

Background: Excessive scar formation is a debilitating complication underlying many diseases of the eye, lungs, and skin.
Results: We identified salinomycin as an inhibitor of TGFβ-dependent cell signaling.
Conclusion: Salinomycin and other polyether ionophores block TGFβ-driven fibroblast proliferation and myofibroblast formation.
Significance: There are few effective therapies to combat excessive scarring, and salinomycin is a novel therapeutic candidate.
Keywords: Collagen, Fibrosis, Myofibroblast, p38 MAPK, SMAD Transcription Factor, MKK6, TGFβ, p38, Polyether Ionophores, Salinomycin
Abstract
Although scarring is a component of wound healing, excessive scar formation is a debilitating condition that results in pain, loss of tissue function, and even death. Many tissues, including the lungs, heart, skin, and eyes, can develop excessive scar tissue as a result of tissue injury, chronic inflammation, or autoimmune disease. Unfortunately, there are few, if any, effective treatments to prevent excess scarring, and new treatment strategies are needed. Using HEK293FT cells stably transfected with a TGFβ-dependent luciferase reporter, we performed a small molecule screen to identify novel compounds with antiscarring activity. We discovered that the polyether ionophore salinomycin potently inhibited the formation of scar-forming myofibroblasts. Salinomycin (250 nm) blocked TGFβ-dependent expression of the cardinal myofibroblast products α smooth muscle actin, calponin, and collagen in primary human fibroblasts without causing cell death. Salinomycin blocked phosphorylation and activation of TAK1 and p38, two proteins fundamentally involved in signaling myofibroblast and scar formation. Expression of constitutively active mitogen activated kinase kinase 6, which activates p38 MAPK, attenuated the ability of salinomycin to block myofibroblast formation, demonstrating that salinomycin targets the p38 kinase pathway to disrupt TGFβ signaling. These data identify salinomycin and other polyether ionophores as novel potential antiscarring therapeutics.
Introduction
Scarring is a consequence of the normal wound healing response. However, scar formation can be exuberant, leading to hypertrophic scarring and/or fibrosis that can ultimately lead to a loss of tissue function (1–4). Although there is a major knowledge gap as to why scarring sometimes proceeds out of control, hypertrophic scarring usually results from physical injury, such as laceration or surgery, or from burns, either induced thermally or chemically or by radiation (5). For example, an unfortunate consequence of severe thermal burns is the development of debilitating hypertrophic scars (6). Chronic inflammation and autoimmune disease can also lead to aberrant tissue reorganization and scarring (7, 8). Thyroid eye disease (TED)2 is an example of an autoimmune disease in which immune cells target the muscle and connective tissue in the ocular orbit, leading to orbital tissue remodeling and excessive scarring (3, 9). Although aberrant scarring is observed in numerous pathologies, there are few, if any, effective therapies that limit or prevent scarring.
The key effector cell in scar formation is the contractile and secretory myofibroblast (10). Myofibroblasts can be derived from tissue-resident fibroblasts, epithelial-mesenchymal transitions, circulating fibrocytes, mesenchymal stem cells, or other progenitor cells (11). Myofibroblasts highly express α-smooth muscle actin (αSMA), an important protein required for wound contraction, and these cells produce large amounts of extracellular matrix material, including collagen, fibronectin, and glycosaminoglycans (12, 13). The contractile properties of myofibroblasts and their excessive production of extracellular matrix material such as collagen result in disruptive tissue remodeling. In addition to their contractile phenotype, myofibroblasts also secrete a variety of cytokines, including IL-6, MCP-1, and TGFβ, that recruit immune cells and lead to further myofibroblast formation (14). Myofibroblasts are fundamentally involved in scar formation, making them ideal targets for new therapeutic options to limit or even reverse scarring.
Myofibroblast differentiation is mainly driven by the cytokine TGFβ, which is produced during the natural healing process (15, 16). TGFβ is also involved in many other cellular responses, including immune suppression, cell migration, and extracellular matrix remodeling (17). Although TGFβ is normally tightly regulated at multiple levels to limit its powerful effects, TGFβ is highly expressed in conditions such as cancer, chronic inflammation, and fibrosis (17). TGFβ regulates numerous cell signaling pathways while driving myofibroblast formation. One key pathway activated by TGFβ is Smad-dependent signaling. Smads are a family of highly conserved transcription factors that regulate the expression of many genes that contain Smad-binding element (SBEs) in their promoter and/or regulatory regions (18). TGFβ binding to its cognate receptor on the cell surface triggers phosphorylation of the Smad family members Smad2 and Smad3. When phosphorylated, these transcription factors bind their binding partner, the cosmad Smad4. This Smad complex is then shuttled into the nucleus, where it activates transcription of myofibroblast genes, including αSMA, calponin, and collagen (19–21).
The goal of this study was to identify a small molecule that could disrupt TGFβ-dependent myofibroblast formation and, thereby, mitigate excessive scarring and fibrosis. Although many attempts have been undertaken, there are few, if any, proven effective therapies for scarring. Our results reveal that the polyether ionophore, salinomycin, is a potent inhibitor of TGFβ-dependent human myofibroblast formation. Therefore, our results provide new evidence that salinomycin and other small molecules of its class are potential antiscarring compounds.
EXPERIMENTAL PROCEDURES
Cell Culture
Primary human fibroblasts were acquired and cultured as described previously (22). HEK293FT cells were obtained from the ATCC and cultured in DMEM supplemented with 10% fetal calf serum (Hyclone) and antibiotics. DMEM, minimum Eagle's medium, and hygromycin were purchased from Invitrogen. Fibroblast growth medium was purchased from Promocell. Other compounds used were salinomycin (Cayman) and narasin, monensin, and clioquinol (Sigma). Recombinant human TGFβ was obtained from R&D Systems and was used at final concentrations of either 1, 5, or 10 ng/ml. Antibodies against αSMA and FLAG were purchased from Sigma. Antibodies against TAK1, phospho-TAK1, and collagen were from Santa Cruz Biotechnologies. Antibodies against Smad2, phosho-Smad2, p38, phospho-p38, cleaved PARP, and β-tubulin were from Cell Signaling Technology.
Development of a TGFβ-responsive Cell Line
The minimal thymidine kinase promoter was amplified by PCR with a forward primer that contained four tandem SBEs (forward, 5′-AGGTACCTACTAAGTCTAGACGGCAGTCTAGACGTACTAAGTCTAGACGGCAGTCTAGACGTAGAGCTCGGCCCCGCCCAGCGTCTTGTC-3′; reverse, 5′-TAAAGCTTCTCGAGATCTGCGGCACGCT-3′), with restriction sites underlined. The resultant PCR product was TOPO cloned (Invitrogen), and the correct insert was verified by DNA sequencing. The construct was digested with SacI-HindIII, and the SBEx4-TK insert was purified and ligated with the pGL4.15 vector (Promega). Clones were verified by restriction digestion and then tested for TGFβ responsiveness in transient transfections of 293FT cells. When the pSBEx4-TK-luc construct was demonstrated to be TGFβ-responsive, the construct was introduced into 293FT cells for stable cell line production. Clones were selected by treatment with 200 μg/ml hygromycin. Twenty clones were selected and screened for TGFβ-induced luciferase activity. One clone that was robustly responsive was subsequently used in the small molecule screen.
Plasmid DNA Transfection
Plasmid DNA was introduced into human fibroblasts by electroporation. Plasmids were electroporated with an Amaxa nucleofector (program U-025) into 1 × 106 cells. The pcDNA3-FLAG MKK6(glu) plasmid, which encodes constitutively active MKK6 kinase S207E and T211E mutations, was obtained from Addgene (plasmid 13518 from the laboratory of Roger Davis). After transfection, cells were cultured for 12–24 h, and the culture medium was subsequently changed to treatment conditions.
Western Blotting
Total protein was isolated from 0.5–2 × 106 cells and lysed in 60 mm Tris (pH 6.8) and 2% SDS containing 1× protease inhibitor mixture (Sigma). The lysates were passed through a 26-gauge needle five to six times to shear genomic DNA. Protein concentrations were determined using the detergent-compatible protein assay (Bio-Rad). Total protein (1–10 μg/lane) was subjected to SDS-PAGE. Protein gels were transferred to a PVDF membrane (Millipore) and probed with antibodies as specified. Western blot band intensities were quantified using ImageLab software (Bio-Rad). Protein expression was normalized to β-tubulin levels.
Collagen Production Assay
Cell culture supernatant was collected and transferred (5–20 μl) to a PVDF membrane using a slot blot device. The membrane was blocked and probed with a goat anti-collagen antibody (1:5000), washed, and incubated with a donkey anti-goat antibody conjugated to HRP. Band intensities were quantified using ImageLab software, and values were normalized to vehicle treatments.
Alamar Blue Viability Assay
5 × 103 cells/well were plated in a 96-well plate (Griener) with 200 μl of culture medium. Vehicle (DMSO), salinomycin, and/or TGFβ (1 ng/ml) were added as indicated, and then 20 μl of Alamar Blue reagent was added to all wells. Cells were incubated for 24 and 48 h, and then the fluorescence of the oxidized Alamar reagent was measured (excitation 470 nm, emission 480 nm). Background fluorescence was subtracted from all wells, and the fluorescence was normalized to vehicle-treated cells. The assay was performed in two different human fibroblast strains, and treatments were performed in triplicate.
BrdU Incorporation Assay
Human fibroblasts were seeded on a 96-well plate at a density of 1 × 105 cells/well. Cells were treated in triplicate. Cell proliferation was determined using the BrdU assay following the instructions of the manufacturer. Briefly, cells were treated with a BrdU label at a 1:2000 dilution for 24 h after the initial 72-h treatment with TGFβ with or without drugs. BrdU incorporation was measured using the BrdU cell proliferation assay kit (Calbiochem, San Diego, CA) at 450–540 nm using a Varioskan microplate reader.
Luciferase Reporter Assays
Primary human fibroblasts were collected and electroporated as described above with pSBEx4-TK-luc and CMV-Renilla luciferase (Promega, Madison, WI). Nucleofected cells were plated and allowed to grow for 12–24 h. DMSO (vehicle), salinomycin (250 nm), and/or TGFβ (1 ng/ml) were added to the cultures for an additional 24–36 h. Following incubation, cells were washed two times in 1× PBS and lysed directly in plates using the Dual-Glo luciferase assay buffer (Promega). Firefly and Renilla luciferase readings were measured on a Varioskan Flash luminescent plate reader (Thermo Fisher) following the instructions of the manufacturer. Luciferase readings were normalized to the control vehicle-treated samples for statistical analysis.
Collagen Contraction Assay
Experiments were conducted using the collagen contraction assay from Cell Biolabs, Inc. following the protocol for the manufacturer. Briefly, 5 × 105 cells and 500 μl of collagen suspension per well were plated in a 24-well plate. After collagen gel polymerization, vehicle (ethanol), salinomycin, or narasin (250 nm) were added for 1 h before TGFβ (5 ng/ml) treatment. Gels were detached from well and photographed at time 0, 24, 48 and 72 h. Contraction was measured by analyzing the gel area at each time point using ImageJ software. Gels treated with vehicle and TGFβ only were set to 100%.
Wound Healing Assay
Six-well plates of human fibroblasts were grown in normal culture medium until they reached confluence. The medium was then replaced with low-serum (0.05%) medium for 24 h, and a scratch wound was made using a micropipette tip. After wounding, cells were treated with either vehicle (ethanol) or salinomycin (250 nm) for 1 h before treatment with TGFβ (5 ng/ml). The wound area was photographed by brightfield microscopy at 0, 24, 48, and 72 h. The area of each wound was measured using ImageJ software. Wound areas at 24, 48, and 72 h were normalized to the area of the wound at time 0.
Statistical Analysis
Data were analyzed using GraphPad Prism software, and Student's t test and One-way analysis of variance were used for statistical analysis. p < 0.05, p < 0.01, and p < 0.001 were considered significant.
RESULTS
Excessive scarring results from the formation of too many myofibroblasts and the production of too much extracellular matrix material such as collagen. Because TGFβ drives the formation of myofibroblasts in part through activating the Smad pathway, we designed a reporter construct that served as a measure of TGFβ-induced Smad activity (Fig. 1A). We inserted four tandem SBEs upstream of the minimal thymidine kinase (tk) promoter. The SBEx4-tk promoter was inserted into the pGL4.15 vector, which contains a destabilized firefly luciferase gene (Luc2P) and the hygromycin resistance gene (Hygro). The pSBEx4-tk-luc-Hygro plasmid (Fig. 1A) was then introduced into HEK293FT cells by lipofection. Stable clones were generated and screened for TGFβ-induced luciferase activity. One clone was selected that consistently gave a 15- to 20-fold induction of luciferase activity when cells were treated with TGFβ for 24 h (Fig. 1B). To find compounds that inhibit TGFβ activity and, therefore, may be novel anti-scarring compounds, we screened the Spectrum collection of 2300 small molecules with the cell line. Several small molecules inhibited TGFβ-dependent luciferase activity (Fig. 1C). One small molecule that was particularly potent at inhibiting luciferase activity was compound L18, which corresponded to the polyether ionophore salinomycin. Further testing of salinomycin showed a dose-dependent decrease in TGFβ-induced luciferase, where 100 nm salinomycin reduced luciferase activity ∼4-fold, and 1 μm salinomycin reduced luciferase activity levels to below baseline levels (Fig. 1D). Salinomycin, an antibiotic produced by Streptomyces albus is used as coccidiostatic agent in animal feed (23). Furthermore, salinomycin has been shown recently to have activity against cancer stem cells (24). Therefore, we proceeded to further investigate the potential of salinomycin as an antiscarring agent.
FIGURE 1.

A small molecule screen identifies salinomycin as a potential antiscarring compound. A, the Smad-dependent reporter construct. Four tandem SBEs were inserted upstream of the minimal tk promoter. Downstream of the promoter is a destabilized version of the firefly luciferase gene (luc2P) present in the pLuc2P-Hygro plasmid that also harbors the hygromycin resistance gene. B, the reporter plasmid was introduced into the HEK293FT cell line, and individual colonies were selected with hygromycin (200 μg/ml) to develop a Smad/TGFβ-dependent luciferase reporter cell line. 1 ng/ml treatment of TGFβ for 24 h resulted in a robust increase of luciferase activity. C, the HEK293FT-luc reporter line was screened with the 2300-compound Spectrum collection of small molecules. Several hits from the initial screen that blocked TGFβ-induced luciferase activity were tested further in a Dual-Luciferase screen including a constitutive Renilla luciferase to normalize the SBE luciferase activity. Compound L18, which corresponded to salinomycin, a polyether ionophore antibiotic, inhibited the reporter construct more than 10-fold. Unt., untreated; Veh, vehicle; comp., compound. D, salinomycin (two-dimensional structure, right) exhibited a dose-dependent decrease in TGFβ-induced SBE-luciferase activity. 1 μm salinomycin reduced SBE luciferase activity to below baseline levels, whereas 100 nm salinomycin reduced SBE luciferase activity more than 3-fold. Experiments were repeated three times in triplicate, and similar results were observed in all tests. *, p < 0.01 versus untreated cells; #, p < 0.01 versus TGFβ with vehicle.
Fibroblasts are sentinel cells that respond to numerous stimuli and serve as key effector cells in many biological processes (25). One crucial function of fibroblasts is their differentiation into scar-forming myofibroblasts that are involved in wound healing but, in excess, cause scarring and fibrosis. TED is a disorder in which myofibroblasts and scar tissue accumulate in the ocular orbit, causing pain, proptosis, and, in severe cases, blindness (7). We have shown previously that human orbital fibroblasts dramatically respond to TGFβ by forming myofibroblasts that express αSMA and calponin and produce high levels of collagen (26). Therefore, TED fibroblasts were an ideal model to test the ability of salinomycin to block TGFβ function. We treated human TED fibroblasts with TGFβ (1 ng/ml) in the presence or absence of 0.1 and 1 μm salinomycin (compound L18 from the Spectrum library) or various doses of other small molecule hits from the Spectrum library for 72 h to allow the formation of myofibroblasts. After 72 h, cells were harvested and analyzed by Western blot. As expected, the fibroblasts robustly responded to TGFβ, inducing expression of αSMA and calponin 36- and 10-fold, respectively, over vehicle-treated cells (Fig. 2A). Interestingly, salinomycin completely blocked the expression of αSMA and calponin in a dose-dependent manner (Fig. 2A). To further characterize the effect of salinomycin, a dose-response experiment was carried out in which human fibroblasts were treated with TGFβ and doses of salinomycin from 10–250 nm for 72 h (Fig. 2B). At 50 nm salinomycin, expression of αSMA and calponin was reduced 4-fold and 3-fold, respectively, over TGFβ treatment alone. Furthermore, at 250 nm salinomycin, αSMA and calponin expression was reduced ∼16-fold and 5.2-fold, respectively, to levels at or below untreated fibroblasts (Fig. 2B). To further investigate the timing of the effect of salinomycin on TGFβ-induced myofibroblast formation, fibroblasts were treated with salinomycin (250 nm) and TGFβ for 0, 24, 48, and 72 h before cell harvest and subsequent analysis of αSMA and β-tubulin by Western blot (Fig. 2C). TGFβ induced αSMA at 24, 48, and 72 h of treatment, with a maximal induction at 72 h (13-fold over vehicle treatment). Salinomycin blocked TGFβ-induced αSMA expression at 24, 48, and 72 h, with more than a 20-fold inhibition at 72 h.
FIGURE 2.
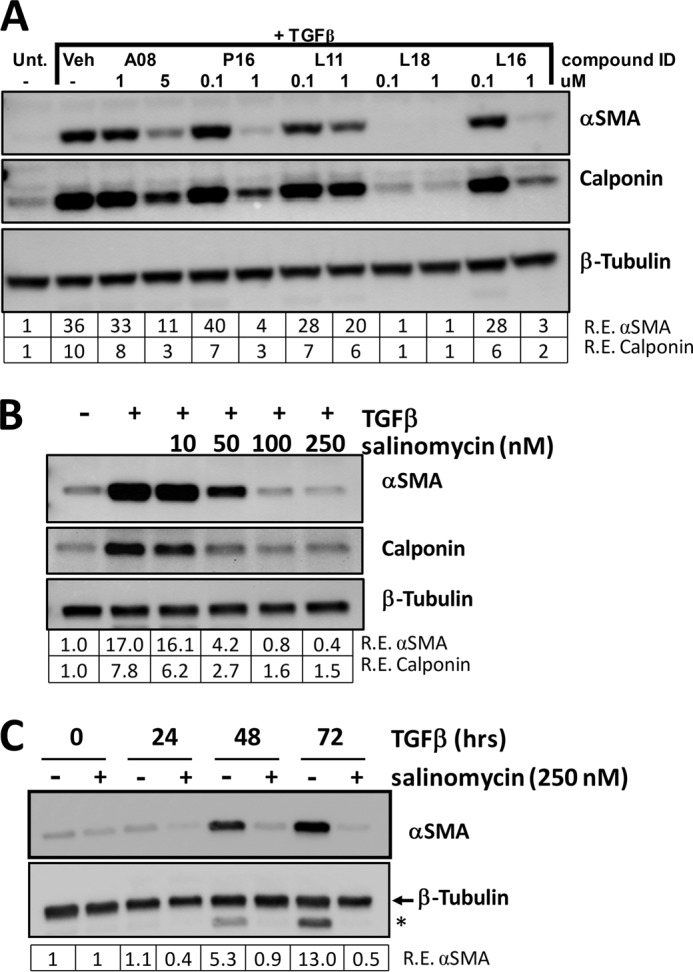
Salinomycin inhibits TGFβ-induced expression of myofibroblast markers in primary human fibroblasts. A, human fibroblasts were treated with two different concentrations of five small molecules from the Spectrum collection and treated with TGFβ (1 ng/ml) for 72 h. The cells were then lyses and analyzed by Western blot for the myofibroblast markers αSMA and calponin. β-tubulin was used as a loading control. Relative expression (R.E.) of αSMA and calponin (both normalized to β-tubulin) as determined by densitometry is shown below each lane. Salinomycin (compound ID L18) dramatically reduced expression of both myofibroblast markers to at or below untreated (Unt.) fibroblast expression levels. Veh, vehicle. B, to determine the minimal dose at which salinomycin effectively inhibited expression of αSMA and calponin, human fibroblasts were treated with salinomycin (10–250 nm) and TGFβ (1 ng/ml) for 72 h, and cells were isolated and analyzed as in A. 250 nm salinomycin inhibited expression of αSMA and calponin by more than 17- and 5-fold, respectively. C, to determine the time points at which salinomycin inhibits myofibroblast marker expression, human fibroblasts were treated with vehicle or 250 nm salinomycin in the presence of 1 ng/ml TGFβ, and then cells were harvested at 0, 24, 48, and 72 h. Cell extracts were collected and analyzed by Western blot for αSMA and β-tubulin. The tubulin band is indicated by the arrow, and the asterisk denotes the signal from αSMA expression. Salinomycin inhibits expression of αSMA at 24, 48, and 72 h. Experiments were repeated using at least three different primary human fibroblast strains, with similar results seen in all strains. A representative experiment is shown.
Another key role of myofibroblasts in scar formation is the production of collagen. To test whether salinomycin could block myofibroblast collagen production, human fibroblasts were treated with TGFβ and salinomycin (10–250 nm) for 72 h, and then the culture medium was collected and analyzed for collagen I levels using a specific collagen I antibody and slot blot analysis (Fig. 3A). As expected, TGFβ-induced myofibroblasts produce high levels of collagen, and salinomycin blocked production of collagen in a dose-dependent manner. Starting at 10 nm salinomycin, collagen production decreased, and, at 100 nm salinomycin, collagen production was back to baseline levels. Finally, at 250 nm salinomycin, collagen production was below baseline levels of untreated fibroblasts (Fig. 3A). Additionally, a time course experiment was performed with fibroblasts treated with TGFβ or TGFβ plus salinomycin (250 nm), and the culture medium was collected at 5, 24, 48, and 72 h. As expected, TGFβ induced collagen production in a time-dependent manner in human fibroblasts (Fig. 3B). Interestingly, salinomycin reduced collagen production at 24, 48, and 72 h, with a more than 4-fold reduction in collagen production at 72 h.
FIGURE 3.
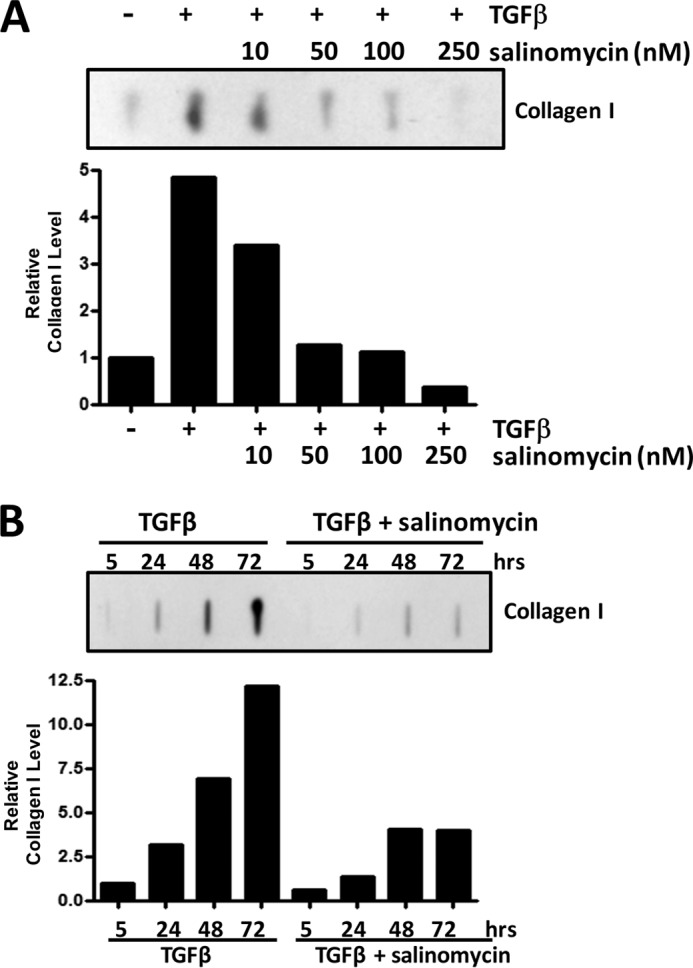
Salinomycin prevents TGFβ-induced collagen production in primary human fibroblasts. A, primary human fibroblasts were treated with salinomycin (10–250 nm) and TGFβ (1 ng/ml) for 72 h, and then cell culture supernatants were collected and analyzed for collagen I levels by slot blot analysis. Salinomycin reduced collagen I levels in a dose-dependent manner, with salinomycin (100 and 250 nm) reducing collagen production to baseline levels. B, fibroblasts were treated with 250 nm salinomycin and TGFβ (1 ng/ml), and cell culture supernatants were collected at 5, 24, 48, and 72 h to analyze production of collagen I. Salinomycin prevented collagen production at 24, 48, and 72 h. Experiments were performed in at least three primary human fibroblast strains, with representative strains shown.
Because salinomycin, a polyether ionophore, prevented formation of myofibroblasts, we next tested other ionophores for anti-myofibroblast activity (Fig. 4A). Narasin is a methylated derivative of salinomycin that is also a coccidiostat used in animal feed (27). Monensin is another polyether ionophore that is used extensively in animal feed to prevent coccidiosis (27). Finally, we chose a fourth compound, clioquinol, which is also an antiprotozoal drug and ionophore but it is structurally unrelated to salinomycin and other polyether ionophores (28). We initially tested the ability of these ionophores to inhibit the luciferase activity of our TGFβ reporter cell line. We observed that, like salinomycin, narasin and monensin inhibited luciferase expression, whereas clioquinol did not (data not shown). To further test these four compounds, we treated human fibroblasts with TGFβ in the absence or presence of 250 nm of salinomycin, narasin, clioquinol, or monensin for 72 h. After treatment, cell lysates were harvested and analyzed for the expression of the myofibroblast markers αSMA and calponin (Fig. 4B). As expected, salinomycin reduced the expression of αSMA and calponin to lower than vehicle levels. Interestingly, both narasin and monensin had a similar activity as salinomycin. However, the structurally unrelated compound clioquinol did not inhibit expression of αSMA or calponin (Fig. 4B). Therefore, these data support that salinomycin and other polyether ionophores specifically block TGFβ-induced myofibroblast formation. To further characterize the ability of narasin to block TGFβ-induced myofibroblast formation, three doses of narasin (50, 100, and 250 nm) and salinomycin (250 nm) were used to block TGFβ effects (Fig. 4C). As expected, TGFβ induced αSMA and calponin expression by more than 10- and 5-fold, respectively, over vehicle-treated cells, and narasin effectively blocked αSMA and calponin expression at 50, 100, and 250 nm (Fig. 4C).
FIGURE 4.

The polyether ionophores salinomycin, narasin, and monensin inhibit myofibroblast formation. A, molecular structures of the ionophores narasin, monensin, and clioquinol. Narasin, a methylated derivative of salinomycin, and monensin are polyether ionophores, whereas clioquinol is an unrelated ionophore. B, human fibroblasts were treated with vehicle (DMSO), TGFβ, or TGFβ plus 250 nm of the indicated compounds for 72 h and then analyzed for myofibroblast markers by Western blot. Salinomycin (SNC), narasin (NAR), and monensin (MNS) all inhibited expression of αSMA and calponin, whereas clioquinol (CLQ) did not. Relative expression (R.E.) of αSMA and calponin (both normalized to β-tubulin) as determined by densitometry is shown below each lane. C, human fibroblasts were treated with 10–250 nm narasin or 250 nm salinomycin and TGFβ (1 ng/ml) for 72 h, and then cells were isolated and analyzed as in B. Salinomycin (250 nm) inhibited expression of αSMA and calponin 10- and 5-fold, respectively, whereas narasin exhibited a dose-dependent decrease in expression of the myofibroblast markers, where 250 nm narasin inhibited expression of αSMA and calponin by more than 10- and 5-fold, respectively. Experiments were repeated in two different strains, with representative results shown.
In addition to showing that salinomycin inhibits expression of αSMA, calponin, and collagen, we next tested the ability of salinomycin and narasin to inhibit myofibroblast contraction of a collagen gel matrix (Fig. 5A). Myofibroblast contraction is an important function of myofibroblasts in wound healing, and excessive contraction of unwanted myofibroblasts leads to debilitating scar formation (14, 29). Salinomycin (250 nm) and narasin (250 nm) significantly inhibited TGFβ-induced myofibroblast contraction by 40–80% after 2–3 days of TGFβ treatment (Fig. 5A). As a second test of myofibroblast function, we measured cell migration after an induced scratch in a monolayer of fibroblasts. After the scratch was made, cells were treated with TGFβ in the presence or absence of salinomycin. After 3 days of TGFβ treatment alone, the open area of the wound was reduced to ∼15% of the original wound size because of the growth and migration of myofibroblasts (Fig. 5B). Salinomycin (250 nm) significantly blocked TGFβ-induced myofibroblast growth and migration more than 3-fold.
FIGURE 5.

Salinomycin blocks TGFβ-induced myofibroblast contraction and migration. A, human fibroblasts were cultured in a collagen gel matrix as described in the text. After collagen gel formation, cells were treated with TGFβ (5 ng/ml) or TGFβ plus either salinomycin (SNC, 250 nm) or narasin (Nar, 250 nm) and allowed to contract for 72 h. Gel matrices were imaged at the 0, 24, 48, and 72 h time points. The gel area was quantified using ImageJ. TGFβ-induced contraction was set to 100%, and all other measurements were normalized to it. Representative images are shown, demonstrating that TGFβ readily induced myofibroblast contraction, whereas salinomycin and narasin inhibit contraction by more than 80% and 90%, respectively, 72 h post-TGFβ. *, p < 0.01 versus TGFβ-treated cells. B, human fibroblasts were cultured in a 12-well culture dish until they reached a confluent monolayer. A scratch wound was then induced in the cultures, and wells were washed to remove cells and debris. Cells were treated with TGFβ (5 ng/ml) or TGFβ plus salinomycin (250 nm) for 72 h to allow myofibroblast migration. Open areas were quantified using ImageJ software. Areas were normalized to time 0 (area = 1.0 at time 0). Images were captured at 0, 24, 48, and 72 h. After 72 h, TGFβ-treated cells had reduced the scratched area to less than 20% of the original area. Salinomycin treatment prevented myofibroblast migration 3- to 4-fold over TGFβ only. *, p < 0.01 versus TGFβ-treated cells. Experiments were repeated in triplicate, with two different strains and representative images shown.
Although salinomycin and other polyether ionophores blocked myofibroblast formation and myofibroblast function, we tested whether these effects were a result of toxicity and/or because of a block of cell proliferation. Our first analysis was to visually inspect fibroblasts treated with vehicle, TGFβ, or TGFβ plus salinomycin (250 nm) for 72 h using brightfield microscopy (Fig. 6A). Representative images (original magnification ×200) from two different primary human fibroblast strains demonstrate that TGFβ induces hallmark morphological changes of fibroblasts into myofibroblasts. However, salinomycin treatment prevented morphological changes induced by TGFβ treatment (Fig. 6A, left panels). Additionally, visualization of salinomycin-treated cells indicated that salinomycin did not lead to cell death, and cells appeared morphologically similar to vehicle-treated fibroblasts. To further quantitatively measure that salinomycin did not affect cell viability, human fibroblasts were treated with vehicle (DMSO), 50–250 nm salinomycin alone, TGFβ, or TGFβ plus 250 nm salinomycin. As a positive control, fibroblasts were treated with the cytotoxic drug puromycin (5 μg/ml). Treated cells were cultured in the presence of Alamar Blue reagent, which measures mitochondrial oxidation-reduction (redox) potential and, therefore, serves as a quantitative viability sensor. After 72 h of culture, Alamar Blue fluorescence was measured to assay cell viability (Fig. 6B). As expected, puromycin treatment resulted in a total loss of cell viability. However, treatment with salinomycin alone or salinomycin plus TGFβ did not result in a loss of viability. These results demonstrate that salinomycin, at the indicated doses, does not affect human fibroblast viability. We also tested whether salinomycin modifies basal or TGFβ-induced fibroblast proliferation. Cells were treated with vehicle or 250 nm of either salinomycin or narasin for 24 h in the presence of BrdU to measure cell proliferation (Fig. 6C). Neither salinomycin nor narasin blocked basal fibroblast proliferation. As expected, TGFβ induced human fibroblast proliferation 9-fold. Remarkably, both salinomycin and narasin were able to beneficially block TGFβ-induced proliferation 5-fold (Fig. 6C). Although we did not observe any decreases in basal fibroblast proliferation in cells treated with salinomycin, we further investigated whether salinomycin may be triggering apoptosis in fibroblasts treated with or without TGFβ. Several recent reports have demonstrated that salinomycin can induce apoptosis in cancer cells at concentrations of over 5 μm (30, 31). To determine whether salinomycin triggers apoptosis in human fibroblasts at the doses needed to block myofibroblast formation, we measured production of cleaved PARP, a hallmark of apoptosis (Fig. 6D). As a positive control, cells were treated with the apoptosis-inducing drug puromycin. As expected, puromycin treatment resulted in a large induction of cleaved PARP production. Salinomycin, at doses of 50–500 nm, did not induce cleaved PARP production in human fibroblasts treated with or without TGFβ. This result suggests that salinomycin is not working through apoptosis to prevent myofibroblast formation.
FIGURE 6.

Salinomycin does not affect viability or basal proliferation of human fibroblasts but does block TGFβ-induced proliferation. A, human fibroblasts were treated with vehicle (DMSO), TGFβ (1 ng/ml), or TGFβ plus 250 nm salinomycin (SNC) and cultured for 72 h. After culture, cell images were taken (original magnification ×200). Two different primary fibroblast strains at two different culture confluence states are shown. TGFβ induces morphology changes in human fibroblasts, indicative of myofibroblast formation. Cotreatment with salinomycin effectively blocked the morphological changes induced by TGFβ, and fibroblasts appeared similar to vehicle-treated fibroblasts. B, human fibroblasts were treated with vehicle (Veh, DMSO); 50, 100, or 250 nm salinomycin; TGFβ (1 ng/ml); and TGFβ plus 250 nm salinomycin or puromycin (P) (5 μg/ml) for 72 h in the presence of the redox-sensitive fluorescent dye Alamar Blue. After 72 h, fluorescence was measured to analyze cellular viability. RFU, relative fluorescent units. Puromycin, which served as a positive control, resulted in a total loss of cell viability, whereas salinomycin did not significantly affect cell viability at the tested doses either in the presence or absence of TGFβ. C, human fibroblasts were treated with vehicle (DMSO), 250 nm salinomycin, 250 nm narasin (Nar), TGFβ alone, or TGFβ plus salinomycin or narasin (250 nm) for 24 h before addition of BrdU. BrdU treatment was carried out for an additional 24 h, and then cells were fixed and stained for BrdU incorporation, which served as a measure of cell proliferation. Salinomycin and narasin did not affect basal fibroblast proliferation (first three columns). However, salinomycin and narasin significantly blocked TGFβ-induced proliferation (last three columns). Results are from a representative experiment performed in triplicate. *, p < 0.01 versus vehicle; #, p < 0.01 versus TGFβ treatment. D, primary human fibroblasts were treated with salinomycin (50–500 nm) in the presence or absence of TGFβ (1 ng/ml) for 72 h, and then cell extracts were collected and analyzed by Western blot for the apoptotic marker cleaved PARP. β-tubulin was used as a loading control. Cells were treated with puromycin as a positive control to induce apoptosis. As expected, puromycin induced cleaved PARP levels. However, salinomycin at the doses used (50–500 nm) did not induce PARP cleavage.
To further characterize the molecular mechanisms by which salinomycin blocks TGFβ-induced myofibroblast formation, we analyzed the activation kinetics of some of the key mediators of TGFβ signaling (18). Because we first identified salinomycin as a putative antiscarring molecule by its ability to block TGFβ-induced Smad activity, we analyzed the levels of phospho-Smad2 by Western blot in cells treated with TGFβ or TGFβ plus salinomycin (Fig. 7A). TGFβ treatment rapidly induced phosphorylation of Smad2 in human fibroblasts. Smad2 phosphorylation was completed ablated in the presence of the known TGFβ receptor inhibitor SB-43152. However, salinomycin treatment did not affect phospho-Smad2 levels at 1 h of TGFβ treatment (Fig. 7A). Because we saw that salinomycin did inhibit αSMA expression at longer time points (24–72 h) and because of the fact that our initial luciferase screen was performed at 24 h of treatment, we analyzed the expression of other key mediators of TGFβ-dependent myofibroblast differentiation at 24, 48, and 72 h. In addition to Smad activation, TGFβ induced phosphorylation and activation of TGFβ-activated kinase 1 (TAK1, also called MAP3K) and the MAPK p38 (Fig. 7, B and C). The effect of salinomycin on acute TGFβ-induced phosphorylation of p38 and TAK1 was analyzed at 10–60 min. Salinomycin blocked TGFβ-induced TAK1 phosphorylation at 10 and 30 min. Phosphorylation of p38 was induced at 60 min by TGFβ, and salinomycin had only modest effects at these acute time points. At longer treatment times, TGFβ treatment induced phospho-TAK1 2.4, 3.9, and 5.8-fold at 24, 48, and 72 h, respectively. Interestingly, salinomycin blocked phosphorylation of TAK1 at these time points, with a more than 3-fold inhibition at 72 h (Fig. 7C). Likewise, TGFβ induced phosphorylation of p38 at 24, 48, and 72 h, whereas salinomycin blocked phosphorylation of p38, with a more than 3-fold block at 72 h. We observed similar effects on TAK1, p38, and SMAD2 phosphorylation using narasin and monensin (data not shown). These data support the concept that salinomycin and other polyether ionophores do not directly target Smad signaling but, rather, target TGFβ-induced MAPK pathways through TAK1 and p38.
FIGURE 7.
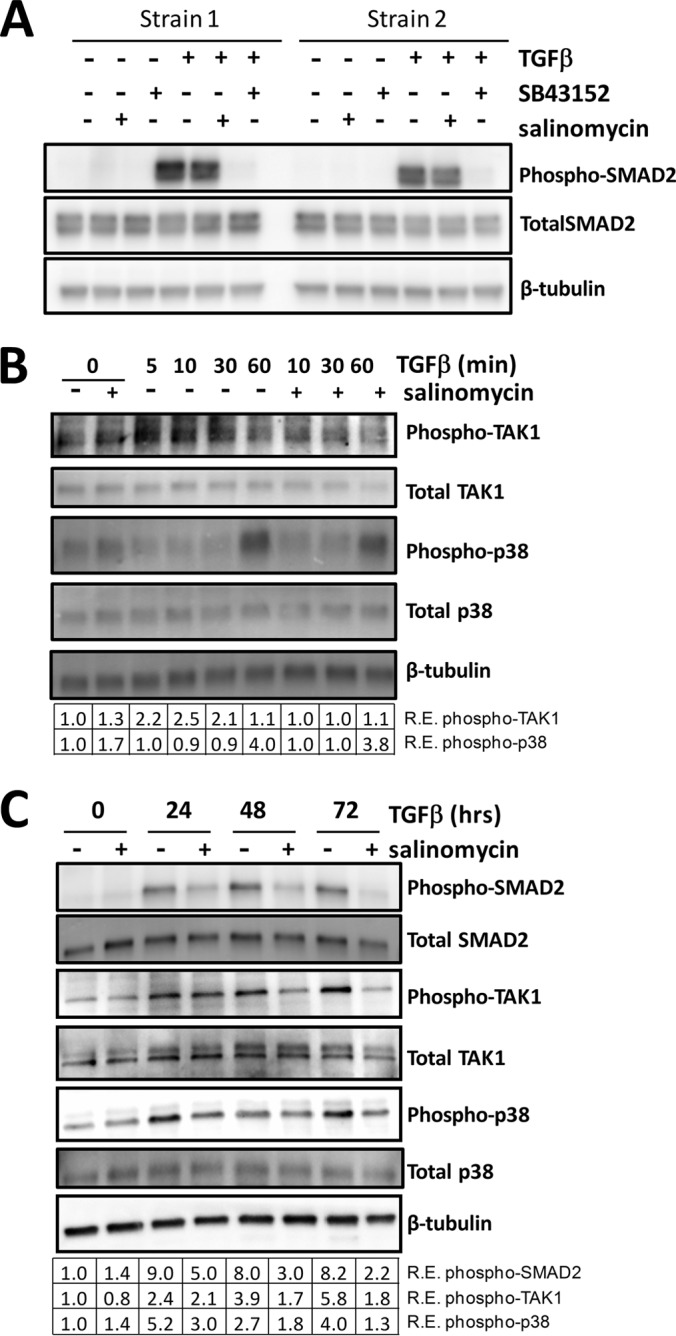
Salinomycin does not directly target TGFβ-induced SMAD2 phosphorylation but inhibits TGFβ-induced TAK1 and p38 phosphorylation. A, human fibroblasts were treated with vehicle, salinomycin, or SB-43152 with or without TGFβ. Cells were harvested at 1 h and analyzed for phospho-SMAD2, total SMAD2, and β-tubulin (loading control) by Western blot. Two representative strains are shown, demonstrating that, at 1 h of treatment, TGFβ induces phosphorylation of SMAD2 and SB-43152 completely ablates phospho-SMAD2, whereas salinomycin has no effect. B, fibroblasts were treated with TGFβ and/or salinomycin, and cells were harvested 0, 5, 10, 30, and 60 min after TGFβ treatment to analyze levels of phospho-TAK1, total TAK1, phospho-p38, and total p38 by Western blot. Salinomycin blocked phospho-TAK production 10 and 30 min after TGFβ treatment. R.E., relative expression. C, Human fibroblasts were treated as in A, except that cells were harvested at the 0, 24, 48, and 72 h time points to analyze levels of phospho-TAK1, totalTAK1, phospho-p38, total p38, phospho-SMAD2, and total SMAD2 by Western blot. The relative expression level of each phosphoprotein (normalized to β-tubulin) as determined by densitometry is shown below each lane. Salinomycin reduced TGFβ-induced phospho-Smad2 levels more than 2-fold at 48 h. Likewise, salinomycin also inhibited phospho-TAK1 levels at 24, 48, and 72 h. Salinomycin blocked p38 phosphorylation at the 24, 48, and 72 h time points, with an 80% reduction at 24 h and more than a 3-fold reduction at 72 h. Data are representative of at least three different experiments performed with different human fibroblasts strains.
To further test whether salinomycin targets the TAK1-p38 pathway to block myofibroblast formation, we used a plasmid encoding a mutant MKK6 (pcDNA3-FLAG MKK6(glu)) protein. MKK6 (also called MAP2K) is activated by TAK1 and phosphorylates p38 in response to extracellular signals such as TGFβ or other environmental stresses. The MKK6(glu) protein, which harbors glutamate mutations at amino acid residues serine 207 and threonine 211, is constitutively active and, therefore, phosphorylates p38 independently of upstream signals such as phospho-TAK1 (32). Therefore, if salinomycin is blocking the TAK1-p38 signaling pathway, then MKK6(glu) should overcome the effect of salinomycin. We first tested MKK6(glu) using our SBE4-TK-luc reporter plasmid. The luciferase reporter plasmid was introduced into human fibroblasts along with the MKK6(glu) plasmid or a control plasmid (pcDNA3-GFP). Cells were then treated with vehicle, TGFβ, or TGFβ plus 250 nm salinomycin. After 24 h, luciferase activity was measured (Fig. 8A). As expected, in human fibroblasts expressing the GFP plasmid, TGFβ induced luciferase activity 2-fold, and salinomycin completely blocked TGFβ-induced luciferase activity. Interestingly, in cells expressing constitutively active MKK6(glu) plasmid, salinomycin was unable to block TGFβ-induced luciferase activity (Fig. 8A). We further tested whether MKK6(glu) could attenuate the ability of salinomycin to inhibit p38 phosphorylation. Control or MKK6(glu) plasmids were introduced into human fibroblasts by electroporation, and then cells were treated with TGFβ for 72 h. Samples were analyzed for expression of FLAG-MKK6(glu), αSMA, calponin, and β-tubulin by Western blot (Fig. 8, B and C). As expected, TGFβ treatment induced expression of αSMA and calponin in both pGFP and pMKK6(glu) treated fibroblasts. Furthermore, in fibroblasts treated with pGFP, salinomycin significantly blocked TGFβ induction of αSMA and calponin (Fig. 8, B and C). Interestingly, expression of MKK6(glu) attenuated the ability of salinomycin to block TGFβ-induced αSMA and calponin 5- and 2-fold, respectively (Fig. 8C). Taken together, these data support the concept that salinomycin blocks the TAK1-p38 signaling pathway to prevent TGFβ induced Smad2/3-dependent signaling and formation of scar-forming myofibroblasts (Fig. 9).
FIGURE 8.

Expression of constitutively active MKK6 attenuates the inhibitory effect of salinomycin on TGFβ. A, a Smad-luc reporter, a constitutive CMV-Renilla reporter, and either a control plasmid (pcDNA-GFP) or the pcDNA3.1 FLAG-MKK6(glu) plasmid were introduced into human fibroblasts by electroporation. After electroporation, cells were treated with vehicle (Veh, DMSO), TGFβ, or TGFβ plus 250 nm salinomycin (SNC). After 24 h, cells were lysed, and luciferase activity was measured. Smad-luc activity was normalized to Renilla activity. As expected, TGFβ induced SBE-luc activity over vehicle treatment in both control plasmid and MKK6(glu) plasmid electroporated samples. Salinomycin blocked TGFβ-induced Smad-luc activity in control plasmid samples. However, the presence of MKK6(glu) attenuated the effect of salinomycin on Smad-luc activity. *, p < 0.01 in vehicle versus TGFβ; #, p < 0.01 in TGFβ versus TGFβ plus salinomycin for control pGFP electroporated cells. The experiment was repeated in triplicate in two different fibroblast strains. B, a control plasmid or the pcDNA3.1 FLAG-MKK6(glu) plasmid, which harbors the cDNA for a constitutively active MKK6 protein, was introduced into human fibroblasts by electroporation. Cells were then treated with vehicle, TGFβ, or TGFβ plus 250 nm salinomycin for 72 h. In cells expressing the control pGFP plasmid, TGFβ-induced αSMA and calponin expression was blocked by salinomycin. However, in cells expressing the MKK6(glu) plasmid, salinomycin did not block αSMA and calponin expression. The experiment was performed in three different fibroblast strains, with a representative experiment shown. WB, Western blot; R.E., relative expression. C, Western blot densitometry of the experiment shown in B. *, p < 0.01 in pGFP versus pMKK6(glu).
FIGURE 9.
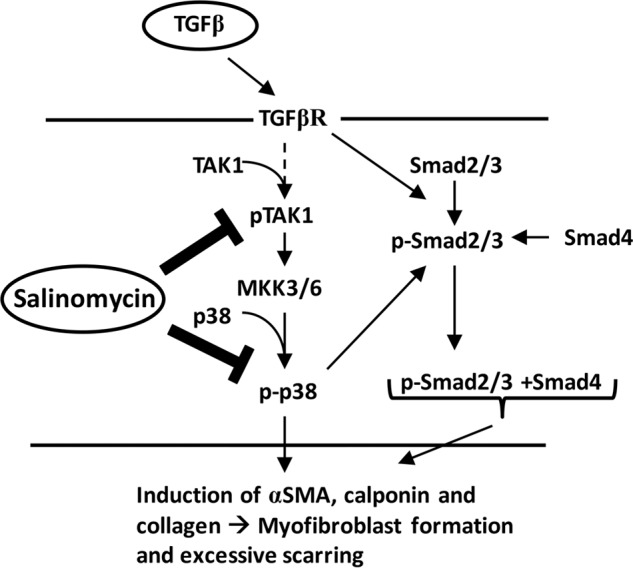
Potential model for the salinomycin mode of action. Active TGFβ, present at high concentrations during wound healing, exuberant scarring, or chronic inflammation, binds to the TGFβ receptor (TGFβR). Activation of the TGFβ receptor triggers a range of cell signaling events, including the phosphorylation and activation of TAK1 and Smad2/3. TAK activates MKK3/6 by phosphorylation, leading to phosphorylation and activation of p38. Active p38 leads to a further increase in Smad2/3 phosphorylation and activation. These signaling pathways reinforce the myofibroblast program, leading to excessive expression of αSMA, calponin, and collagen to promote myofibroblast generation and scar formation. Salinomycin or other polyether ionophores can block the activation and phosphorylation of TAK1 and p38, thereby limiting the activation of Smad2/3 and blocking myofibroblast formation.
DISCUSSION
Exuberant TGFβ signaling and excessive development of scar-forming myofibroblasts is at the root of disorders such as thyroid eye disease and idiopathic pulmonary fibrosis, which involve abnormal tissue remodeling and excessive scar formation. Unfortunately, therapeutic options to treat excessive scarring are limited, and most are not proven to be effective (33). The results presented in this report identify salinomycin and other polyether ionophores as novel small molecules that block myofibroblast formation. Salinomycin potently blocked TGFβ-induced expression of αSMA, calponin, and collagen, all of which are hallmarks of myofibroblasts. Salinomycin also effectively blocked myofibroblast function without affecting cell viability. The ability of salinomycin to inhibit myofibroblast function and block production of these myofibroblast markers has not been recognized previously. The knowledge gained from these studies highlights the potential of salinomycin and other polyether ionophores to serve as the basis for new anti-scarring drugs.
Heightened TGFβ signaling is observed in cancer, fibrosis, hypertrophic scarring, and TED (3, 7, 34). Additionally, TED fibroblasts, which are key effector cells in the disease, both proliferate at a faster rate and more readily form myofibroblasts in response to TGFβ than normal, non-TED fibroblasts (26, 36). Therefore, TGFβ signaling is paramount to the aberrant myofibroblast formation, collagen production, contraction, and ocular tissue remodeling underlying TED pathophysiology. Unfortunately, besides surgical removal of excessive ocular tissue, there are few, if any, effective therapies to combat the disease (7, 37, 38). Here we show that salinomycin blocks TGFβ-induced transformation of primary human TED fibroblasts to scar-forming myofibroblasts. Furthermore, salinomycin blocked TGFβ-induced collagen production, cell proliferation, contraction, and migration, key features of tissue remodeling and fibrosis (39, 40). Therefore, salinomycin, which we show acts robustly to target TGFβ signaling in human fibroblasts, could serve as the basis for new therapeutic options for the treatment of TED.
In our study, we screened a TGFβ-dependent Smad reporter cell line with a small molecule library consisting of bioactive drugs. Because Smad transcription factors are directly downstream of the TGFβ receptor, we expected to identify small molecules that could act directly on the TGFβ receptor or Smad proteins, such as the small molecule inhibitor of the TGFβ receptor, SB-43152 (41). Our data support the hypothesis that salinomycin indirectly targets the Smad pathway by working through the TAK1-p38 MAPK pathway. The TAK1-p38 pathway functions in myofibroblast formation in part by providing a feedforward loop to stimulate Smad2/3 phosphorylation and activation (Fig. 9) (42). Additionally, TAK1 has been shown recently to be essential for myofibroblast formation, and increased activity of TAK1 has been associated with kidney fibrosis (43, 44). These results are consistent with our new data showing that salinomycin blocks myofibroblast formation by impairing phosphorylation of TAK1 acutely and TAK1, p38, and Smad2 phosphorylation at later time points. Salinomycin is therefore distinguished from other TGFβ pathway inhibitors by not affecting SMAD2 phosphorylation at early time points. The TAK1-p38 pathway has been implicated in fibrosis and excessive scarring in numerous organ systems, including the eyes, kidney, lungs, and heart (45–49). Recently, a p38 inhibitor, Esbriet (pirfenidone) (50), was approved for treatment of idiopathic pulmonary fibrosis in Europe, indicating the efficacy of targeting the pathway in fibrosis. Although pirfenidone does slow the disease progression of pulmonary fibrosis, it does not stop it, indicating the need for more efficacious drugs.
The activity of p38 is regulated by the upstream kinases MKK3 and MKK6 (32, 51). Although these two kinases are very similar in sequence and structure, they can often have differential effects on the activation of p38 (52). Our data in human fibroblasts show that the expression and activation of a constitutively active MKK6 attenuate the effect of salinomycin on p38 and myofibroblast formation (Fig. 8). Although our data point to the ability of salinomycin to inhibit TAK1 phosphorylation and activity to prevent MKK3/6 and p38 activation, future studies aimed at dissecting the role of salinomycin in altering TAK1 signaling will be required to fully understand its mechanism of action. Likewise, although the TAK1-p38 pathways appear to be crucial targets of salinomycin in myofibroblast formation, other pathways, such as wnt/β-catenin signaling, may also be involved because other studies have shown that salinomycin modulates wnt signaling (53).
Salinomycin, its derivative narasin, and the related polyether ionophore monensin all appear to have powerful anti-myofibroblast activity (Fig. 4). That all three possess this ability opens up the possibility that polyether ionophore function is required. Polyether ionophores preferentially bind monovalent cations such as sodium and potassium (54). Although this may be important for their coccidiostat properties, it is unclear whether this is required to block TGFβ function. However, because only nanomolar amounts of these ionophores are required and because there are micromolar levels or more of sodium and potassium ions in culture medium, it supports an alternative property of this family of molecules. Development of new analogs of salinomycin or other polyether ionophores that alter the polyether moieties is required to fully understand the nature of their anti-TGFβ properties.
Recent publications have reported the exciting possibility that salinomycin is a powerful therapeutic agent in combating cancer stem cells (24, 35, 55, 56). The concept that salinomycin may target highly proliferative cells as opposed to other, more slowly growing cells is also supported by our data showing that salinomycin is not toxic to human fibroblasts at the levels needed to blunt myofibroblast formation. In addition to driving myofibroblast formation, another consequence of high TGFβ levels in fibrosis are the unwanted fibroproliferative effects (34). Interestingly, salinomycin does not affect basal proliferation of human fibroblasts but does prevent TGFβ-induced proliferation (Fig. 4). Furthermore, although salinomycin induces apoptosis of cancer cells, we saw no indication that salinomycin induces apoptosis in human fibroblasts and myofibroblasts at the doses used in our experiments (50–500 nm).
Interestingly, myofibroblasts present in the tumor microenvironment also play a protective role for malignant cells and aid in metastasis (25). Our results show that salinomycin can disrupt myofibroblast formation. In light of this, another potential benefit of salinomycin is that it may alter the tumor microenvironment by decreasing the number of myofibroblasts, therefore making tumor growth less favorable. Another aspect of our work is that the effects of salinomycin are mimicked by the polyether ionophores narasin and monensin. Given that these small molecules all disrupt myofibroblast formation, it may be that the polyether ionophore family of small molecules is effective in targeting cancer cells. Future studies aimed at testing whether salinomycin and other polyether ionophores alter the tumor microenvironment to promote cancer cell death may further highlight the potential of these small molecules.
In summary, our work identifies salinomycin and other polyether ionophores as novel therapeutics for prevention of excessive scarring. Future studies using animal model(s) of excessive and/or abnormal scarring are required to determine the pharmacokinetics, stability, and efficacy of these drugs in an in vivo setting. In addition to their potential use to treat a variety of fibrotic and scarring diseases such as thyroid eye disease, idiopathic pulmonary fibrosis, and hypertrophic scarring, another future use of salinomycin may be to disrupt the tumor microenvironment by destroying or preventing myofibroblast formation in the tumor region.
Acknowledgments
We thank Dr. Alan Smrcka and Michael Burroughs for help with setting up and performing the high-throughput screen at the University of Rochester HTS core.
This work was supported, in whole or in part, by National Institutes of Health Grants ES-023032 and EY-023239, and ES-001247. This work was also supported by an unrestricted grant from the Research to Prevent Blindness and by a research grant from the Rochester/Finger Lakes Eye and Tissue Bank.
- TED
- thyroid eye disease
- αSMA
- α-smooth muscle actin
- SBE
- Smad-binding element
- DMSO
- dimethyl sulfoxide
- PARP
- poly(ADP-ribose) polymerase.
REFERENCES
- 1. Friedlander M. (2007) Fibrosis and diseases of the eye. J. Clin. Invest. 117, 576–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Noble P. W., Barkauskas C. E., Jiang D. (2012) Pulmonary fibrosis: patterns and perpetrators. J. Clin. Invest. 122, 2756–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bahn R. S. (2010) Graves' ophthalmopathy. N. Engl. J. Med. 362, 726–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hinz B. (2007) Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 [DOI] [PubMed] [Google Scholar]
- 5. Niessen F. B., Spauwen P. H., Schalkwijk J., Kon M. (1999) On the nature of hypertrophic scars and keloids: a review. Plast. Reconstr. Surg. 104, 1435–1458 [DOI] [PubMed] [Google Scholar]
- 6. Gauglitz G. G., Korting H. C., Pavicic T., Ruzicka T., Jeschke M. G. (2011) Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol. Med. 17, 113–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lehmann G. M., Feldon S. E., Smith T. J., Phipps R. P. (2008) Immune mechanisms in thyroid eye disease. Thyroid 18, 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phan S. H. (2002) The myofibroblast in pulmonary fibrosis. Chest 122, 286S–289S [DOI] [PubMed] [Google Scholar]
- 9. Kuriyan A. E., Phipps R. P., Feldon S. E. (2008) The eye and thyroid disease. Curr. Opin. Ophthalmol. 19, 499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hinz B., Phan S. H., Thannickal V. J., Galli A., Bochaton-Piallat M. L., Gabbiani G. (2007) The myofibroblast: one function, multiple origins. Am. J. Pathol. 170, 1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Desmoulière A., Chaponnier C., Gabbiani G. (2005) Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 13, 7–12 [DOI] [PubMed] [Google Scholar]
- 12. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., Gabbiani G. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith R. S., Smith T. J., Blieden T. M., Phipps R. P. (1997) Fibroblasts as sentinel cells: synthesis of chemokines and regulation of inflammation. Am. J. Pathol. 151, 317–322 [PMC free article] [PubMed] [Google Scholar]
- 14. Micallef L., Vedrenne N., Billet F., Coulomb B., Darby I. A., Desmoulière A. (2012) The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair 5, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bourlier V., Sengenès C., Zakaroff-Girard A., Decaunes P., Wdziekonski B., Galitzky J., Villageois P., Esteve D., Chiotasso P., Dani C., Bouloumié A. (2012) TGFβ family members are key mediators in the induction of myofibroblast phenotype of human adipose tissue progenitor cells by macrophages. PloS ONE 7, e31274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. George S. J. (2009) Regulation of myofibroblast differentiation by convergence of the Wnt and TGF-β1/Smad signaling pathways. J. Mol. Cell. Cardiol. 46, 610–611 [DOI] [PubMed] [Google Scholar]
- 17. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weiss A., Attisano L. (2013) The TGFβ superfamily signaling pathway. Wiley Interdiscip. Rev. Developmental Biology 2, 47–63 [DOI] [PubMed] [Google Scholar]
- 19. Usuki J., Matsuda K., Azuma A., Kudoh S., Gemma A. (2012) Sequential analysis of myofibroblast differentiation and transforming growth factor-β1/Smad pathway activation in murine pulmonary fibrosis. J. Nippon Med. Sch. 79, 46–59 [DOI] [PubMed] [Google Scholar]
- 20. Carthy J. M., Garmaroudi F. S., Luo Z., McManus B. M. (2011) Wnt3a induces myofibroblast differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PloS ONE 6, e19809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gu L., Zhu Y. J., Yang X., Guo Z. J., Xu W. B., Tian X. L. (2007) Effect of TGF-β/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol. Sin. 28, 382–391 [DOI] [PubMed] [Google Scholar]
- 22. Lehmann G. M., Woeller C. F., Pollock S. J., O'Loughlin C. W., Gupta S., Feldon S. E., Phipps R. P. (2010) Novel anti-adipogenic activity produced by human fibroblasts. Am. J. Physiol. Cell Physiol. 299, C672–C681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knirschová R., Nováková R., Fecková L., Timko J., Turna J., Bistáková J., Kormanec J. (2007) Multiple regulatory genes in the salinomycin biosynthetic gene cluster of Streptomyces albus CCM 4719. Folia Microbiol. 52, 359–365 [DOI] [PubMed] [Google Scholar]
- 24. Naujokat C., Steinhart R. (2012) Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotechnol. 2012, 950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baglole C. J., Ray D. M., Bernstein S. H., Feldon S. E., Smith T. J., Sime P. J., Phipps R. P. (2006) More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol. Investig. 35, 297–325 [DOI] [PubMed] [Google Scholar]
- 26. Kuriyan A. E., Woeller C. F., O'Loughlin C. W., Phipps R. P., Feldon S. E. (2013) Orbital fibroblasts from thyroid eye disease patients differ in proliferative and adipogenic responses depending on disease subtype. Invest. Ophthalmol. Vis. Sci. 54, 7370–7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mortier L., Daeseleire E., Van Peteghem C. (2005) Determination of the ionophoric coccidiostats narasin, monensin, lasalocid and salinomycin in eggs by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 19, 533–539 [DOI] [PubMed] [Google Scholar]
- 28. Schimmer A. D., Jitkova Y., Gronda M., Wang Z., Brandwein J., Chen C., Gupta V., Schuh A., Yee K., Chen J., Ackloo S., Booth T., Keays S., Minden M. D. (2012) A phase I study of the metal ionophore clioquinol in patients with advanced hematologic malignancies. Clin. Lymphoma Myeloma Leuk. 12, 330–336 [DOI] [PubMed] [Google Scholar]
- 29. Montesano R., Orci L. (1988) Transforming growth factor β stimulates collagen-matrix contraction by fibroblasts: implications for wound healing. Proc. Natl. Acad. Sci. U.S.A. 85, 4894–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fuchs D., Heinold A., Opelz G., Daniel V., Naujokat C. (2009) Salinomycin induces apoptosis and overcomes apoptosis resistance in human cancer cells. Biochem. Biophys. Res. Commun. 390, 743–749 [DOI] [PubMed] [Google Scholar]
- 31. Gupta P. B., Onder T. T., Jiang G., Tao K., Kuperwasser C., Weinberg R. A., Lander E. S. (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raingeaud J., Whitmarsh A. J., Barrett T., Dérijard B., Davis R. J. (1996) MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 16, 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klingberg F., Hinz B., White E. S. (2013) The myofibroblast matrix: implications for tissue repair and fibrosis. J. Pathol. 229, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akhurst R. J., Hata A. (2012) Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Naujokat C., Fuchs D., Opelz G. (2010) Salinomycin in cancer: a new mission for an old agent. Mol. Med. Rep. 3, 555–559 [DOI] [PubMed] [Google Scholar]
- 36. Guo N., Woeller C. F., Feldon S. E., Phipps R. P. (2011) Peroxisome proliferator-activated receptor γ ligands inhibit transforming growth factor-β-induced, hyaluronan-dependent, T cell adhesion to orbital fibroblasts. J. Biol. Chem. 286, 18856–18867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuriyan A. E., Phipps R. P., O'Loughlin C. W., Feldon S. E. (2008) Improvement of thyroid eye disease following treatment with the cyclooxygenase-2 selective inhibitor celecoxib. Thyroid 18, 911–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sundaresh V., Brito J. P., Wang Z., Prokop L. J., Stan M. N., Murad M. H., Bahn R. S. (2013) Comparative effectiveness of therapies for Graves' hyperthyroidism: a systematic review and network meta-analysis. J. Clin. Endocrinol. Metab. 98, 3671–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gabbiani G. (2003) The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 [DOI] [PubMed] [Google Scholar]
- 40. Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., Brown R. A. (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 [DOI] [PubMed] [Google Scholar]
- 41. Laping N. J., Grygielko E., Mathur A., Butter S., Bomberger J., Tweed C., Martin W., Fornwald J., Lehr R., Harling J., Gaster L., Callahan J. F., Olson B. A. (2002) Inhibition of transforming growth factor (TGF)-β1-induced extracellular matrix with a novel inhibitor of the TGF-β type I receptor kinase activity: SB-431542. Mol. Pharmacol. 62, 58–64 [DOI] [PubMed] [Google Scholar]
- 42. Yang Y., Wang Z., Yang H., Wang L., Gillespie S. R., Wolosin J. M., Bernstein A. M., Reinach P. S. (2013) TRPV1 potentiates TGFβ-induction of corneal myofibroblast development through an oxidative stress-mediated p38-SMAD2 signaling loop. PloS ONE 8, e77300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shi-wen X., Parapuram S. K., Pala D., Chen Y., Carter D. E., Eastwood M., Denton C. P., Abraham D. J., Leask A. (2009) Requirement of transforming growth factor β-activated kinase 1 for transforming growth factor β-induced α-smooth muscle actin expression and extracellular matrix contraction in fibroblasts. Arthritis Rheum. 60, 234–241 [DOI] [PubMed] [Google Scholar]
- 44. Choi M. E., Ding Y., Kim S. I. (2012) TGF-β signaling via TAK1 pathway: role in kidney fibrosis. Semin. Nephrol. 32, 244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fan H. N., Wang H. J., Ren L., Ren B., Dan C. R., Li Y. F., Hou L. Z., Deng Y. (2013) Decreased expression of p38 MAPK mediates protective effects of hydrogen sulfide on hepatic fibrosis. Eur. Rev. Med. Pharmacol. Sci. 17, 644–652 [PubMed] [Google Scholar]
- 46. Li J., Campanale N. V., Liang R. J., Deane J. A., Bertram J. F., Ricardo S. D. (2006) Inhibition of p38 mitogen-activated protein kinase and transforming growth factor-β1/Smad signaling pathways modulates the development of fibrosis in adriamycin-induced nephropathy. Am. J. Pathol. 169, 1527–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang S., Weinheimer C., Courtois M., Kovacs A., Zhang C. E., Cheng A. M., Wang Y., Muslin A. J. (2003) The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J. Clin. Invest. 111, 833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsuoka H., Arai T., Mori M., Goya S., Kida H., Morishita H., Fujiwara H., Tachibana I., Osaki T., Hayashi S. (2002) A p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L103–L112 [DOI] [PubMed] [Google Scholar]
- 49. Saika S. (2006) TGFβ pathobiology in the eye. Lab. Invest. 86, 106–115 [DOI] [PubMed] [Google Scholar]
- 50. Moran N. (2011) p38 kinase inhibitor approved for idiopathic pulmonary fibrosis. Nat. Biotechnol. 29, 301. [DOI] [PubMed] [Google Scholar]
- 51. Enslen H., Raingeaud J., Davis R. J. (1998) Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 273, 1741–1748 [DOI] [PubMed] [Google Scholar]
- 52. Tanaka N., Kamanaka M., Enslen H., Dong C., Wysk M., Davis R. J., Flavell R. A. (2002) Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Rep. 3, 785–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu D., Choi M. Y., Yu J., Castro J. E., Kipps T. J., Carson D. A. (2011) Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. U.S.A. 108, 13253–13257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Antoszczak M., Popiel K., Stefańska J., Wietrzyk J., Maj E., Janczak J., Michalska G., Brzezinski B., Huczyński A. (2014) Synthesis, cytotoxicity and antibacterial activity of new esters of polyether antibiotic: salinomycin. Eur. J. Med. Chem. 76, 435–444 [DOI] [PubMed] [Google Scholar]
- 55. Huczynski A. (2012) Salinomycin: a new cancer drug candidate. Chem. Biol. Drug Des. 79, 235–238 [DOI] [PubMed] [Google Scholar]
- 56. Wang Y. (2011) Effects of salinomycin on cancer stem cell in human lung adenocarcinoma A549 cells. Med. Chem. 7, 106–111 [DOI] [PubMed] [Google Scholar]