

Abstract
A general aim of studies of signal transduction is to identify mediators of specific signals, order them into pathways, and understand the nature of interactions between individual components and how these interactions alter pathway behavior. Despite years of intensive study and its central importance to animal development and human health, our understanding of the Hedgehog (Hh) signaling pathway remains riddled with gaps, question marks, assumptions, and poorly understood connections. In particular, understanding how interactions between Hh and Patched (Ptc), a 12-pass integral membrane protein, lead to modulation of the function of Smoothened (Smo), a 7-pass integral membrane protein, has defied standard biochemical characterization. Recent structural and biochemical characterizations of Smoothened domains have begun to unlock this riddle, however, and lay the groundwork for improved cancer therapies.
Keywords: Cholesterol, Development, G Protein-coupled Receptor (GPCR), Hedgehog Signaling Pathway, Sterol, Sonic Hedgehog (Shh), Patched (Ptc), Smoothened (Smo), Cell Signaling, Cancer
Introduction
Members of the Hedgehog (Hh)3 family of secreted signaling proteins are present in most metazoans and owe their name to the effects that loss of Hh function has on Drosophila embryos, which lose their normal segmented pattern and develop a uniform coat of bristles reminiscent of the coats of hedgehogs (1). As presaged by this phenotype, Hh proteins mediate essential tissue patterning events during many stages of animal development (2), and abnormal Hh function is associated with birth defects and cancer (3). Hh proteins are also involved in tissue maintenance and wound repair in adult animals (4). Hh proteins achieve their patterning effects by functioning as classical morphogens (5). That is, Hh proteins form gradients of decreasing concentration from sites of secretion and induce concentration-dependent differentiation of distinct cell types (6, 7). As befits a morphogen, Hh expression, release, diffusion, and signal reception are tightly regulated by multiple factors (8).
Classical and modern genetic techniques have identified several cell-surface proteins and glycans involved in receiving or modifying Hh signals (9). The core components of this process, conserved in all organisms known to have active Hh signaling, are Patched (Ptc) and Smoothened (Smo) (Fig. 1) (10–13). Ptc functions upstream of Smo and has been genetically and biochemically defined as a primary component of the Hh receptor (14, 15). Ptc is a 12-pass integral membrane protein with distant homology to bacterial resistance-nodulation-cell division (RND) transporters (16, 17). Transmembrane helices 2–6 of Ptc are also homologous to sterol-sensing domains, which are found in diverse integral membrane proteins and regulate activity in response to levels of free cellular sterols (18). Smo is a member of the Frizzled family (class F) of G-protein coupled receptors (GPCRs) (19), and contains an N-terminal, ∼14-kDa extracellular cysteine-rich domain (CRD) connected via a linker to 7 membrane-spanning helices (7TM) and an extended (∼200 amino acids, human; ∼450 amino acids, Drosophila) C-terminal tail.
FIGURE 1.

Major transmembrane components of Hh signal reception and transduction. Ptc (left) represses Smo (right) through an unknown, indirect mechanism. The interaction of Sonic hedgehog N-terminal domain (ShhN) with Ptc relieves Ptc-mediated repression of Smo. The sterol-sensing domain of Ptc (TM II–TM VI) is colored blue. For Smo, the 8 cysteines mediating 4 disulfide bonds in the Smo ECLs are shown in green; D473H, a Vismodegib resistance mutation, is in blue; W535L, a constitutively activating mutation, is in red; and C-tail sites of serine and threonine phosphorylation (indicated by pS/pT) are in orange.
Hh signaling responses are modulated by additional cell-surface components including CDO, BOC, Gas1, Hedgehog-interacting protein, and glypicans in vertebrates and Ihog, BOI, and the glypican Dally-like protein in flies (20–29). These factors either lack intracellular regions because of glycophosphatidylinositol anchors (Gas1, glypicans) or have intracellular regions that are not implicated in Hh signaling and do not appear to transmit Hh signals across the cell membrane directly (14). Instead, transmission of Hh signals across the membrane appears to be mediated by Smo, the most downstream cell-surface component of the Hh signaling pathway. Consistent with this role, the cytoplasmic tail of Smo becomes heavily phosphorylated and likely changes disposition when the Hh pathway is active (30–32). These changes are coupled to intracellular signaling events that ultimately converge on members of the Gli family of transcription factors, active forms of which translocate to the nucleus and up-regulate expression of target genes (33).
Recent discovery of the importance of Ptc and Smo localization for normal Hh signaling has added additional complexity to Hh pathway regulation. In vertebrates, Sonic Hh (Shh) and Hh pathway agonists result in movement of Smo from the plasma membrane to the primary cilium, a nonmotile flagellar-like organelle present on most cells, and dispersal of Ptc from its previous localization at the base of the primary cilium (34). Although movement of Smo to the primary cilium appears essential for normal Hh signaling in vertebrates (35), this movement is neither sufficient for signaling (36) nor conserved in flies (37), and a core signaling capacity that is independent of ciliary localization must be present in Smo. This minireview will focus on recent advances in structural and biochemical characterization of Smo, and readers are encouraged to consult other sources for background on additional Hh pathway components.
Patched and Smoothened
In unstimulated Hh-responsive cells, Ptc functions upstream of Smo to inhibit its activity (2). Hh triggers signaling responses by interacting with Ptc to relieve this inhibition, but both how Ptc inhibits Smo and how Hh relieves this inhibition remain unclear. As a small amount of Ptc is sufficient to inhibit a large stoichiometric excess of Smo (16), Ptc does not appear to inhibit Smo through a direct interaction. Rather, the homology of Ptc to transporters and the ability of Smo activity to be modulated by small molecules have led to the widespread belief that Ptc controls Smo through transport of a small molecule intermediary (16). Indeed, the ability of Smo to bind and be inhibited by the plant sterol cyclopamine led to the development of compounds targeting the cyclopamine-binding site for the treatment of cancers with abnormally active Hh signaling (38, 39). As some Smo-binding compounds function as Hh pathway agonists, it has been tempting to speculate that an endogenous cyclopamine-like compound modulates Smo activity (40). Indeed, the sterol vitamin D3 has been proposed to function as a Ptc-dependent inhibitor of Smo (41), although this observation awaits confirmation.
Smoothened: 7TM Region
The absence of knowledge of the physiological factors responsible for Smo activation (or inhibition) has presented a frustrating barrier to understanding Hh pathway regulation, but several recent results have begun to clarify this issue. Firstly, Stevens and colleagues (42–44) have determined atomic resolution crystal structures of the 7TM region of human Smo complexed with five different small molecules, including cyclopamine. These landmark structures show that, despite sharing less than 10% sequence identity with class A GPCRs such as rhodopsin and the β2-adrenergic receptor (β2AR), the Smo7 TM region adopts an overall conformation very similar to that of inactive class A GPCRs (Fig. 2A) (45). As discussed in more detail below (46), this structural homology couples with the observation that activating mutations in Smo occur at sites that appear to stabilize the inactive state of class A GPCRs to suggest that the 7TM region of Smo is likely to undergo GPCR-like conformational changes during its activity cycle (45). Such a conformational cycle would also be consistent with the ability of Smo to signal through G-proteins in certain circumstances (47–52).
FIGURE 2.

Structures of the Smo 7TM domain. A, β2AR in complex with Carazolol (Protein Data Bank (PDB): 2RH1) and Smo in complex with LY2940680 (PDB: 4JKV) colored by GPCR helix number. Key Smo residues are shown in spheres (ECL disulfides are green; D473H is light blue; W535L is orange; and arrows mark D473H and W535L). B, the five Smo 7TM crystal structures with bound ligands shown in spheres. (From left to right, PDB: 4O9R, 4QIN, 4QIM, 4JKV, and 4N4W.)
Although the overall fold of its 7TM bundle is conserved with other GPCRs, Smo has additional features including an extension of extracellular loops (ECLs). All of the co-crystallized compounds bind Smo in a long narrow pocket formed by the ECL extensions and upper portions of the transmembrane domains (Fig. 2) (42–44). The drug-binding pocket is exposed to the extracellular space, suggesting that drugs and any endogenous ligands access the pocket from the extracellular surface. This extracellular accessibility contrasts with a class A GPCR lipid receptor where the extracellular loops form a closed cage and ligand is thought to access its binding site from within the membrane (53). Although the CRD was deleted from the crystallized Smo 7TM domain, the majority of the residues of the extracellular linker between the CRD and the first TM domain are present and adopt an ordered structure. Disulfide bonds both within the linker and between the linker and the second extracellular loop appear to stabilize the linker structure (Fig. 2A), and disruption of these disulfides results in increased Smoothened activity (54). In addition, the extracellular linker interacts with the extended extracellular loop connecting TMs VI and VII (ECL3), which forms a cap over the drug-binding pocket. This ordered linker region suggests that the CRD may be directly coupled to the 7TM region and influence its conformation.
The five compounds crystallized in the Smo-binding pocket include an agonist (SAG1.5) and four antagonists (LY2940680, SANT1, ANTA XV, and cyclopamine) (see Fig. 4A). All ligands bind in the pocket with their long axes perpendicular to the plane of the membrane but vary in their depth relative to the extracellular outlet (Fig. 2B). At the extremes, cyclopamine interacts predominantly with the extracellular loops, whereas another antagonist, SANT-1, binds deep within the pocket, which spans 28 Å from the top of cyclopamine to the bottom of SANT1. Asp-473, a residue that when mutated to histidine confers resistance to the anti-cancer agent Vismodegib (GDC-0449) (55, 56), lines the drug-binding pocket but interacts differently with different antagonists and does not confer universal drug resistance (43). Asp-473 does not directly contact LY2940680, for example, and the D473H substitution does not affect the activity of LY2940680 (57). The variable susceptibility of individual drugs to resistance mutations suggests that second generation drugs or combination therapies may prolong the time to development of resistance.
FIGURE 4.

Smoothened-interacting small molecules. A, 7TM-targeting small molecules. B, CRD-targeting small molecules. C, other Smo-targeting small molecules. Activating small molecules are noted by green type.
LY2940680, cyclopamine, ANTA XV, and the agonist SAG1.5 contact the Smoothened extracellular loops lining the top of the ligand-binding cavity, but SANT1 binds more deeply in the pocket and only contacts ECL2, which is positioned within the 7TM region. In contrast to cyclopamine, which binds more tightly to Smo than to a constitutively active Smo variant bearing a single-site substitution (SmoM2), SANT1 binds both Smo and SmoM2 with equal potency (40). How the position of SANT1 deep within the 7TM bundle correlates to its ability to inhibit both Smo and SmoM2, whose W535L substitution occurs at the base of TM VII, is not clear. Also of interest are the variable effects Smo antagonists have on Smo localization. SANT1, LY2940680, and cyclopamine all inhibit Smo function, but only cyclopamine promotes the translocation of a still inactive Smo to the primary cilium, indicating that translocation and activation are separable functions.
The failure of Smo to adopt an active-like conformation when bound to the agonist SAG1.5 is curious but not unprecedented for agonist-bound GPCRs (58). Binding of an agonist to an apparently inactive state may reflect a low energetic barrier between active and inactive states, conformational flexibility of the active state (59), and/or the effects of truncation of Smo N- and C-terminal regions. SAG1.5 binds in the same region of the binding pocket as LY2940680, ANTA XV, and cyclopamine, and Smo with SAG1.5 bound displays only slight alterations in binding pocket residues. Larger conformational changes associated with active state GPCRs, such as the movements of TMs VI and VII to accommodate G-protein binding, are not seen in the Smo-SAG1.5 structure. Crystallization of an active state of Smo may require adding back the CRD or portions of the C-terminal tail or co-crystallization with active conformation-specific nanobodies (60). Interesting features of the effects of these different drugs on the conformational equilibria of intact Smo and their relation to Smo function clearly remain to be worked out.
Smoothened: Cysteine-rich Domain
A second major insight into Smo regulation emerged when three groups independently showed that oxysterols, oxidized derivatives of cholesterol, bind specifically to the Smo CRD and activate the Hh signaling pathway (61–63). Oxysterol binding by the Smo CRD is functionally as well as physically separable from small molecule binding to the 7TM site as deletion of the Smo CRD results in loss of oxysterol activation of Smo but does not affect the function of agonists and antagonists that target the 7TM region (61). It had previously been shown that oxysterols could modulate Hh signaling by affecting Smo function (64–66). The site of oxysterol action was not characterized at that time, although oxysterols did not appear to compete with cyclopamine for binding to Smo (66).
The new studies all show that 20(S)-hydroxycholesterol (20(S)-OHC) (see Fig. 4B) activates Smo by binding to the CRD. Additionally, the Rohatgi and Siebold groups (63) were able to determine the crystal structure of the zebrafish Smo CRD (Fig. 3B). All groups mapped the site of sterol action on the CRD via mutagenesis and in silico modeling to a hydrophobic groove that is homologous to the site at which the palmitoyl group of Wnt binds to the Frizzled CRD (Fig. 3A) (61–63, 67), confirming an earlier prediction based on structural homology that this region of Smo and Frizzled CRDs would bind lipophilic molecules (68). Curiously, the Drosophila Smo CRD does not bind to 20(S)-OHC (63), but it and human Smo CRD were recently shown to bind to the glucocorticoid budesonide (Fig. 4B), suggesting that sterol binding by the Smo CRD may be a conserved feature of Hh signaling (69). Glucocorticoids represent an interesting class of Smo modulators as both inhibitors and activators of the Hh pathway have been found with glucocorticoid scaffolds, and budesonide inhibits WT Smo, SmoD473H, and SmoM2 equally well, ideal features for a Smo-targeting drug (70).
FIGURE 3.
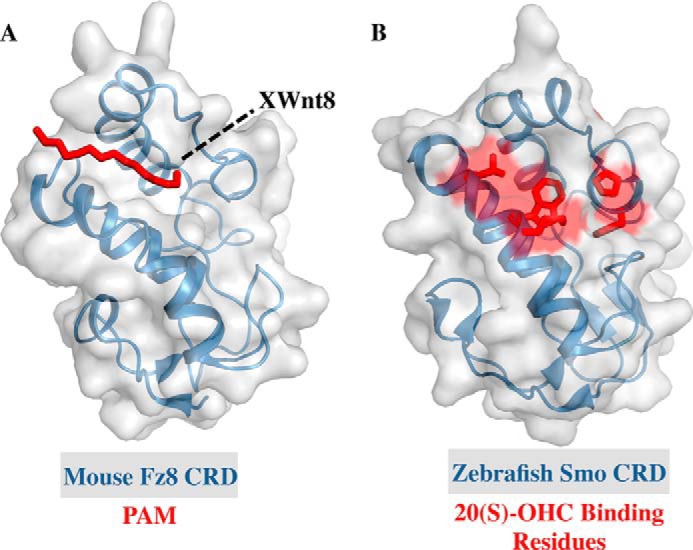
Structures of class F GPCR CRDs. A, the structure of the mouse Frizzled-8 CRD (Fz8 CRD) shown with the palmitoleic acid moiety (PAM) in red (PDB: 4F0A). The position of Xenopus Wnt8 loop to which PAM is attached is noted by a dashed black line. B, the structure of the zebrafish Smoothened CRD with residues implicated in binding 20(S)-OHC shown in red (PDB: 4C79).
Variable specificity for 20(S)-OHC among Smo CRDs is perhaps not surprising given that the absence of a cellular sterol hydroxylase known to produce it makes it unlikely to be an endogenous ligand (61). Assuming that endogenous ligands for Smo CRDs exist, the question naturally arises of what that ligand is. A survey of oxysterols for Smo modulatory activity found that 7-keto-27-OHC and 7-keto-25-OHC, both metabolites of 7-ketocholesterol, are able to stimulate Hh signaling in a manner that depends on the presence of the CRD (61). Compounds that bind the CRD and inhibit (azasterols, e.g. 22-azacholesterol) (Fig. 4B) or partially agonize (20(R)-yne, 20-keto-yne) Smo activity validate the Smo CRD as a potential drug target and raise the possibility that an endogenous ligand for the Smo CRD may be an inhibitor rather than an activator (62, 63). More work is needed to identify and validate potential CRD ligands, but it seems likely that such ligands exist, and their discovery and characterization will take our understanding of Hh pathway regulation in new directions.
Two immediately exciting prospects stimulated by the discovery of a specific and functionally important sterol-binding site on the Smo CRD were that it might be the route by which Ptc modulates Smo activity or that it might rationalize why cholesterol depletion reduces Hh signaling (71). Defying Occam's razor, however, oxysterol binding by the Smo CRD cannot fully account for either of these processes. Deletion of the CRD from Smo (ΔCRDSmo) alters but does not abolish Shh-mediated pathway activation (61–63). Although varying levels of responsiveness of ΔCRDSmo to Shh have been reported, this is likely due to varying tags and expression systems. Rohatgi and colleagues (63) showed that oxysterol-binding mutants of Smo retain negative regulation by Ptc and respond to Shh. Beachy and colleagues (61) showed that deletion of the CRD increases basal Smo activity, but this activity can be reduced by co-expression with Ptc and restored by addition of Shh, indicating that Ptc can exert its effects on Smo independent of the CRD. Higher basal activity and Shh responsiveness of ΔCRDSmo were also reduced by cyclodextrin depletion, which reduces wild-type Smo activity (71), suggesting that the CRD is also not essential for this process but rather that cholesterol within the cell membrane is needed for normal Smo function. Indeed, a specific role for membrane-localized cholesterol in Smo modulation has been suggested (61), although no ordered cholesterol molecules were identified in the Smo crystal structures. Modulation of Smo activity independent of the CRD or cyclopamine-binding pocket is not unprecedented as Itraconazole (Fig. 4C) acts on Smo at a site distinct from both the canonical 7TM pocket and the CRD to inhibit Hh pathway activity (72).
Cholesterol binding to the 7TM region of GPCRs is also not unprecedented. The structure of β2AR bound to cholesterol and the partial inverse agonist timolol led Stevens and colleagues (73) to propose a cholesterol consensus motif (CCM) in class A GPCRs. The CCM comprises 3 residues predictive of cholesterol binding: an aromatic residue (Trp or Tyr) at position 4.50, a positively charged residue at or about position 4.43 that interacts with the cholesterol hydroxyl group, and a hydrophobic residue at position 4.46. The positions here refer to the Ballesteros-Weinstein numbering for GPCRs (74), which allows cross comparison of topologically equivalent residues in GPCR TMs and was recently extended to class F GPCRs (42). Interestingly, Smo Trp-3654.50 overlays well with β2AR Trp-1584.50, which stacks against the sterol ring of cholesterol in the β2AR structure with cholesterol bound. Although Smo does not have a positively charged residue at position 4.43, Smo residue His-3614.46 maps to the hydrophobic position 4.46 of the CCM. A nitrogen on the imidazole ring of His-3614.46 is within 3.6 Å of the cholesterol hydroxyl group from cholesterol-bound β2AR structure (73). Whether these highly conserved class F residues, Trp-3654.50 and His-3614.46, act as an alternative cholesterol-binding motif presents an intriguing possibility.
Targeting Smoothened in the Clinic
Hh pathway-activating mutations in the gene encoding Ptc, and less commonly the gene encoding Smo, are found in subsets of several cancers, most notably basal cell carcinoma (BCC) and pediatric medulloblastomas (46, 75). Constitutively active mutants of Smo found in sporadic BCC (W535L7.55 “SmoM2”) and more recently in meningiomas and ameloblastomas (W535L7.55, L412F5.51) are resistant to Vismodegib treatment (46, 76–78). Superscripts refer to Ballesteros-Weinstein numbering. Trp-5357.55 is absolutely conserved in class F GPCRs and maps to the intracellular tip of TM VII, a region structurally homologous to the NPXXY motif in class A GPCRs (79, 80). Trp-5357.55 overlays with the Tyr7.53 of the NPXXY motif, which undergoes rearrangement in inactive versus active structures of class A GPCRs (60, 81, 82), Leu-4125.51 is highly conserved across class F GCPRs and also appears in a conformationally labile region of GPCRs. In class A GPCRs, residue 5.51 is one of a group of conserved hydrophobic and aromatic residues (3.40, 5.51, 6.44, 6.48) thought to constitute a “transmission switch” that rearranges when agonist binds (45, 83). Collectively, these constitutively active mutants bolster the notion that Smo cycles through canonical GPCR inactive-active states.
Vismodegib is a Smo inhibitor that binds to the 7TM pocket (Fig. 4) and has been approved for the treatment of advanced BCC. Resistance to Vismodegib usually appears within a few months, however (84). Cancers with active Hh signaling are often driven by inactivating Ptc mutations, but resistance mutations often appear in Smo, the target of the drug. The Vismodegib resistance mutation originally found in medulloblastoma, D473H (55), disrupts Vismodegib binding to Smo but does not result in Smo activation or loss of Smo regulation by physiological levels of Ptc. Additional drug resistance mutations in Smo were found in a mouse model of medulloblastoma where treatment with NVP-LDE225, a Smo 7TM antagonist, led to resistance mutations in Smo that predominantly localize to the 7TM-binding pocket and result in phenotypes similar to D473H (85). Several unique Smo resistance mutations (W281L2.57, V321M3.32) were also recently found in BCC after treatment with Vismodegib (86). W281L2.57 localizes to the base of the 7TM-binding pocket within 3.7 Å of the base of the LY2940680 ligand. V321M3.32 is further buried at the base of the binding pocket and 5.8 Å from SANT1 at its closest point. It is not known whether these mutations function to disrupt binding of Vismodegib to Smo or to activate Smo, but its position in the Smo structure suggests that W281L is more likely to interfere with ligand binding than V321M. Given the rapid resistance to drugs targeting the Smo 7TM pocket, antagonists that bind the Smo CRD hold out the hope that drugs targeting the CRD may prove more effective or less susceptible to resistance when used either alone or in combination with compounds targeting the Smo 7TM pocket (62, 63).
Any discussion of the Smo 7TM and CRD regions naturally leads to questions concerning how these components interact and how their interplay affects the Smo C-terminal tail. Little is known about the structure of the Smo C-tail alone or with the Smo 7TM bundle, but its low complexity and high hydrophilicity suggest that it does not adopt a rigid globular structure. The Smo C-tail is phosphorylated in response to pathway activation, although the identities of the kinases responsible for phosphorylation differ between vertebrates and invertebrates (31, 87). A conformational change of the Drosophila Smo C-tail has been proposed to stem from C-tail phosphorylation altering interactions between positively charged clusters of Arg residues and negatively charged clusters of Asp residues (32), but the vertebrate Smo C-tail does not possess the Arg clusters. A C-tail conformational change in vertebrates has also been proposed, however (88).
Conclusion
Multiple inputs (oxysterol binding to the CRD, small molecule binding to the 7TM pocket, and sterols within the cell membrane) are all capable of modulating Smo activity and presumably conformation. Sorting out what the endogenous inputs are, which of these inputs are important in specific instances, how multiple inputs are integrated, how best to exploit various ways of modulating Smo for anticancer therapies, and the role of Ptc in modulating these inputs present exciting challenges. Recent results have helped clarify the nature and sites of these inputs, however, and provided a framework for understanding how each of the parts fit together to achieve remarkable biological results.
Acknowledgments
We thank Ben Myers, Jennifer Kavran, and Matthew Ward for comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HD055545 (to D. J. L.).
- Hh
- Hedgehog
- Ptc
- Patched
- Smo
- Smoothened
- GPCR
- G-protein coupled receptor
- CRD
- cysteine-rich domain
- 7TM
- 7-transmembrane
- Shh
- sonic hedgehog
- β2AR
- β2-adregenergic receptor
- ECL
- extracellular loop
- ANTA XV
- 2-(6-(4-(4-benzylphthalazin-1-yl)piperazin-1-yl)pyridin-3-yl)propan-2-ol
- 20(S)-OHC
- 20(S)-hydroxycholesterol
- 22(R)-yne
- alkyne derivative of 22(R)-hydroxycholesterol
- 20-keto-yne
- derivative of 20(S)-OHC with the hydroxyl group converted to a ketone
- CCM
- cholesterol consensus motif
- BCC
- basal cell carcinoma.
REFERENCES
- 1. Nüsslein-Volhard C., Wieschaus E. (1980) Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801 [DOI] [PubMed] [Google Scholar]
- 2. Ingham P. W., Nakano Y., Seger C. (2011) Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 12, 393–406 [DOI] [PubMed] [Google Scholar]
- 3. Briscoe J., Thérond P. P. (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 [DOI] [PubMed] [Google Scholar]
- 4. Arwert E. N., Hoste E., Watt F. M. (2012) Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 12, 170–180 [DOI] [PubMed] [Google Scholar]
- 5. Heemskerk J., DiNardo S. (1994) Drosophila hedgehog acts as a morphogen in cellular patterning. Cell 76, 449–460 [DOI] [PubMed] [Google Scholar]
- 6. Gradilla A.-C., Guerrero I. (2013) Hedgehog on the move: a precise spatial control of Hedgehog dispersion shapes the gradient. Curr. Opin. Genet. Dev. 23, 363–373 [DOI] [PubMed] [Google Scholar]
- 7. Ribes V., Briscoe J. (2009) Establishing and interpreting graded Sonic Hedgehog signaling during vertebrate neural tube patterning: the role of negative feedback. Cold Spring Harb. Perspect. Biol. 1, a002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thérond P. P. (2012) Release and transportation of Hedgehog molecules. Curr. Opin. Cell Biol. 24, 173–180 [DOI] [PubMed] [Google Scholar]
- 9. Ryan K. E., Chiang C. (2012) Hedgehog secretion and signal transduction in vertebrates. J. Biol. Chem. 287, 17905–17913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakano Y., Guerrero I., Hidalgo A., Taylor A., Whittle J. R., Ingham P. W. (1989) A protein with several possible membrane-spanning domains encoded by the Drosophila segment polarity gene patched. Nature 341, 508–513 [DOI] [PubMed] [Google Scholar]
- 11. Stone D. M., Hynes M., Armanini M., Swanson T. A., Gu Q., Johnson R. L., Scott M. P., Pennica D., Goddard A., Phillips H., Noll M., Hooper J. E., de Sauvage F., Rosenthal A. (1996) The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384, 129–134 [DOI] [PubMed] [Google Scholar]
- 12. Alcedo J., Ayzenzon M., Von Ohlen T., Noll M., Hooper J. E. (1996) The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the Hedgehog signal. Cell 86, 221–232 [DOI] [PubMed] [Google Scholar]
- 13. van den Heuvel M., Ingham P. W. (1996) smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 382, 547–551 [DOI] [PubMed] [Google Scholar]
- 14. Zheng X., Mann R. K., Sever N., Beachy P. A. (2010) Genetic and biochemical definition of the Hedgehog receptor. Genes Dev. 24, 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuse N., Maiti T., Wang B., Porter J. A., Hall T. M. T., Leahy D. J., Beachy P. A. (1999) Sonic hedgehog protein signals not as a hydrolytic enzyme but as an apparent ligand for Patched. Proc. Natl. Acad. Sci. U.S.A. 96, 10992–10999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taipale J., Cooper M. K., Maiti T., Beachy P. A. (2002) Patched acts catalytically to suppress the activity of Smoothened. Nature 418, 892–897 [DOI] [PubMed] [Google Scholar]
- 17. Tseng T. T., Gratwick K. S., Kollman J., Park D., Nies D. H., Goffeau A., Saier M. H. (1999) The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1, 107–125 [PubMed] [Google Scholar]
- 18. Kuwabara P. E., Labouesse M. (2002) The sterol-sensing domain: multiple families, a unique role? Trends Genet. 18, 193–201 [DOI] [PubMed] [Google Scholar]
- 19. Ayers K. L., Thérond P. P. (2010) Evaluating Smoothened as a G-protein-coupled receptor for Hedgehog signalling. Trends Cell Biol. 20, 287–298 [DOI] [PubMed] [Google Scholar]
- 20. Beachy P. A., Hymowitz S. G., Lazarus R. A., Leahy D. J., Siebold C. (2010) Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 24, 2001–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao S., Lum L., Beachy P. (2006) The Ihog cell-surface proteins bind Hedgehog and mediate pathway activation. Cell 125, 343–357 [DOI] [PubMed] [Google Scholar]
- 22. McLellan J. S., Yao S., Zheng X., Geisbrecht B. V., Ghirlando R., Beachy P. A., Leahy D. J. (2006) Structure of a heparin-dependent complex of Hedgehog and Ihog. Proc. Natl. Acad. Sci. U.S.A. 103, 17208–17213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McLellan J. S., Zheng X., Hauk G., Ghirlando R., Beachy P. A., Leahy D. J. (2008) The mode of Hedgehog binding to Ihog homologues is not conserved across different phyla. Nature 455, 979–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bosanac I., Maun H. R., Scales S. J., Wen X., Lingel A., Bazan J. F., de Sauvage F. J., Hymowitz S. G., Lazarus R. A. (2009) The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat. Struct. Mol. Biol. 16, 691–697 [DOI] [PubMed] [Google Scholar]
- 25. Bishop B., Aricescu A. R., Harlos K., O'Callaghan C. A., Jones E. Y., Siebold C. (2009) Structural insights into hedgehog ligand sequestration by the human hedgehog-interacting protein HHIP. Nat. Struct. Mol. Biol. 16, 698–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams E. H., Pappano W. N., Saunders A. M., Kim M.-S., Leahy D. J., Beachy P. A. (2010) Dally-like core protein and its mammalian homologues mediate stimulatory and inhibitory effects on Hedgehog signal response. Proc. Natl. Acad. Sci. U.S.A. 107, 5869–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim M.-S., Saunders A. M., Hamaoka B. Y., Beachy P. A., Leahy D. J. (2011) Structure of the protein core of the glypican Dally-like and localization of a region important for hedgehog signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 13112–13117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Izzi L., Lévesque M., Morin S., Laniel D., Wilkes B. C., Mille F., Krauss R. S., McMahon A. P., Allen B. L., Charron F. (2011) Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev. Cell 20, 788–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allen B. L., Song J. Y., Izzi L., Althaus I. W., Kang J.-S., Charron F., Krauss R. S., McMahon A. P. (2011) Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev. Cell 20, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang C., Williams E. H., Guo Y., Lum L., Beachy P. A. (2004) Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc. Natl. Acad. Sci. U.S.A. 101, 17900–17907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jia J., Tong C., Wang B., Luo L., Jiang J. (2004) Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045–1050 [DOI] [PubMed] [Google Scholar]
- 32. Zhao Y., Tong C., Jiang J. (2007) Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 450, 252–258 [DOI] [PubMed] [Google Scholar]
- 33. Hui C.-C., Angers S. (2011) Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27, 513–537 [DOI] [PubMed] [Google Scholar]
- 34. Rohatgi R., Milenkovic L., Scott M. P. (2007) Patched1 regulates Hedgehog signaling at the primary cilium. Science 317, 372–376 [DOI] [PubMed] [Google Scholar]
- 35. Huangfu D., Liu A., Rakeman A. S., Murcia N. S., Niswander L., Anderson K. V. (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87 [DOI] [PubMed] [Google Scholar]
- 36. Rohatgi R., Milenkovic L., Corcoran R. B., Scott M. P. (2009) Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. U.S.A. 106, 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Denef N., Neubüser D., Perez L., Cohen S. M. (2000) Hedgehog induces opposite changes in turnover and subcellular localization of Patched and Smoothened. Cell 102, 521–531 [DOI] [PubMed] [Google Scholar]
- 38. Cooper M. K., Porter J. A., Young K. E., Beachy P. A. (1998) Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280, 1603–1607 [DOI] [PubMed] [Google Scholar]
- 39. Taipale J., Chen J. K., Cooper M. K., Wang B., Mann R. K., Milenkovic L., Scott M. P., Beachy P. A. (2000) Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406, 1005–1009 [DOI] [PubMed] [Google Scholar]
- 40. Chen J. K., Taipale J., Young K. E., Maiti T., Beachy P. A. (2002) Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. U.S.A. 99, 14071–14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bijlsma M. F., Spek C. A., Zivkovic D., van de Water S., Rezaee F., Peppelenbosch M. P. (2006) Repression of Smoothened by Patched-dependent (Pro-)vitamin D3 Secretion. PLoS Biol. 4, e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang C., Wu H., Katritch V., Han G. W., Huang X.-P., Liu W., Siu F. Y., Roth B. L., Cherezov V., Stevens R. C. (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang C., Wu H., Evron T., Vardy E., Han G. W., Huang X.-P., Hufeisen S. J., Mangano T. J., Urban D. J., Katritch V., Cherezov V., Caron M. G., Roth B. L., Stevens R. C. (2014) Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 5, 4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weierstall U., James D., Wang C., White T. A., Wang D., Liu W., Spence J. C. H., Doak R. B., Nelson G., Fromme P., Fromme R., Grotjohann I., Kupitz C., Zatsepin N. A., Liu H., Basu S., Wacker D., Han G. W., Katritch V., Boutet S., Messerschmidt M., Williams G. J., Koglin J. E., Seibert M. M., Klinker M., Gati C., Shoeman R. L., Barty A., Chapman H. N., Kirian R. A., Beyerlein K. R., Stevens R. C., Li D., Shah S. T. A., Howe N., Caffrey M., Cherezov V. (2014) Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 46. Xie J., Murone M., Luoh S.-M., Ryan A., Gu Q., Zhang C., Bonifas J. M., Lam C.-W., Hynes M., Goddard A., Rosenthal A., Epstein E. H., Jr, de Sauvage F. J. (1998) Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92 [DOI] [PubMed] [Google Scholar]
- 47. Riobo N. A., Saucy B., DiLizio C., Manning D. R. (2006) Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. U.S.A. 103, 12607–12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Philipp M., Caron M. G. (2009) Hedgehog signaling: is Smo a G protein-coupled receptor? Curr. Biol. 19, R125–R127 [DOI] [PubMed] [Google Scholar]
- 49. Polizio A. H., Chinchilla P., Chen X., Kim S., Manning D. R., Riobo N. A. (2011) Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J. Biol. Chem. 286, 19589–19596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Polizio A. H., Chinchilla P., Chen X., Manning D. R., Riobo N. A. (2011) Sonic Hedgehog activates the GTPases Rac1 and RhoA in a Gli-independent manner through coupling of Smoothened to Gi proteins. Sci. Signal. 4, pt7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shen F., Cheng L., Douglas A. E., Riobo N. A., Manning D. R. (2013) Smoothened is a fully competent activator of the heterotrimeric G protein Gi. Mol. Pharmacol. 83, 691–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carbe C. J., Cheng L., Addya S., Gold J. I., Gao E., Koch W. J., Riobo N. A. (2014) Gi proteins mediate activation of the canonical hedgehog pathway in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 307, H66–H72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hanson M. A., Roth C. B., Jo E., Griffith M. T., Scott F. L., Reinhart G., Desale H., Clemons B., Cahalan S. M., Schuerer S. C., Sanna M. G., Han G. W., Kuhn P., Rosen H., Stevens R. C. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carroll C. E., Marada S., Stewart D. P., Ouyang J. X., Ogden S. K. (2012) The extracellular loops of Smoothened play a regulatory role in control of Hedgehog pathway activation. Development 139, 612–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yauch R. L., Dijkgraaf G. J. P., Alicke B., Januario T., Ahn C. P., Holcomb T., Pujara K., Stinson J., Callahan C. A., Tang T., Bazan J. F., Kan Z., Seshagiri S., Hann C. L., Gould S. E., Low J. A., Rudin C. M., de Sauvage F. J. (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326, 572–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robarge K. D., Brunton S. A., Castanedo G. M., Cui Y., Dina M. S., Goldsmith R., Gould S. E., Guichert O., Gunzner J. L., Halladay J., Jia W., Khojasteh C., Koehler M. F. T., Kotkow K., La H., Lalonde R. L., Lau K., Lee L., Marshall D., Marsters J. C., Jr., Murray L. J., Qian C., Rubin L. L., Salphati L., Stanley M. S., Stibbard J. H. A., Sutherlin D. P., Ubhayaker S., Wang S., Wong S., Xie M. (2009) GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 19, 5576–5581 [DOI] [PubMed] [Google Scholar]
- 57. Bender M. H., Hipskind P. A., Capen A. R., Cockman M., Credille K. M., Gao H., Bastian J. A., Clay J. M., Lobb K. L., Sall D. J., Thompson M. L., Wilson T., Wishart G. N., Patel B. K. R. (2011) Abstract 2819: Identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated hedgehog signaling. Cancer Res. 71, 2819 [Google Scholar]
- 58. Warne T., Moukhametzianov R., Baker J. G., Nehmé R., Edwards P. C., Leslie A. G. W., Schertler G. F. X., Tate C. G. (2011) The structural basis for agonist and partial agonist action on a β1-adrenergic receptor. Nature 469, 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K. (2013) The dynamic process of β2-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ring A. M., Manglik A., Kruse A. C., Enos M. D., Weis W. I., Garcia K. C., Kobilka B. K. (2013) Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Myers B. R., Sever N., Chong Y. C., Kim J., Belani J. D., Rychnovsky S., Bazan J. F., Beachy P. A. (2013) Hedgehog pathway modulation by multiple lipid binding sites on the Smoothened effector of signal response. Dev. Cell 26, 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nedelcu D., Liu J., Xu Y., Jao C., Salic A. (2013) Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat. Chem. Biol. 9, 557–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nachtergaele S., Whalen D. M., Mydock L. K., Zhao Z., Malinauskas T., Krishnan K., Ingham P. W., Covey D. F., Siebold C., Rohatgi R. (2013) Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. Elife 2, e01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Corcoran R. B., Scott M. P. (2006) Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 103, 8408–8413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dwyer J. R., Sever N., Carlson M., Nelson S. F., Beachy P. A., Parhami F. (2007) Oxysterols are novel activators of the Hedgehog signaling pathway in pluripotent mesenchymal cells. J. Biol. Chem. 282, 8959–8968 [DOI] [PubMed] [Google Scholar]
- 66. Nachtergaele S., Mydock L. K., Krishnan K., Rammohan J., Schlesinger P. H., Covey D. F., Rohatgi R. (2012) Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 8, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Janda C. Y., Waghray D., Levin A. M., Thomas C., Garcia K. C. (2012) Structural basis of Wnt recognition by Frizzled. Science 337, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bazan J. F., de Sauvage F. J. (2009) Structural ties between cholesterol transport and morphogen signaling. Cell 138, 1055–1056 [DOI] [PubMed] [Google Scholar]
- 69. Rana R., Carroll C. E., Lee H.-J., Bao J., Marada S., Grace C. R. R., Guibao C. D., Ogden S. K., Zheng J. J. (2013) Structural insights into the role of Smoothened cysteine-rich domain in Hedgehog signaling. Nat. Commun. 4, 2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang Y., Davidow L., Arvanites A. C., Blanchard J., Lam K., Xu K., Oza V., Yoo J. W., Ng J. M. Y., Curran T., Rubin L. L., McMahon A. P. (2012) Glucocorticoid compounds modify Smoothened localization and Hedgehog pathway activity. Chem. Biol. 19, 972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cooper M. K., Wassif C. A., Krakowiak P. A., Taipale J., Gong R., Kelley R. I., Porter F. D., Beachy P. A. (2003) A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 33, 508–513 [DOI] [PubMed] [Google Scholar]
- 72. Kim J., Aftab B. T., Tang J. Y., Kim D., Lee A. H., Rezaee M., Kim J., Chen B., King E. M., Borodovsky A., Riggins G. J., Epstein E. H., Jr., Beachy P. A., Rudin C. M. (2013) Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 23, 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V.-P., Chien E. Y. T., Velasquez J., Kuhn P., Stevens R. C. (2008) A specific cholesterol binding site is established by the 2.8 Ä structure of the human β2-adrenergic receptor. Structure 16, 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 75. Goodrich L. V., Milenković L., Higgins K. M., Scott M. P. (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113 [DOI] [PubMed] [Google Scholar]
- 76. Sweeney R. T., McClary A. C., Myers B. R., Biscocho J., Neahring L., Kwei K. A., Qu K., Gong X., Ng T., Jones C. D., Varma S., Odegaard J. I., Sugiyama T., Koyota S., Rubin B. P., Troxell M. L., Pelham R. J., Zehnder J. L., Beachy P. A., Pollack J. R., West R. B. (2014) Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat. Genet. 46, 722–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brastianos P. K., Horowitz P. M., Santagata S., Jones R. T., McKenna A., Getz G., Ligon K. L., Palescandolo E., Van Hummelen P., Ducar M. D., Raza A., Sunkavalli A., Macconaill L. E., Stemmer-Rachamimov A. O., Louis D. N., Hahn W. C., Dunn I. F., Beroukhim R. (2013) Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 45, 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Clark V. E., Erson-Omay E. Z., Serin A., Yin J., Cotney J., Ozduman K., Avar T., Li J., Murray P. B., Henegariu O., Yilmaz S., Günel J. M., Carrión-Grant G., Yilmaz B., Grady C., Tanrikulu B., Bakircioğlu M., Kaymakçalan H., Caglayan A. O., Sencar L., Ceyhun E., Atik A. F., Bayri Y., Bai H., Kolb L. E., Hebert R. M., Omay S. B., Mishra-Gorur K., Choi M., Overton J. D., Holland E. C., Mane S., State M. W., Bilgüvar K., Baehring J. M., Gutin P. H., Piepmeier J. M., Vortmeyer A., Brennan C. W., Pamir M. N., Kiliç T., Lifton R. P., Noonan J. P., Yasuno K., Günel M. (2013) Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339, 1077–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Barak L. S., Ménard L., Ferguson S. S., Colapietro A. M., Caron M. G. (1995) The conserved seven-transmembrane sequence NP(X)2,3Y of the G-protein-coupled receptor superfamily regulates multiple properties of the β2-adrenergic receptor. Biochemistry 34, 15407–15414 [DOI] [PubMed] [Google Scholar]
- 80. Rosenbaum D. M., Rasmussen S. G. F., Kobilka B. K. (2009) The structure and function of G-protein-coupled receptors. Nature 459, 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Deupi X., Edwards P., Singhal A., Nickle B., Oprian D., Schertler G., Standfuss J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. U.S.A. 109, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P. M., Christopoulos A., Felder C. C., Gmeiner P., Steyaert J., Weis W. I., Garcia K. C., Wess J., Kobilka B. K. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Deupi X., Standfuss J. (2011) Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr. Opin. Struct. Biol. 21, 541–551 [DOI] [PubMed] [Google Scholar]
- 84. Chang A. L. S., Oro A. E. (2012) Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch. Dermatol. 148, 1324–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Buonamici S., Williams J., Morrissey M., Wang A., Guo R., Vattay A., Hsiao K., Yuan J., Green J., Ospina B., Yu Q., Ostrom L., Fordjour P., Anderson D. L., Monahan J. E., Kelleher J. F., Peukert S., Pan S., Wu X., Maira S.-M., García-Echeverría C., Briggs K. J., Watkins D. N., Yao Y.-M., Lengauer C., Warmuth M., Sellers W. R., Dorsch M. (2010) Interfering with resistance to Smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2, 51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Brinkhuizen T., Reinders M. G., van Geel M., Hendriksen A. J. L., Paulussen A. D. C., Winnepenninckx V. J., Keymeulen K. B., Soetekouw P. M. M. B., van Steensel M. A. M., Mosterd K. (2014) Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J. Am. Acad. Dermatol. 71, 1005–1008 [DOI] [PubMed] [Google Scholar]
- 87. Chen W. (2004) Activity-Dependent Internalization of Smoothened mediated by β-arrestin 2 and GRK2. Science 306, 2257–2260 [DOI] [PubMed] [Google Scholar]
- 88. Chen Y., Sasai N., Ma G., Yue T., Jia J., Briscoe J., Jiang J. (2011) Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of Smoothened. PLoS Biol. 9, e1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]