trafficking, it can be speculated that prion infection can abolish the role of nSMase2 in exosome protein packaging by modifying nSMase2 trafficking, diverting it away from the plasma membrane and endosomal compartments. Similarly, nSMase1 could also contribute toward PrPSc packaging into exosomes as a result of altered subcellular trafficking due to cellular stress (Fig. 8). ER stress has previously been reported in prion-infected cells, and this could give rise to relocation of nSMase1 from the ER to the lysosome via the endosomal system (45, 46). In addition, nSMase1 is also reported to display sphingomyelinase activity in vivo under stressed conditions, consistent with the observed role of nSMase1 in regulating PrP packaging into exosomes in prion-infected cells.
Additional support for the proposed role of the nSMase pathway in PrP packaging into exosomes can be taken from Naslavsky et al., in which decreasing sphingomyelin levels were linked to increasing PrPSc levels (57). Based on the results presented here, decreasing sphingomyelin levels will impair the nSMase pathway and thus reduce exosome biogenesis and packaging of PrPSc. Consequently, PrPSc will accumulate inside the cell and stimulate further prion conversion, thereby increasing PrPSc levels. Packaging and release of PrPSc via exosomes could therefore be considered as beneficial for the cell. In fact, neuron-derived exosomes have been reported to be internalized and degraded by microglia, which suggests that exosomes could represent an efficient mechanism for clearance of PrPSc (58). Furthermore, exosomes could also facilitate clearance of other disease-associated proteins, such as Aβ, as exosomes generated in an nSMase2-dependent manner has been found to promote clearance of Aβ by microglia (28). Since PrPC has been proposed to function as a receptor for Aβ, it can be speculated that exosome-mediated clearance of extracellular Aβ could be facilitated by nSMase2-regulated incorporation of PrPC into exosomes.
Together, these data have provided further support for a role for the nSMase pathway in exosome biogenesis and also demonstrated a novel role for this pathway in regulating PrPC and PrPSc packaging into exosomes. We also show that while nSMase2 is the predominant isoform mediating this process, other nSMase isoforms could also contribute, depending on the prion-infected status of the cell. Comprehensive analyses of all the nSMase isoforms in non- and prion-infected cells is thus required to fully characterize the roles of each isoform in exosome biogenesis and protein packaging. Understanding how the nSMase pathway modulates this process not only has important implications for understanding the unique transmissible nature of prions, but will also have important implications for other diseases such as Alzheimer's disease.
Acknowledgments
We thank Dr. Victoria Lawson (The University of Melbourne) for kindly providing mouse brain homogenates, and Dr. Bradley Coleman (The University of Melbourne) for imaging the exosomes at the Bio21 Electron Microscopy Unit (The University of Melbourne). We also thank Dr. Peter Kaub and Dr. Simon Hawke (University of Sydney) for kindly providing the monoclonal L3 antibody, and Prof. John Collinge for the gift of monoclonal antibody ICSM-18. We thank Metabolomics Australia (Bio21 Institute) for performing the lipidomic analyses.
This study was funded by grants from the Australian National Health and Medical Research Council (NHMRC; Grants 628946 and 1041413). This work was also supported by an Australian Postgraduate Award and Faculty of Medicine, Dentistry and Health Sciences (MDHS) Top-Up Scholarship (to B. B. G.); a Bellberry Indigenous Health Fellowship (MDHS, The University of Melbourne) (to S. A. B.); an Australian Research Council (www.arc.gov.au) Future Fellowship (Grant FT100100560) (to A. F. H.). A.F.H is a shareholder of D-Gen Limited, which supplies an antibody used in the study.
- ILV
- intraluminal vesicles
- ESCRT
- endosomal sorting complexes required for transport
- KD
- knockdown
- nSMase
- neutral sphingomyelinase
- MVB
- multivesicular body
- AChE
- acetylcholinesterase.
REFERENCES
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kanu N., Imokawa Y., Drechsel D. N., Williamson R. A., Birkett C. R., Bostock C. J., Brockes J. P. (2002) Transfer of scrapie prion infectivity by cell contact in culture. Curr. Biol. 12, 523–530 [DOI] [PubMed] [Google Scholar]
- 3. Gousset K., Schiff E., Langevin C., Marijanovic Z., Caputo A., Browman D. T., Chenouard N., de Chaumont F., Martino A., Enninga J., Olivo-Marin J. C., Männel D., Zurzolo C. (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nature Cell Biology. 11, 328–336 [DOI] [PubMed] [Google Scholar]
- 4. Fevrier B., Vilette D., Archer F., Loew D., Faigle W., Vidal M., Laude H., Raposo G. (2004) Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 101, 9683–9688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mattei V., Barenco M. G., Tasciotti V., Garofalo T., Longo A., Boller K., Löwer J., Misasi R., Montrasio F., Sorice M. (2009) Paracrine diffusion of PrP(C) and propagation of prion infectivity by plasma membrane-derived microvesicles. PLoS One 4, e5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Théry C., Zitvogel L., Amigorena S. (2002) Exosomes: composition, biogenesis and function. Nature Reviews Immunology 2, 569–579 [DOI] [PubMed] [Google Scholar]
- 7. Simons M., Raposo G. (2009) Exosomes: vesicular carriers for intercellular communication. Curr. Opin. Cell Biol.. 21, 575–581 [DOI] [PubMed] [Google Scholar]
- 8. Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J. J., Lötvall J. O. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature Cell Biology. 9, 654–659 [DOI] [PubMed] [Google Scholar]
- 9. Bellingham S. A., Coleman B. M., Hill A. F. (2012) Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. 40, 10937–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coleman B. M., Hanssen E., Lawson V. A., Hill A. F. (2012) Prion-infected cells regulate the release of exosomes with distinct ultrastructural features. Faseb J. 26, 4160–4173 [DOI] [PubMed] [Google Scholar]
- 11. Beaudoin A. R., Grondin G. (1991) Shedding of vesicular material from the cell surface of eukaryotic cells: different cellular phenomena. Biochim. Biophys. Acta 13, 203–219 [DOI] [PubMed] [Google Scholar]
- 12. Wubbolts R., Leckie R. S., Veenhuizen P. T., Schwarzmann G., Möbius W., Hoernschemeyer J., Slot J. W., Geuze H. J., Stoorvogel W. (2003) Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 278, 10963–10972 [DOI] [PubMed] [Google Scholar]
- 13. Vella L. J., Sharples R. A., Lawson V. A., Masters C. L., Cappai R., Hill A. F. (2007) Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211, 582–590 [DOI] [PubMed] [Google Scholar]
- 14. Rajendran L., Honsho M., Zahn T. R., Keller P., Geiger K. D., Verkade P., Simons K. (2006) Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharples R. A., Vella L. J., Nisbet R. M., Naylor R., Perez K., Barnham K. J., Masters C. L., Hill A. F. (2008) Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. Faseb J. 22, 1469–1478 [DOI] [PubMed] [Google Scholar]
- 16. Emmanouilidou E., Melachroinou K., Roumeliotis T., Garbis S. D., Ntzouni M., Margaritis L. H., Stefanis L., Vekrellis K. (2010) Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saman S., Kim W., Raya M., Visnick Y., Miro S., Jackson B., McKee A. C., Alvarez V. E., Lee N. C., Hall G. F. (2012) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gomes C., Keller S., Altevogt P., Costa J. (2007) Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neuroscience Letters 428, 43–46 [DOI] [PubMed] [Google Scholar]
- 19. Izquierdo-Useros N., Naranjo-Gómez M., Archer J., Hatch S. C., Erkizia I., Blanco J., Borràs F. E., Puertas M. C., Connor J. H., Fernández-Figueras M. T., Moore L., Clotet B., Gummuluru S., Martinez-Picado J. (2009) Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 113, 2732–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bellingham S. A., Guo B. B., Coleman B. M., Hill A. F. (2012) Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 3, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hicke L. (2001) Protein regulation by monoubiquitin. Nature Reviews Molecular Cell Biology 2, 195–201 [DOI] [PubMed] [Google Scholar]
- 22. Raiborg C., Stenmark H. (2009) The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452 [DOI] [PubMed] [Google Scholar]
- 23. de Gassart A., Geminard C., Fevrier B., Raposo G., Vidal M. (2003) Lipid raft-associated protein sorting in exosomes. Blood 102, 4336–4344 [DOI] [PubMed] [Google Scholar]
- 24. Fang Y., Wu N., Gan X., Yan W., Morrell J. C., Gould S. J. (2007) Higher-order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS Biology 5, 1267–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perez-Hernandez D., Gutiérrez-Vazquez C., Jorge I., López-Martin S., Ursa A., Sánchez-Madrid F., Vázquez J., Yáñez-Mó M. (2013) The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries to exosomes. J. Biol. Chem. 288, 11649–11661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trajkovic K., Hsu C., Chiantia S., Rajendran L., Wenzel D., Wieland F., Schwille P., Brügger B., Simons M. (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 27. Kosaka N., Iguchi H., Yoshioka Y., Takeshita F., Matsuki Y., Ochiya T. (2010) Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 285, 17442–17452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yuyama K., Sun H., Mitsutake S., Igarashi Y. (2012) Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 287, 10977–10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu B. X., Clarke C. J., Hannun Y. A. (2010) Mammalian neutral sphingomyelinases: regulation and roles in cell signaling responses. Neuromolecular Medicine 12, 320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hofmann K., Tomiuk S., Wolff G., Stoffel W. (2000) Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc. Natl. Acad. Sci. U.S.A. 97, 5895–5900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krut O., Wiegmann K., Kashkar H., Yazdanpanah B., Krönke M. (2006) Novel tumor necrosis factor-responsive mammalian neutral sphingomyelinase-3 is a C-tail-anchored protein. J. Biol. Chem. 281, 13784–13793 [DOI] [PubMed] [Google Scholar]
- 32. Wu B. X., Rajagopalan V., Roddy P. L., Clarke C. J., Hannun Y. A. (2010) Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 285, 17993–18002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawson V. A., Vella L. J., Stewart J. D., Sharples R. A., Klemm H., Machalek D. M., Masters C. L., Cappai R., Collins S. J., Hill A. F. (2008) Mouse-adapted sporadic human Creutzfeldt-Jakob disease prions propagate in cell culture. Int. J. Biochem. Cell Biol. 40, 2793–2801 [DOI] [PubMed] [Google Scholar]
- 34. Bellingham S. A., Coleman L. A., Masters C. L., Camakaris J., Hill A. F. (2009) Regulation of Prion Gene Expression by Transcription Factors SP1 and Metal Transcription Factor-1. J. Biol. Chem. 284, 1291–1301 [DOI] [PubMed] [Google Scholar]
- 35. Ellman G. L., Courtney K. D., Andres V., Featherstone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 [DOI] [PubMed] [Google Scholar]
- 36. Cantin R., Diou J., Bélanger D., Tremblay A. M., Gilbert C. (2008) Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J. Immunol. Methods 338, 21–30 [DOI] [PubMed] [Google Scholar]
- 37. Rabesandratana H., Toutant J. P., Reggio H., Vidal M. (1998) Decay-accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) are released within exosomes during in vitro maturation of reticulocytes. Blood 91, 2573–2580 [PubMed] [Google Scholar]
- 38. Cheng L., Sun X., Scicluna B. J., Coleman B. M., Hill A. F. (2014) Characterization and deep sequencing analysis of exosomal and non-exosomal miRNA in human urine. Kidney Int. 86, 433–444 [DOI] [PubMed] [Google Scholar]
- 39. Botté C. Y., Yamaryo-Botté Y., Rupasinghe T. W., Mullin K. A., MacRae J. I., Spurck T. P., Kalanon M., Shears M. J., Coppel R. L., Crellin P. K., Maréchal E., McConville M. J., McFadden G. I. (2013) Atypical lipid composition in the purified relict plastid (apicoplast) of malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 110, 7506–7511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mellon P. L., Windle J. J., Goldsmith P. C., Padula C. A., Roberts J. L., Weiner R. I. (1990) Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5, 1–10 [DOI] [PubMed] [Google Scholar]
- 42. Schätzl H. M., Laszlo L., Holtzman D. M., Tatzelt J., DeArmond S. J., Weiner R. I., Mobley W. C., Prusiner S. B. (1997) A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 71, 8821–8831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sokolova V., Ludwig A. K., Hornung S., Rotan O., Horn P. A., Epple M., Giebel B. (2011) Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids and Surfaces B: Biointerfaces 87, 146–150 [DOI] [PubMed] [Google Scholar]
- 44. Garza-Licudine E., Deo D., Yu S., Uz-Zaman A., Dunbar W. B. (2010) Portable nanoparticle quantization using a resizable nanopore instrument - the IZON qNano. Conference proceedings: Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Conference 2010, 5736–5739 [DOI] [PubMed] [Google Scholar]
- 45. Tonnetti L., Verí M. C., Bonvini E., D'Adamio L. (1999) A role for neutral sphingomyelinase-mediated ceramide production in T cell receptor-induced apoptosis and mitogen-activated protein kinase-mediated signal transduction. J. Exp. Med. 189, 1581–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yabu T., Imamura S., Yamashita M., Okazaki T. (2008) Identification of Mg2+ -dependent neutral sphingomyelinase 1 as a mediator of heat stress-induced ceramide generation and apoptosis. J. Biol. Chem. 283, 29971–29982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alais S., Simoes S., Baas D., Lehmann S., Raposo G., Darlix J. L., Leblanc P. (2008) Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol. Cell 100, 603–615 [DOI] [PubMed] [Google Scholar]
- 48. Luberto C., Hassler D. F., Signorelli P., Okamoto Y., Sawai H., Boros E., Hazen-Martin D. J., Obeid L. M., Hannun Y. A., Smith G. K. (2002) Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J. Biol. Chem. 277, 41128–41139 [DOI] [PubMed] [Google Scholar]
- 49. Choi S. I., Ju W. K., Choi E. K., Kim J., Lea H. Z., Carp R. I., Wisniewski H. M., Kim Y. S. (1998) Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathologica 96, 279–286 [DOI] [PubMed] [Google Scholar]
- 50. Sisková Z., Mahad D. J., Pudney C., Campbell G., Cadogan M., Asuni A., O'Connor V., Perry V. H. (2010) Morphological and functional abnormalities in mitochondria associated with synaptic degeneration in prion disease. Am. J. Pathol. 177, 1411–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mattei V., Matarrese P., Garofalo T., Tinari A., Gambardella L., Ciarlo L., Manganelli V., Tasciotti V., Misasi R., Malorni W., Sorice M. (2011) Recruitment of cellular prion protein to mitochondrial raft-like microdomains contributes to apoptosis execution. Mol. Biol. Cell 22, 4842–4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Choi H. S., Choi Y. G., Shin H. Y., Oh J. M., Park J. H., Kim J. I., Carp R. I., Choi E. K., Kim Y. S. (2014) Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem. Biophys. Res. Commun. 448, 157–162 [DOI] [PubMed] [Google Scholar]
- 53. Tomiuk S., Zumbansen M., Stoffel W. (2000) Characterization and subcellular localization of murine and human magnesium-dependent neutral sphingomyelinase. J. Biol. Chem. 275, 5710–5717 [DOI] [PubMed] [Google Scholar]
- 54. Milhas D., Clarke C. J., Idkowiak-Baldys J., Canals D., Hannun Y. A. (2010) Anterograde and retrograde transport of neutral sphingomyelinase-2 between the Golgi and the plasma membrane. Biochim. Biophys. Acta 1801, 1361–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marchesini N., Osta W., Bielawski J., Luberto C., Obeid L. M., Hannun Y. A. (2004) Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J. Biol. Chem. 279, 25101–25111 [DOI] [PubMed] [Google Scholar]
- 56. Marchesini N., Luberto C., Hannun Y. A. (2003) Biochemical properties of mammalian neutral sphingomyelinase 2 and its role in sphingolipid metabolism. J. Biol. Chem. 278, 13775–13783 [DOI] [PubMed] [Google Scholar]
- 57. Naslavsky N., Shmeeda H., Friedlander G., Yanai A., Futerman A. H., Barenholz Y., Taraboulos A. (1999) Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J. Biol. Chem. 274, 20763–20771 [DOI] [PubMed] [Google Scholar]
- 58. Fitzner D., Schnaars M., van Rossum D., Krishnamoorthy G., Dibaj P., Bakhti M., Regen T., Hanisch U. K., Simons M. (2011) Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Science 124, 447–458 [DOI] [PubMed] [Google Scholar]