

Background: The protective antigen adenylate cyclase toxin (ACT) has not been included in current pertussis vaccines partly due to incomplete understanding of its protective epitopes.
Results: The repeat-in-toxin (RTX) domain is immunodominant in mice and contains neutralizing epitopes.
Conclusion: The RTX domain induces similar neutralizing antibody responses as ACT.
Significance: The RTX domain may be an alternative to ACT for inclusion in future vaccines.
Keywords: Antibody Engineering, Antigen, Bacterial Toxin, Bordetella pertussis, Protein Engineering, CyaA, Adenylate Cyclase, Immunogen Engineering, Immunogenicity, Structural Vaccinology
Abstract
The adenylate cyclase toxin (ACT) is a multifunctional virulence factor secreted by Bordetella species. Upon interaction of its C-terminal hemolysin moiety with the cell surface receptor αMβ2 integrin, the N-terminal cyclase domain translocates into the host cell cytosol where it rapidly generates supraphysiological cAMP concentrations, which inhibit host cell anti-bacterial activities. Although ACT has been shown to induce protective immunity in mice, it is not included in any current acellular pertussis vaccines due to protein stability issues and a poor understanding of its role as a protective antigen. Here, we aimed to determine whether any single domain could recapitulate the antibody responses induced by the holo-toxin and to characterize the dominant neutralizing antibody response. We first immunized mice with ACT and screened antibody phage display libraries for binding to purified ACT. The vast majority of unique antibodies identified bound the C-terminal repeat-in-toxin (RTX) domain. Representative antibodies binding two nonoverlapping, neutralizing epitopes in the RTX domain prevented ACT association with J774A.1 macrophages and soluble αMβ2 integrin, suggesting that these antibodies inhibit the ACT-receptor interaction. Sera from mice immunized with the RTX domain showed similar neutralizing activity as ACT-immunized mice, indicating that this domain induced an antibody response similar to that induced by ACT. These data demonstrate that RTX can elicit neutralizing antibodies and suggest it may present an alternative to ACT.
Introduction
Whooping cough is a highly infectious disease caused primarily by the bacteria Bordetella pertussis. While disease incidence has dropped dramatically due to the initiation of widespread vaccination programs using killed bacteria in the 1940s, in recent years rates have rebounded, reaching a 60-year high in the United States in 2012 (1–3). This trend is especially troubling for unimmunized infants, who are most susceptible to the disease and exhibit the highest rates of morbidity and mortality. Modified vaccination strategies, including booster immunization of adolescents, adults, and pregnant women, have been implemented to reduce transmission to neonates.
This increase in disease incidence coincides with the switch from whole cell to acellular vaccines in the 1990s, and has been attributed to several factors, including increased awareness, mismatch between vaccine and circulating strains, a Th1/Th2 immune response instead of the more effective Th1 response, and a shorter duration of protection conferred by acellular vaccines (4). Recently, Warfel et al. (5) demonstrated that acellular vaccines protect against disease symptoms but not subclinical infection or transmission in a novel non-human primate model. Taken together, these data provide a compelling argument for modification of the current vaccine.
Currently licensed acellular vaccines contain chemically detoxified pertussis toxin and up to four surface adhesins, including filamentous hemagglutinin, pertactin, and fimbriae 2/3. Exciting approaches in development to enhance vaccine-mediated protective immunity include a genetically attenuated B. pertussis for intranasal delivery (6), nanoparticle formulations, including purified antigens and novel adjuvant formulations (7), as well as inclusion of additional highly conserved antigens in the current vaccine (8, 9). A strong candidate for inclusion in any of these is the adenylate cyclase toxin (ACT),2 which aids in immune evasion and is produced by three closely related Bordetella species, including B. pertussis, Bordetella parapertussis, and Bordetella bronchiseptica (10, 11).
ACT-deficient Bordetella strains have shown significantly compromised colonization and persistence in various mouse models (12–14), whereas some hypervirulent strains express higher ACT levels (15). Moreover, active or passive immunization with polyclonal anti-ACT antibodies protected mice against lethal respiratory challenges by B. pertussis and B. parapertussis (15) and shortened the period of bacterial colonization in the respiratory tract (16). Finally, natural infection of humans results in a strong anti-ACT antibody response (17).
ACT is a large ∼177-kDa protein consisting of two functionally discrete regions as follows: the catalytic domain (residues 1–385) and a pore-forming or hemolysin region that is part of the larger repeat-in-toxin (RTX) family, represented in >250 bacterial strains (Fig. 1A). After translocation into the cytosol, the catalytic domain binds eukaryotic calmodulin with low nanomolar affinity (18) and rapidly converts available ATP to cAMP via its adenylate cyclase activity (19). The resulting supraphysiological cAMP levels disrupt signaling and bactericidal activities in phagocytic cells (20–22). The C-terminal ∼1300 residues exhibit homology to the Escherichia coli α-hemolysin. This region consists of a hydrophobic domain capable of forming a cation-selective transmembrane channel (residues 525–715) (23), a modification region bearing two acylation sites at residues Lys-860 and Lys-983 (24), the RTX domain (residues 1006–1600), consisting of ∼40 calcium binding sites formed by glycine- and aspartate-rich nonapeptide repeats, and finally, a C-terminal secretion signal (residues 1600–1706). The RTX region also harbors the receptor-binding site, with specificity for the αMβ2 integrin (also called CR3, Mac-1, and CD11b/CD18) present on phagocytic leukocytes (25, 26). Both post-translational acylation by the co-expressed enzyme CyaC and calcium ion-mediated structural changes are essential for receptor binding, cAMP intoxication, and pore forming activities (24, 27).
FIGURE 1.
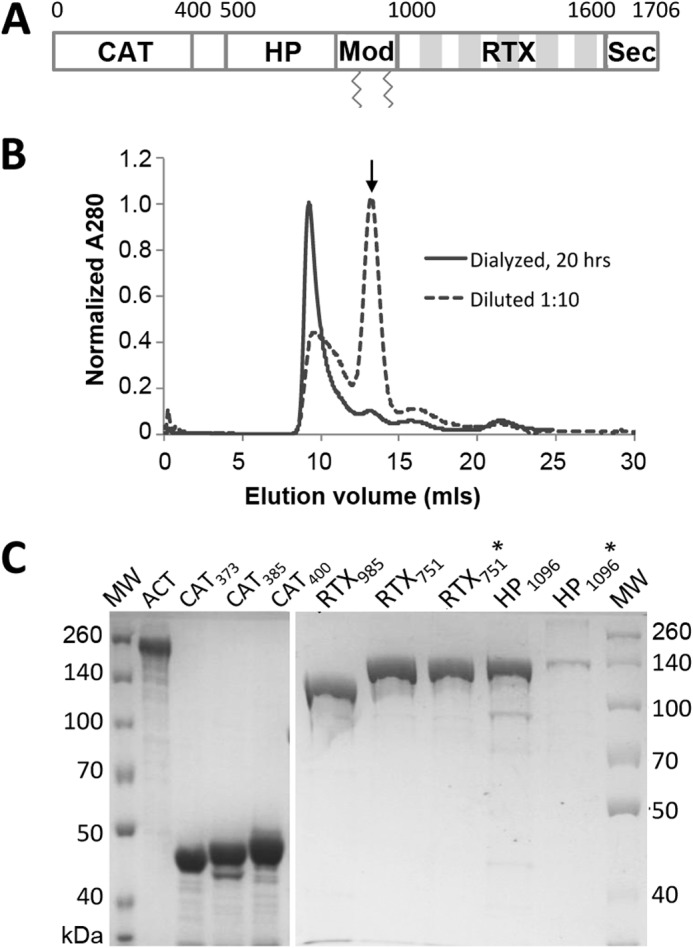
Expression and purification of intact ACT and domains. A, adenylate cyclase toxin domain architecture. ACT is a 177-kDa protein toxin, consisting of five sequential domains as follows: the catalytically active N-terminal adenylate cyclase (CAT) domain, the central hydrophobic (HP) domain, modification (Mod) region carrying two acylation sites, Lys-860 and Lys-983, the RTX domain, and finally a C-terminal secretion signal (Sec). The five shaded blocks in the RTX region represent tandem Gly-Asp-rich repeats. B, ACT forms high molecular mass species (>600 kDa) when urea is removed by dialysis or dilution. Size exclusion chromatograms of ∼120 μg of ACT using a Superdex 200 column were collected directly after a 1:10 dilution (final urea concentration 0.8 m) or overnight dialysis into HBSC with 1 m urea. Arrow indicates the size expected for monomer, 177 kDa. C, SDS-polyacrylamide gel comparing full-length ACT with different domain constructs after purification from E. coli. Three versions of the CAT domain (residues 1–373 (CAT373), 1–385 (CAT385), and 1–400 (CAT400)) and three versions of the RTX domain (residues 985–1706 (RTX985), 751–1706 (RTX751), and acylated 751–1706 (RTX751*)) were expressed from plasmid pET28a with an N-terminal His6 tag. The HP domains (residues 399–1096 (HP1096) and acylated 399–1096 (HP1096*)) were expressed from plasmid pMalc-5x with an N-terminal MBP fusion for enhanced solubility and C-terminal His6 tag.
Despite evidence indicating ACT is a protective antigen, few neutralizing antibodies have been described, and the location of neutralizing epitopes remains unclear. Moreover, ACT is prone to aggregation and degradation when produced by Bordetella or recombinantly by E. coli, precluding its inclusion in current acellular vaccine formulations (28). Therefore, we aimed to identify neutralizing antibodies and their domain specificity and to determine whether any single domain, possessing desirable expression and protein stability characteristics, can recapitulate the antibody responses induced by the holo-toxin.
EXPERIMENTAL PROCEDURES
Molecular Cloning of ACT Domains for Soluble Expression
Individual ACT domains were expressed in the bacterial cytoplasm of E. coli with His6 tags. To generate plasmids expressing only the catalytic domain (residues 1–373, 1–385, or 1–400), the corresponding coding regions were amplified from pT7CACT3 (29) by PCR, with the common forward primer 5′-aggaaacaCATATGcagcaatcgcatcaggctgg-3′ and reverse primers 5′-actaGAATTCttacgaacgtccgctcggcacg-3′, 5′-cacaGAATTCttacgccggcaccgtttccagtacatc-3′, and 5′-cataGAATTCttactggcgttccactgcgcc-3′, respectively (restriction sites in uppercase and underlined). The amplified fragments were gel-purified and double-digested with NdeI and EcoRI and ligated into similarly digested pET28a vectors. To generate plasmids expressing the RTX domains (residues 751–1706 or 985–1706), DNA fragments encoding these regions were amplified using forward primers 5′-tcacgaaCATATGgccaattcggacg-3′ and 5′-ctacggcCATATGacggagaatgtcca-3′, and common reverse primer 5′-ataGGATCCtcagcgccagttgacag-3′. The resulting PCR products were gel-purified, double-digested with NdeI and BamHI, and ligated into similarly digested pET28a vector.
To enhance folding and solubility, the hydrophobic domain, encompassing the region between the catalytic and RTX domains (residues 399–1096), was cloned into pMalc-5x vector (New England Biolabs) between NdeI and BamHI sites, downstream of the maltose-binding protein (MBP). The primers for PCR were 5′-gggcgcaCATATGcgccaggattccggct-3′ and 5′-atcggcGGATCCttaatggtgatgatggtgatgggcgctggcctcggaaggctggtgcac-3′; with the boldface nucleotides encoding a C-terminal His6-tag. The cyaC gene was inserted downstream of the hydrophobic domain between the BamHI and HindIII sites, with an upstream ribosome binding to allow for co-expression.
ACT and Domain Expression and Purification
Full-length ACT was expressed from the plasmid pT7CACT3 with co-expression of the palmitoylating enzyme CyaC in E. coli strain XL-1 Blue (29). The holo-toxin was purified using a single-step calmodulin-agarose affinity chromatography as described by Sebo and co-workers (29). Purified ACT was stored in 50 mm Tris, 8 m urea, 2 mm EDTA, pH 8.0, at 4 °C for short term or −80 °C for long term storage. The protein concentration was determined by absorbance at 280 nm using a molecular extinction coefficient of 143,590 m−1 cm−1 as calculated from its amino acid sequence (30). ACT from BEI Resources was used as a reference for purity and toxicity.
The catalytic and RTX domains of ACT were expressed in E. coli strain BL21(DE3). Briefly, 250 ml of TB media were inoculated from starter cultures to an absorbance at 600 nm (A600) of 0.05, grown at 37 °C until A600 = 0.3–0.6, at which time 0.4 mm isopropyl β-d-thiogalactopyranoside (IPTG) was added to induce expression. After 4 h of growth at room temperature, the cells were harvested, resuspended in Buffer A (50 mm Hepes, 250 mm NaCl, 2 mm CaCl2, 40 mm imidazole, pH 8.0), and lysed with a French press (Thermo Scientific). After a 20-min centrifugation step at 20,000 rpm (JA-20 rotor), the supernatant was applied to a HisTrap column on an ÅKTA FPLC (GE Healthcare), followed by elution with a linear gradient of Buffer B (Buffer A + 500 mm imidazole). The hydrophobic domain was expressed in E. coli strain BL21 as above, and purification included immobilized metal affinity chromatography (IMAC) resin followed by an MBPTrap affinity column (GE Healthcare) and elution with 10 mm maltose.
Biophysical Characterization
To assess the oligomeric status of ACT and its domains, fractions eluted from HisTrap or IMAC were supplemented with either 100 μl of 100 mm EGTA or 100 mm HBSC (HBS + 2 mm CaCl2), incubated on ice for 1 h, and loaded onto Superdex S200 column (except Superdex75 for CAT400) equilibrated with HBS (50 mm Hepes, 150 mm NaCl, pH 7.8) or HBSC, respectively. Running buffers were HBS or HBSC to ensure the absence or presence of calcium ion. To assess secondary structure characteristics, purified ACT domains were dialyzed into 10 mm Tris-H2SO4, pH 8.0, and the concentrations were adjusted to 100 μg/ml. To observe the effect of calcium ions on protein conformation, CaCl2 were added to a final concentration of 2 mm and incubated at room temperature for 1 h. CD spectra between 180 and 260 nm were collected on a J-815 CD spectrometer (Jasco) at 25 °C, at 1-nm intervals using a 1-mm rectangular cell. Each spectrum represents the average of three scans subtracted with the spectrum of buffers (with or without CaCl2). Data were fitted with the CDSSTR program on the DichroWeb server to estimate the percentage of secondary structures (31).
Murine Immunization
All protocols were approved by the University of Texas at Austin IACUC (protocol number 2012-00068), and all mice were handled in accordance with IACUC guidelines. As a source for antibody libraries, two 6-week-old BALB/c mice were primed intraperitoneally with 17 μg of ACT (dialyzed against PBS to remove urea) in complete Freud's adjuvant. Four weeks later, the mice were bled through a tail vein and boosted subcutaneously with the same amount of PBS-dialyzed ACT in incomplete Freund's adjuvant. Two weeks later, the mice were sacrificed, and blood was collected by cardiac puncture. Spleens were removed sterilely, sliced into pieces, and immediately immersed in 1 ml of cold RNAlater solution. After soaking overnight at 4 °C, the solution was removed, and the spleens were stored at −80 °C.
To assess the immunogenicity of individual ACT domains, 4–6 BALB/c mice per group were immunized subcutaneously with equal moles of ACT and individual domains (10 μg for ACT, 2.6 μg for CAT400, 6.7 μg for HP1096*, and 4.4 μg for RTX985) in complete Freund's adjuvant. Four weeks later, the mice were boosted subcutaneously with the same amount of antigen in incomplete Freud's adjuvant, a process that was repeated at 6 and 8 weeks. Blood was collected before immunization, 4 weeks after the first injection, and 2 weeks after each boost. Anti-ACT antibody titers were determined by ELISA (described below), with titer defined as the 50% effective concentration (EC50) from a four-parameter logistic fitting to the ELISA data. Neutralization of ACT-induced cAMP intoxication of J774A.1 macrophage cells (ATCC number TIB-67) was tested with a 1:400 sera dilution.
Phage Display Antibody Library Construction
Total RNA was extracted from frozen spleens with TRIzol (Invitrogen) and the RNeasy mini kit (Qiagen) or PureLink RNA kit (Invitrogen) according to the manufacturers' instructions. The quality and concentration of total RNA were assessed by agarose gel electrophoresis and A230:260:280 ratio (∼1:2:1 for pure RNA) measured by NanoDrop 2000 (Thermo Scientific). For first-strand cDNA synthesis, 5 μg of total RNA was used. To maximize diversity, two separate reactions were performed using combinations of Superscript II + d(T)23 VN primer or Superscript III (Invitrogen)+ random hexamer (Thermo Scientific), following the manufacturers' instructions. The two sets of cDNA were pooled as template for amplification of the VL and VH repertoires using the primer sets and PCR conditions described by Krebber et al. (32). The PCR products were gel-purified, with 10 ng each of VL and VH used as template in an overlap PCR to generate VL-linker-VH fragments (scFv). This product was gel-purified and digested overnight with SfiI prior to directional ligation with similarly SfiI-digested pMopac24 vector (33). Ten individual electroporations were performed to transform XL1-Blue cells. The transformants were pooled, and an aliquot was 10-fold serially diluted and plated to count library size; the rest were plated on eight 150-mm 2×YT agar plates (10 μg/ml tetracycline, 200 μg/ml ampicillin, and 2% glucose). After incubation overnight at 37 °C, the bacterial lawns were scraped off in 2×YT medium and pooled to form the master library.
Phage Production, Purification, and Panning
Aliquots of the master library were used to inoculate 250 ml of 2×YT medium with 10 μg/ml tetracycline, 200 μg/ml ampicillin, and 2% glucose in 1-liter flasks to an A600 of ∼0.1. The cultures were grown at 37 °C for 2–3 h until the A600 reached ∼0.6, induced, and rescued by adding 1 mm IPTG and M13KO7 helper phage (multiplicity of infection of ∼20), incubated for 30 min without shaking at 37 °C, and then returned to a shaking incubator at room temperature. Three hours after adding helper phage, the culture was supplemented with 50 μg/ml kanamycin prior to overnight incubation with shaking. Phage were then purified by double precipitation with 0.2 volume of precipitation solution (2.5 m NaCl, 20% PEG-8000). The concentration of viable phage was assessed as colony-forming units (cfu), with serially diluted phage added to log-phase XL1-Blue cells, followed by plating on 2×YT agar plate with 200 μg/ml ampicillin, and enumeration of colonies after overnight incubation.
Two rounds of panning were performed using ACT as bait. Eight ELISA plate wells (Costar) were coated with 50 μl of 2 and 1 μg/ml ACT in PBS at 4 °C overnight for the first and second rounds, respectively. Input phage (100 μl) were diluted into 900 μl of 5% nonfat milk in PBST (PBS, 0.05% Tween 20) and incubated for 1 h before transferring 100 μl to each of the 8 wells. After a 1-h incubation at room temperature, followed by five (or 10 for round 2) washes with PBST, bound phage were eluted with 100 μl per well of 0.1 n HCl for 10 min at room temperature. The eluted phage was pooled and immediately neutralized with 48 μl of 2 m Tris base. Half of the output phages was added into 5 ml of log-phase XL1-Blue culture grown with 10 μg/ml tetracycline at 37 °C to retain the F plasmid, incubated for 30 min without shaking and 1 h with shaking at 225 rpm in 37 °C, spun down, and then plated on six 150-mm 2YT agar plates (200 μg/ml ampicillin, 10 μg/ml tetracycline, and 2% glucose). After overnight incubation at 37 °C, colonies or lawn were scraped, pooled, and mixed thoroughly, and aliquots were used for second round of panning as described above.
Input and output phage titers (colony-forming units) were determined by infecting and plating E. coli as described above. Sequence diversity was monitored throughout all steps by performing colony PCR of random colonies on the phage titration plates, followed by BstNI fingerprinting and agarose gel electrophoresis. Clones with unique fingerprints were confirmed by DNA sequencing.
To produce monoclonal phage clones from panning outputs, single colonies from output plates were inoculated into sterile 96-well plates containing 100 μl of 2YT medium (2% glucose, 200 μg/ml ampicillin, and 10 μg/ml tetracycline) and grown at 37 °C overnight with shaking. The next morning, 10 μl of the overnight culture was inoculated into another plate with 90 μl per well of fresh medium containing 0.25% glucose and antibiotics, and grown at 37 °C for 3 h, then 50 μl of 2YT (200 μg/ml ampicillin, 3 mm IPTG, and M13KO7 helper phage) was added and then shaken at room temperature for 3 h before adding 50 μl of 2YT (200 μg/ml ampicillin, 1 mm IPTG, 200 μg/ml kanamycin). The plate was then shaken at room temperature overnight.
Antibody Expression and Purification
To convert phage-displayed scFvs to soluble single chain antibody fragments (scAbs), consisting of a variable light chain domain (VL) connected by a flexible (Gly4Ser)2 to a variable heavy chain domain (VH) and followed by a human κ constant domain to enhance expression and solubility, the scFv region was removed from pMopac24 phagemid vector by SfiI digestion and directionally ligated into SfiI-digested pMopac54 plasmid (34). For scAb production, 100 ml of TB supplemented with 200 μg/ml ampicillin and 1% glucose were inoculated at A600 = 0.02 and grown overnight at room temperature. The next morning, cells were pelleted at 5000 × g for 10 min at room temperature, resuspended in 100 ml of TB medium with ampicillin but no glucose, and grown at room temperature for 1 h before induction with 1 mm IPTG. After another 4 h, cells were harvested by centrifugation at 5000 × g for 10 min at 4 °C. Osmotic shock was performed as described (34). scAbs in the dialyzed shockates were purified by IMAC resin followed by size exclusion chromatography with a Superdex 200 column on FPLC (GE Healthcare). Protein concentrations were measured by BCA assays (Pierce) using a BSA standard with purity assessed by SDS-PAGE.
To convert scAbs into full-length IgG with enhanced stabilities and in vivo half-lives, the VL and VH genes were subcloned onto Igκ-Abvec and IgG-Abvec vectors as described by Smith et al. (35). For IgG production, paired Igκ-Abvec and IgG-Abvec plasmids were transiently transfected into CHO-K1 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Culture media were collected at 1–2-day intervals, neutralized with 1 Tris, pH 8.0, and pooled, and IgG was purified by ammonium sulfate precipitation followed by HiTrap protein A column. The purity and presence of aggregates were assessed by SDS-PAGE and size exclusion chromatography using a Superdex S200 column. The concentration was determined by A280 using extinction coefficients calculated from deduced amino acid sequences (30).
Analysis of Antibody Binding by ELISA
For monoclonal phage screening, the 96-well phage production plates described above were spun at 3000 × g for 20 min with 40 μl of supernatant transferred to a coated (2 μg/ml ACT in PBS) and blocked (5% nonfat milk in PBST, M-PBST) ELISA plate, containing 60 μl of M-PBST per well, and incubated at room temperature for 1 h. After four washes with PBST, 50 μl of 1:2000 HRP-conjugated anti-M13 antibody (GE Healthcare) was added and incubated for1 h at room temperature. The plate was washed four times, and 50 μl/well 3,3′,5,5′-tetramethylbenzidine substrate was added and incubated at room temperature. The reaction was quenched by adding 1 m HCl to the 50 μl/well, and the absorbance at 450 nm recorded with a SpectraMax M5 (Molecular Devices). Wells with absorbance higher than 2-fold of background were identified for further characterization.
Binding assays to assess domain specificity and relative affinity of soluble scAb or IgG proteins were performed in a similar manner. Purified ACT (1 μg/ml) or domains (equimolar with ACT) were coated on ELISA plates, followed by blocking and serial dilutions of purified antibodies, and finally detection with goat anti-mouse IgG HRP-conjugated antibody (to detect murine antibodies), goat anti-human κ chain HRP-conjugated antibody (to detect scAbs), or goat anti-human IgG (Fc-specific) HRP-conjugated antibody (to detect recombinant IgG). For competition ELISA, the antibody of interest was used at a fixed concentration determined to yield 70–80% of the maximal signal, and mixed with an equal volume of serially diluted competitor antibody, followed by detection as above.
To assess the reactivity of sera from humans exposed to B. pertussis, purified ACT (1 μg/ml) or domains (equimolar concentrations as ACT) were coated on ELISA plates. Plates were blocked as above; human sera were serially diluted in M-PBST, and bound antibodies were detected with goat anti-human IgG (Fc-specific) HRP-conjugated antibody. Nine randomly selected samples were tested in duplicate. The absorbance value for each sample binding to a domain was normalized to that sample's signal on an ACT-coated well at a 100-fold dilution (in the linear dose-response range) as follows: (A450 (domain coated well) − A450(uncoated well))/(A450 (ACT coated well) − A450(uncoated well)). Human sera were obtained from Vanderbilt University Medical Center under a protocol approved by the local institutional review board (IRB 061262, 070258, and 090806). Use of the samples was approved by the University of Texas at Austin (2009-05-0096). The study was conducted in accordance with the Declaration of Helsinki, with written informed consent obtained from each participant prior to study entry. The original consent forms allowed for sample use in subsequent studies. CaCl2 levels in all ELISAs were maintained at >2 mm unless indicated, and each assay was performed at least twice.
In Vitro cAMP Intoxication and Neutralization Assay
J774A.1 cells were grown in DMEM (Sigma) supplemented with 10% fetal bovine serum, 1 mm sodium pyruvate, and penicillin/streptomycin. To measure the cAMP intoxication of J774A.1 cells by ACT or antibody neutralization, J774A.1 cells were seeded at 4 × 104/cm2 in 24-well plates (Costar) 1 day before the assay. ACT alone or with antibodies was diluted in DMEM without supplements in a final volume of 1 ml; base DMEM contains 1.8 mm CaCl2. ACT was used at 125 ng/ml in all assays unless otherwise specified; antibodies were present at 160-fold molar excess. ACT and antibody mixtures were incubated at room temperature for 30 min, and cells were washed twice with plain DMEM prior to addition of 480 μl of antibody/ACT solution to duplicate wells. The plate was incubated at 37 °C for 30 min in a CO2 incubator, followed by two washes with cold PBS. Lysis solution (500 μl; 0.1 n HCl, 0.1% Triton X-100) was added into each well, and the plate was rocked on ice for 10 min. The lysates were transferred into 1.5-ml tubes and boiled for 10 min to inactivate ACT and cAMP-hydrolyzing enzymes. To assess cAMP intoxication in CHO cells, ACT was used at 250 ng/ml, with all other assay conditions held constant.
The resulting cellular lysates were clarified by centrifugation at 13,000 × g for 5 min. The supernatant was diluted 6-fold with 200 mm Hepes, 150 mm NaCl, 0.05% Tween, pH 8.0, prior to cAMP measurement using a competition ELISA as described by Karimova and Ladant (36). All assays were performed at least in duplicate, with cAMP concentrations normalized to total protein concentration in the lysates as measured by BCA assay (Pierce).
To evaluate ACT neutralization in the context of the whole bacterium, B. pertussis Tohama I was grown on a Bordet Gengou agar plate supplemented with 15% defibrinated sheep blood (BD Biosciences) at 37 °C for 4 to 5 days. Bacteria were then inoculated into modified synthetic Stainer-Scholte medium and grown at 37 °C with shaking at 225 rpm for 20–24 h to an A600 of 0.7–1.0. Bacteria were pelleted by centrifugation at 5000 × g for 10 min, resuspended in PBS, and diluted to an A600 of 0.4 in DMEM + 10% heat-inactivated FBS, and then mixed with an equal volume of serially diluted neutralizing or control IgGs in DMEM + 10% heat-inactivated FBS, and incubated at room temperature for 20 min before addition to adherent J774A.1 cells and incubation of 1 h at 37 °C. Intracellular cAMP level was then measured as above.
J774A.1 Cell Lysis Assay
Cell lysis was monitored by enzymatic activity of lactate dehydrogenase released into the medium upon cell lysis. J774A.1 cells were seeded 105 cells/well in 96-well round-bottom plates 1 day before the assay. 100 μl each of ACT alone (0.25 μg/ml) or ACT preincubated with scAbs (10 μg/ml, 160-fold molar excess) in plain DMEM were added to triplicate wells and incubated for 2 h at 37 °C. The plate was then centrifuged at 250 × g for 5 min, and lactate dehydrogenase activity in the supernatants was measured by a colorimetric assay with the CytoTox 96 kit (Promega, WI). After subtracting the background signal from control wells without ACT, the sample absorbance at 490 nm was normalized to ACT-only controls as follows: 100%·(A490 (ACT+antibody) − A490 (no ACT))/(A490 (ACT only) − A490 (no ACT)).
Analysis of ACT-Integrin Binding by ELISA
Recombinant murine αMβ2 integrin (R&D Systems) was coated at 1 μg/ml in PBS at 4 °C overnight and blocked with M-PBST. ACT or purified domains were serially diluted and incubated for 1 h at room temperature, followed by detection with polyclonal rabbit anti-ACT antibody and HRP-conjugated goat anti-rabbit antibody. To assess the effect of antibodies on ACT binding to integrin, ACT (1 μg/ml) was mixed with an equal volume of serially diluted antibody (5-, 1.6-, 0.5-, and 0-fold molar excess) and incubated for 1 h at room temperature before transferring to a blocked αMβ2 ELISA plate. The bound ACT was detected as described above.
Flow Cytometry Analysis of ACT Binding to Cells
ACT was dialyzed against HBSC (50 mm Hepes, 150 mm NaCl, 2 mm CaCl2, pH 8.0) to remove urea and then biotinylated with 100-fold molar excess of EZ-Link Sulfo-NHS-LC-Biotin (Thermo Scientific) at room temperature for 2–3 h before quenching with 1 m Tris, pH 8.0, and dialysis against HBSC overnight in 4 °C. Biotinylated ACT (210 μl of 0.8 μg/ml) was incubated with an equal volume containing 120 μg/ml purified scAb (M2B10, M1H5, M1F11, and M1C12) in DMEM + 1% BSA at room temperature for 30 min. Then 200 μl of the incubated mixtures was added to 4 × 105 washed J774A.1 cells in duplicate and incubated on ice for 30 min to allow ACT binding but not internalization. After two washes with FACS buffer (HBSC + 2% FBS), 200 μl of 1:500 phycoerythrin (PE)-conjugated streptavidin (BioLegend) was used to detect cell-associated biotinylated ACT. After 20 min of incubation on ice and three washes, the cells were finally resuspended in 600 μl of FACS buffer and analyzed on an LSR Fortessa II. Data analysis was performed with Flowjo software (Version 10).
RESULTS
ACT Is Prone to Aggregation and Proteolysis
The native ACT holotoxin secreted from B. pertussis readily aggregates at the bacterial surface and is prone to proteolytic degradation (28), while recombinant toxin expressed in the E. coli cytoplasm forms inclusion bodies (37–39). To increase the yield of purified protein, 8 m urea was used to extract the aggregated protein from bacterial cell pellets; even so, the protein is highly susceptible to proteolysis with early reports observing enzymatic activity in 43- and 45-kDa fragments (40). Efforts to remove urea, such as dialysis and dilution, result in significant aggregation and fragmentation (41).
As a result, standard purification protocols include solubilization of the cell pellet with urea, followed by calmodulin affinity or sequential anionic and hydrophobic interaction chromatographic steps followed by storage in 8 m urea. Assays using the toxin call for dialysis or dilution of urea-solubilized ACT into assay media immediately before experimentation, most likely resulting in an ensemble of fully and partially folded ACT molecules and uncertainty regarding the exact concentration of active toxin molecules. ACT purified in our laboratory exhibited similar in vitro activity when incubated with J774A.1 cells as described by Eby et al. (42): 125 ng/ml produced 10,000-∼40,000 pmol of cAMP per mg of total cellular protein in 30 min at 37 °C.
However, dialysis or dilution of ACT into buffer without urea led to protein that was poorly behaved and retained by the SEC column (<5% of protein applied to the column was eluted). In contrast, overnight dialysis into HBSC containing 1 m urea resulted in high molecular weight aggregates that eluted off the SEC column at a size corresponding to ∼600 kDa. An alternative refolding approach, rapid 10-fold dilution into HBSC (final urea concentration 0.8 m) followed immediately by SEC, yielded a broader aggregate peak and a smaller peak corresponding to the expected size for monomer (Fig. 1B). The different monomer yields may reflect a time-dependent aggregation process or the presence of folding intermediates with different aggregation propensities in the two refolding procedures. A recent report reiterated the challenges of refolding ACT (43).
Individual ACT Domains Are Biophysically Superior to ACT
To identify which, if any, ACT domains are predominantly recognized by polyclonal antibody responses, we expressed individual domains in E. coli with affinity tags to facilitate purification (Table 1). Based on prior reports (18, 29, 44–46), the N-terminal catalytic domains (residues 1–373 (CAT373), 1–385 (CAT385), and 1–400 (CAT400)) and C-terminal RTX domains (residues 751–1706 (RTX751) and 985–1706 (RTX985)) were cloned into the pET28a vector for cytoplasmic expression with N-terminal His6 tags to facilitate purification. In our hands, RTX(482–1706) was poorly soluble and purified inefficiently; instead we selected RTX985 as the largest fragment to exclude both acylation sites but retaining the N terminus before the first Gly-Asp rich repeat. To enhance solubility, the hydrophobic domain (residues 399–1096 (HP1096), encompassing the region between the catalytic and RTX domains) was fused downstream of MBP, with a C-terminal His6 tag and di-cistronic expression of the specific acylating enzyme CyaC (indicated by *). After cytoplasmic expression of each construct and cell lysis, a one-step affinity chromatography with a HisTrap column yielded ∼5–80 mg of protein per liter of culture with >90% purity as determined by SDS-PAGE (Fig. 1C). For HP-MBP fusion proteins, a second chromatographic step with an MBPTrap column was required to reach a similar level of purity, although purity, proteolysis, and solubility issues persisted.
TABLE 1.
Biophysical analysis of ACT constructs
ACT and various derivatives were purified and subjected to size exclusion chromatography in the presence of 2 mm calcium or an excess of EGTA to chelate free calcium ions. The expected molecular mass for each construct is noted, as is the observed size and thermal melting temperature of the major peak. Constructs noted with * were co-expressed with CyaC to acylate residues Lys-860 and Lys-983. ND means not determined.
| Domain | Theoretical mass | SEC calculated mass |
Melting temperature | |
|---|---|---|---|---|
| +Ca | +EGTA | |||
| kDa | kDa | °C | ||
| ACT | 177 | >600 | ND | ND |
| CAT400 | 45 | 40 | 40 | 43.3 |
| HP1096 | 118 | >600 | >600 | ND |
| HP1096* | 118 | >600 | >600 | ND |
| RTX985 | 77 | 78 | 380 | 40.7 |
| RTX751 | 102 | 108 | 115 | 57.7 |
| RTX751* | 102 | 91 | >600 | 56.8 |
To determine whether the purified domains exhibited native-like structure and expected calcium-dependent structural changes, SEC was used to assess the oligomeric state, and circular dichroism (CD) spectroscopy was used to assess the secondary structure content. The catalytic domains eluted as a single peak of expected size (40 kDa) with the estimated composition of secondary structures (56% helix, 14% strands, 13% turns, and 17% unordered) similar to that determined by x-ray crystallography (Fig. 2A) (18). Although the catalytic domain formally encompasses residues 1–373, the construct encompassing residues 1–400 was selected for further use to include the neutralizing epitope recognized by antibody 3D1 (47).
FIGURE 2.

ACT domain oligomeric state and secondary structure. Purified domains were separated by size exclusion chromatography (Superdex200 column, except Superdex75 for CAT400), with far UV circular dichroism spectra (Jasco J-815) used to assess secondary structure in the presence of 2 mm CaCl2 or the absence of calcium ions. A, catalytic domain, spanning residues 1–400 (CAT400), eluted as a single peak of expected size. The secondary structure is similar to that observed in the CAT373 crystal structure (56% helix, 14% strands, 13% turns, and 17% unordered). B, HP domain, spanning residues 399–1096, with an N-terminal MBP fusion protein eluted off SEC as high molecular weight aggregates whether acylated (*) or nonacylated. C, RTX985 formed high molecular weight aggregates in the absence of calcium ions but eluted as two peaks, one corresponding to the expected molecular mass of 78 kDa in the presence of calcium. Circular dichroism revealed significant conformational change upon addition of 2 mm CaCl2 corresponding to an increase in β-strand content. D, RTX751 domain exhibits a similar calcium-dependent delay in elution volume, which is more pronounced when the protein is acylated (*), and under these conditions it yields a single monomer peak. Inset, SDS-polyacrylamide gels show proteins present in the indicated peaks, with arrows indicating the expected monomer size.
The hydrophobic fusion proteins eluted as broad aggregate peaks when acylated or nonacylated. In the absence of acylation, multiple smaller peaks were observed, suggesting that acylation may stabilize folding of this domain and protect against proteolysis. In both cases, the CD spectra were not characteristic of unfolded or aggregated proteins (Fig. 2B). The solubility and CD spectra of these constructs may be dominated by the MBP fusion partner, but we did not attempt to remove it, as a shorter hydrophobic domain was reported to further aggregate under these circumstances (48).
The RTX domain includes ∼40 calcium binding Gly-Asp repeats, grouped into five blocks, and separated by non-RTX flanking regions (Fig. 1A). Structural data for RTX-containing proteins suggest the repeats fold into parallel β-helix structures. In the presence of calcium ions, the protein converts from an intrinsically disordered domain into a compact β-roll structure with an altered CD spectrum and a reduced hydrodynamic radius that appears to be further stabilized by acylation (43, 49, 50). RTX985 exhibited a shift from largely monomer in the presence of calcium (78 kDa) to a mixture of oligomers (∼380 kDa) upon the addition of EGTA to chelate calcium ions. These structural changes are captured by CD, which shows a more ordered state in the presence of calcium ions (Fig. 2C), consistent with that observed with a similar construct also lacking the acylation sites (residues 1006–1706) (51). SDS-PAGE indicates the two peaks observed with calcium have the same molecular weight suggesting that RTX985 forms two stable states with different hydrodynamic radii (Fig. 2C).
Theorizing that these two forms are a consequence of the missing acylation sites, we generated a larger construct to include both sites. RTX751 expressed without CyaC eluted as a single peak of expected size (∼110 kDa) in the presence or absence of calcium. When co-expressed with CyaC, presumably resulting in acylation at residues Lys-860 and Lys-983, RTX751* exhibited a calcium-dependent conversion from a compact monomer (∼90 kDa) to a soluble higher molecular weight aggregate (∼600 kDa) after depletion of calcium (Fig. 2D).
The catalytic and RTX domains retain much of the expected structural behavior, with RTX751* appearing to better stabilize the monomeric form than RTX985. Similar to previous reports on ACT behavior (26, 27, 51–53), the presence of calcium and acylation appears to stabilize the RTX monomers. Anecdotally, the RTX domains were stable for at least 6 months at 4 °C with minimal aggregation or degradation (RTX751 and RTX751* were slightly more stable than RTX985), as measured by SDS-PAGE and SEC to assess the monomeric fraction. Although the CAT400 domain also remains monomeric, it starts to degrade after 3 months under the same conditions, as measured by SDS-PAGE. The compact β-roll structure RTX domains adopt in the presence of calcium may contribute to their overall higher melting temperature and resistance to proteolysis than CAT400 (Table 1).
ACT Domains Are Biochemically Similar to ACT
To determine whether our domain constructs retain structural elements present in ACT, we screened a panel of nine previously characterized monoclonal antibodies for binding to ACT and individual domains by ELISA (47). All nine antibodies tested recognized only the expected domain and did not distinguish between acylated and nonacylated domains (Table 2), supporting the notion that the domains are properly folded. One exception is 2B12, whose epitope includes residues 888–1006, but did not recognize HP1096. This may be due to incomplete folding of HP1096 or the binding site may require additional residues distal to residue 1006 not present in this construct.
TABLE 2.
Biochemical analysis of ACT constructs
Binding of previously characterized anti-ACT murine antibodies (47) to ACT and various derivatives described in this work in ELISAs. ACT constructs were coated on ELISA plates, and thee antibodies were titrated and detected with anti-mouse IgG-HRP.
| ACT domain | 3D1 epitope 373–399 | 2A12 epitope 399–828 | 10A1 epitope 624–780 | 2B12 epitope 888–1006 | 6E1 epitope 1320–1489 | 9D4 epitope 1156–1489 | 7C7 epitope 1320–1627 | 1H6 epitope 1590–1706 | 10A8 epitope 1590–1706 |
|---|---|---|---|---|---|---|---|---|---|
| CAT400 | +++ | ||||||||
| HP1096 | +++ | + | |||||||
| HP1096* | +++ | + | |||||||
| RTX985 | ++ | ++ | ++ | +++ | +++ | ||||
| RTX751 | + | ++ | ++ | + | +++ | +++ | |||
| RTX751* | + | ++ | ++ | + | +++ | +++ | |||
| ACT | +++ | +++ | ++ | ++ | +++ | +++ | + | ++ | +++ |
As the RTX domain harbors the receptor-binding site between residues 1166 and 1281 (26), we assessed the ability of our RTX constructs to bind purified murine extracellular αMβ2 receptor, a known ACT cell surface receptor (25). Although ACT and RTX751* both bound the murine receptor when acylated and in the presence of calcium (apparent EC50 ∼20 nm; Fig. 3), ACT exhibited considerable nonspecific binding to wells without the receptor. This is similar to the sticky behavior observed when ACT without urea was applied to and retained by the SEC column and likely reflects the presence of solvent-exposed hydrophobic patches in misfolded ACT molecules. Monomeric RTX985 did not bind the αMβ2 receptor, consistent with prior studies showing that post-translational acylation is essential for receptor binding (29, 54). In summary, the individual domains were readily purified, with yields of CAT400 at ∼80 mg/liter culture, nonacylated RTX and HP domains at ∼5 mg/liter culture, and the acylated domains at <2 mg/liter culture. The CAT and RTX domains share many structural features with ACT, although the HP domain is mostly aggregated.
FIGURE 3.
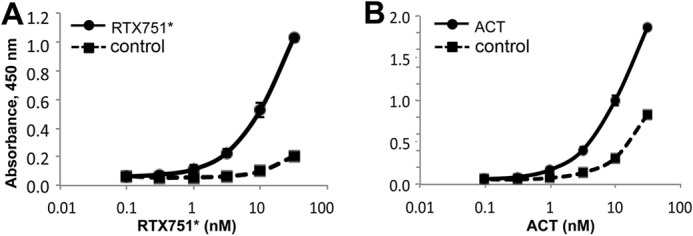
ACT and RTX domains bind purified αMβ2 receptor. Soluble murine αMβ2 receptor was coated onto ELISA plates and blocked, and ACT or domains were serially diluted in M-PBST. Bound protein was detected with polyclonal rabbit anti-ACT antibody and goat anti-rabbit HRP. To assess nonspecific binding, control wells were not coated with αMβ2 receptor but blocked with M-PBST only. A, RTX751*, and B, acylated ACT* showed receptor-dependent binding, although ACT also exhibited significant nonspecific binding. All other domains showed no specific or nonspecific binding.
Anti-ACT Antibodies Primarily Recognize RTX
To determine whether a single ACT domain dominates the immune response, we used phage display to analyze murine antibody repertoires after ACT immunization. Two mice were immunized intraperitoneally with 17 μg of ACT in complete Freund's adjuvant and were boosted subcutaneously once with incomplete Freund's adjuvant. The resulting sera neutralized the toxic activities of ACT at a 1:400 dilution in an in vitro cAMP intoxication assay (data not shown). The spleens were harvested, and each was used to construct an antibody phage display library, each containing ∼107 total transformants. After two rounds of panning against ACT, 90 individual clones from each library were grown in 96-well plates and assessed for ACT binding by phage ELISA. Of these, 57 and 60 clones from the two libraries respectively yielded signals 2-fold above background, with 29 and 21 expressing unique sequences, as determined by BstNI fingerprinting and DNA sequencing.
To determine the domain specificities of unique antibodies, monoclonal phage were assessed for binding to individual domains in ELISA (Fig. 4A). Few antibodies bound the catalytic domain; none bound the hydrophobic domain, whereas the rest (27 of 29 and 20 of 21, respectively) bound the RTX985 domain. This observed high frequency of RTX985-specific antibodies concurs with previous reports by Lee et al. (47) that the majority of antibodies discovered using hybridomas recognized the RTX domain and by Betsou et al. (29) that this domain may be immunodominant.
FIGURE 4.
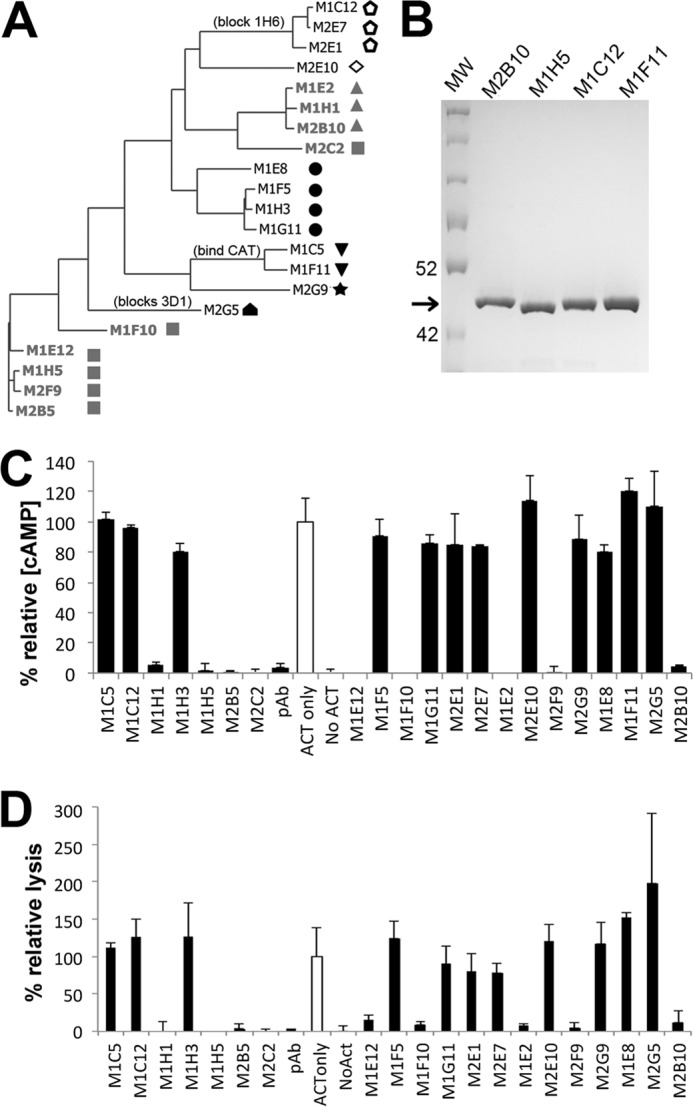
ACT immunization induces a diverse antibody response. A, phylogenetic tree depicting antibody sequence relatedness was generated using the light and heavy variable region amino acid sequences. Neutralizing scAbs are colored gray, with unique shapes denoting recognition of distinct epitopes among this antibody group as determined by competition ELISA. Open symbols denote antibodies whose binding does not depend on the presence of calcium. Antibodies competing with previously characterized monoclonal antibodies are indicated; all antibodies bind RTX except M1C5, M1F11, and M2G5, which bind CAT400. B, representative SDS-PAGE of scAbs after purification by IMAC and Superdex S200. Arrow indicates expected size of ∼40 kDa, 2 μg each of M1F11, M1C12, M1H5, and M2B10 scAbs were loaded. C, 21 unique scAbs identified from the immune phage libraries were tested for the ability to neutralize ACT-mediated increases in intracellular cAMP concentration. ACT was incubated with a 160-fold molar excess of scAb protein before adding to J774A.1 cells. Data are reported as the percent relative cAMP, calculated from the total cAMP concentration in the cellular lysate as determined by cAMP ELISA, divided by the protein concentration of the lysate, and normalized to control cells treated only with ACT (open bar). Error bars indicate range of duplicate assays. D, 21 scAbs were evaluated for their ability to rescue J774A.1 macrophages from ACT-induced lysis, using a similar protocol as for cAMP neutralization. Cell lysis was measured via lactate dehydrogenase release using the Cytotox 96 kit (Promega), normalized to control cells treated only with ACT (empty bar), and reported as the percent relative lysis. Error bars indicate standard deviation of triplicate assays.
Anti-RTX Antibodies Can Neutralize ACT
Next, we screened unique antibodies identified from phage libraries for the ability to neutralize ACT activities using an in vitro cAMP assay. There are several steps during ACT intoxication of cells that are susceptible to antibody-mediated neutralization, including receptor binding, membrane insertion, and translocation, and interference with any of these will be reflected in decreased intracellular cAMP accumulation in or reduced lysis of target cells. For this assay, we employed the murine macrophage cell line J774A.1 bearing the αMβ2 integrin.
For the neutralization assay, antibodies were expressed as recombinant scAbs, composed of the variable light chain (VL) joined to a flexible (Gly4Ser)2 linker and the variable heavy chain (VH), followed by a C-terminal human κ chain constant region to increase solubility and serve as a detection handle (9). Based on multiple sequence alignments, 31 antibodies with unique complementary determining regions were selected for scAb expression (Fig. 4B). Ten scAbs either expressed poorly (<200 μg/liter culture) or bound ACT weakly (concentration >238 nm required for saturation) and were not tested further.
To identify antibodies neutralizing ACT function, ACT and individual scAbs were incubated at a 1:160 molar ratio before addition to adherent J774A.1 cells and determination of intracellular cAMP levels by competition ELISA. Of the 21 scAbs tested, nine reduced the cAMP level by more than 90%, as compared with cells treated with ACT alone, which we consider highly neutralizing in this assay (Fig. 4C). We also determined the ability of these scAbs to rescue J774A.1 cells from lysis using a lactate dehydrogenase release assay, observing a strong correlation with cAMP neutralization (Fig. 4D). This is in agreement with findings by Basler et al. (55) that intracellular ATP depletion is sufficient to promote cell lysis. Notably, all neutralizing antibodies identified recognize the RTX domain.
Two Novel Neutralizing Epitopes in the RTX Domain
We next sought to classify neutralizing antibodies based on their recognition of unique or overlapping epitopes. Here, we used a competitive binding ELISA, in which a single phage-displayed antibody was mixed with buffer or a second antibody in the scAb format, added to an ELISA well coated with ACT, and followed by detection of bound phage remaining in the well. Reduced signal in the presence of a second antibody compared with phage antibody alone indicates competition between the two antibodies for the same or overlapping epitopes. Using this approach, the nine neutralizing antibodies were divided into two groups binding nonoverlapping epitopes, although non-neutralizing antibodies recognized four unique epitopes, for a total of six epitopes represented in this study (Fig. 4A).
One representative antibody binding each neutralizing epitope was selected based on sequence uniqueness, expression level, and binding affinity for conversion into a full-length chimeric immunoglobulin, with human IgG1 and κ constant domains (Fig. 5A). ELISAs with the two RTX constructs helped to further define the epitopes recognized by these two antibodies, named M1H5 and M2B10. Both antibodies bound RTX751* with almost the identical affinity as full-length ACT (Fig. 5B), whereas M1H5 bound the shorter RTX985 domain weakly (Fig. 5C), suggesting that its epitope is not fully contained or properly presented in this construct.
FIGURE 5.

Two novel neutralizing epitopes are present in the RTX domain. A, two representative neutralizing scAbs were converted to chimeric IgG1/κ antibodies, in which the murine variable regions are appended by human constant domains. These were transiently expressed in CHO-K1 cells and purified by (NH4)2SO4 precipitation and protein A affinity chromatography. SDS-PAGE (4–20% gradient gel, 2 μg loaded) shows high purity and expected size of the recombinant IgG as follows: 25 kDa (light chain) and 50 kDa (heavy chain) when reduced and 150 kDa under nonreducing conditions. ELISA demonstrates the ACT domain specificity of the M2B10 (B) and M1H5 (C) IgG antibodies. Microtiter plates were coated with ACT or ACT domains at equimolar concentrations, followed by serial dilution of antibody from 1 nm, and followed by detection with anti-human Fc antibody-HRP conjugate. D, competition ELISA determined that the M2B10 and M1H5 antibodies bind novel nonoverlapping epitopes. A 200-fold molar excess (20 nm) of previously described murine mAbs (3D1, 2A12, 10A1, 2B12, 9D4, 6E1, 7C7, and 1H6) (47) or scAb versions of M1H5 and M2B10 were mixed with M2B10 and M1H5 IgG (0.1 nm) and incubated on ACT-coated ELISA plate, with bound M2B10 or M1H5 detected as above. The absorbance was normalized to that of M2B10 or M1H5 with no competitor; absorbance significantly <1.0 indicates competition between the antibody pair.
To determine whether the antibodies identified here bind epitopes overlapping with those of previously defined murine monoclonal antibodies (47), we performed a second set of competition ELISAs (Fig. 5D and data not shown). None of the murine antibodies competed with M2B10 or M1H5 for ACT binding, demonstrating that these antibodies recognize previously undescribed neutralizing epitopes in the RTX region.
Together, we observed antibodies binding six nonoverlapping epitopes on RTX, two neutralizing and four non-neutralizing, and all but two required the presence of calcium (Fig. 4A). A representative antibody binding one epitope, M1C12, competes with antibody 1H6, one of the four non-neutralizing RTX binding antibodies discovered by Lee et al. (47). Their remaining three anti-RTX antibodies bind unique epitopes, suggesting a total of at least nine distinct epitopes in the RTX domain. Identification of new epitopes is not unexpected, because neither hybridoma nor phage display technology is exhaustive, and each has trade-offs; the former is low throughput and labor-intensive, and the latter does not preserve the pairing between the light and heavy chains and preferentially selects for antibodies with high bacterial expression levels. Even recently described repertoire mining approaches based on high throughput sequencing of antibodies from individual B cells do not identify the same sequences as phage display (56).
Antibodies Binding Novel RTX-neutralizing Epitopes Disrupt ACT-αMβ2 Integrin Binding
ACT primarily targets cells bearing the αMβ2 receptor under conditions in which the RTX domain assumes a receptor-binding competent conformation mediated by the presence of calcium ions and post-translational acylation (26). Combining this with our observation that the M2B10 and M1H5 antibodies showed a much weaker neutralizing effect at the same 160-fold molar excess when CHO-K1 cells lacking this receptor were used than when J774A.1 cells expressing the receptor were used (Fig. 6A), we hypothesized that these two antibodies act by blocking the interaction between ACT and the αMβ2 integrin.
FIGURE 6.

M2B10 and M1H5 antibodies block the ACT-αMβ2 integrin interaction. A, antibody neutralization of cAMP intoxication using J774A.1 presenting the αMβ2 receptor and CHO-K1 cells lacking the receptor. Both assays were performed with a 160-fold molar excess of scAb over ACT and analyzed as in Fig. 4. B, antibody blockade of ACT binding to J774A.1 cells assessed by FACS. Biotinylated ACT was incubated with a 300-fold molar excess of scAb and added to 4 × 105 J774A.1 suspension cells on ice. After washing, bound biotinylated ACT was detected with streptavidin-PE and analyzed by FACS (mean fluorescence noted next to each peak). Controls include untreated cells (Cells only) and cells treated with nonbiotinylated ACT followed by streptavidin-PE (ACT). C, antibody blockade of ACT binding to soluble αMβ2 integrin by ELISA. ACT (0.5 μg/ml) was incubated with serial dilutions of M2B10, M1H5, 3D1 and 2A12 antibodies at 5-, 1.6-, and 0.5-fold molar excess, before transfer to an ELISA plate coated with murine αMβ2 integrin. Bound ACT was detected with rabbit anti-ACT polyclonal antibody followed by HRP-conjugated goat anti-rabbit IgG antibody. D, antibody neutralization of ACT secreted by B. pertussis. Antibodies at 10, 2.5, 0.63, and 0.16 μg/ml were incubated with live B. pertussis (A600 = 0.2) before adding to adherent J774A.1 cells. The resulting intracellular cAMP concentrations were measured, normalized to total protein concentration, and expressed as % relative cAMP.
To test this hypothesis, we used flow cytometry to monitor ACT bound to J774A.1 cells in the presence or absence of the M2B10 and M1H5 scAbs. Biotinylated ACT was incubated with a 300-fold molar excess of neutralizing or non-neutralizing scAbs, added to J774A.1 cells and detected with phycoerythrin-conjugated streptavidin by FACS. The M2B10 and M1H5 antibodies significantly reduced binding of ACT-biotin to J774A.1 cells, whereas two non-neutralizing control scAbs (M1F11 and M1C12) had no significant effect (Fig. 6B). This difference was not due to affinity, as all four scAbs have similar affinities (EC50 = 0.3–0.7 nm; data not shown).
To further confirm that the diminished binding of ACT to J774A.1 cells was due to interference with a specific receptor, a competition ELISA with soluble αMβ2 integrin was performed. ACT (0.5 μg/ml) was incubated with the M2B10, M1H5, M1F11, or 3D1 antibodies in molar ratios ranging from 5 to 0.5 and transferred to an αMβ2 integrin-coated plate. The result was consistent with the flow cytometry assay; M2B10 and M1H5 reduced ACT binding to immobilized αMβ2 integrin in a dose-dependent manner, >90% at a 2-fold molar excess (Fig. 6C). In contrast, 3D1, a neutralizing IgG that blocks translocation of the catalytic domain (57), and 2A12, a neutralizing antibody with unclear mode-of-action, had no effect. Minimal nonspecific binding was observed under these assay conditions.
To determine whether ACT neutralization occurs in the context of the whole bacterium, we repeated this assay with live B. pertussis instead of purified ACT. According to Gray et al. (28), newly synthesized ACT is responsible for intoxication. Therefore, B. pertussis was washed in PBS to remove any secreted ACT. Bacteria (A600 = 0.2) added to J774A.1 cells resulted in cAMP levels similar to that induced by 125 ng/ml purified ACT. When the M2B10 and M1H5 but not the non-neutralizing M1F11 or 7C7 (47) antibodies were added with the bacteria, they resulted in dose-dependent reduction of cAMP levels (Fig. 6D). This suggests these antibodies may be able to neutralize ACT in the context of active infection.
RTX Domain Is Immunodominant and Elicits Neutralizing Antibodies
To determine whether any ACT domain dominates the immune response, we immunized mice with ACT and tested the resulting sera 4 weeks after primary immunization for binding to different ACT domains. Interestingly, strong responses were observed for ACT, RTX985, and RTX751, but no responses were observed for the CAT400 or HP1096* domains (Fig. 7A).
FIGURE 7.
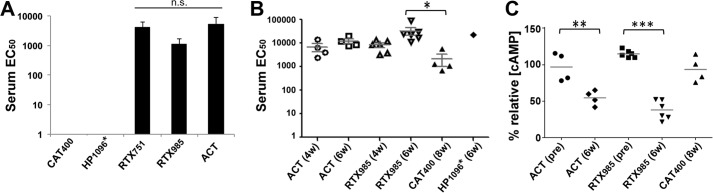
RTX domain is immunodominant and elicits neutralizing antibodies. A, immunization with ACT yields sera preferentially recognizing the RTX domain. Purified domains were coated at equal moles on microtiter plates, with sera serially diluted starting at 1:200. The average EC50 for individual domains is shown. Error bars are the standard deviations of the EC50 value among four mice. B, immunogenicity of purified domains. Mice were immunized with intact ACT or individual domains, with the serum EC50 for ACT measured by ELISA after the first boost (6 weeks) or second boost (8 weeks). C, sera from mice immunized with the ACT and RTX985 domain neutralize cAMP intoxication similarly. Sera from each immunization group at a 1:400 dilution were incubated with 125 ng/ml ACT in DMEM before adding to J774A.1 cells. Intracellular cAMP levels were measured by cAMP ELISA, divided by the total protein concentrations, and normalized to cells treated with ACT alone, as in Fig. 4. pre indicates baseline sera collected prior to immunization. Statistical significance was determined by one-way analysis of variance with Tukey's test; **, p ≤ 0.01; ***, p ≤ 0.001. For all panels, * indicates an acylated domain. n.s. indicates not significant (p > 0.05).
To determine whether this was the result of RTX immunodominance or a lack of immunogenicity by the CAT and HP domains, we immunized additional groups of mice with CAT400, HP1096*, or RTX985. Here, RTX985 was selected because it shares many features with RTX751*, including recognition by at least one neutralizing antibody, yet it lacks the acylation sites rendering it simpler to produce and less likely to engage the native receptor. The calcium concentration in extracellular fluid in the body is 2.2–2.7 mm (59), which is sufficient to support bacterial secretion and folding of ACT during an infection and is expected to support proper folding during immunization.
Mice immunized with ACT or RTX985 showed high anti-ACT titers 4 weeks after the first injection, which increased after boosting (weeks 6 and 8, Fig. 7B). On the contrary, the catalytic domain was much less immunogenic, reaching a detectable anti-ACT titer of ∼1500 only after two boosts (week 8; Fig. 7B). This weak response may reflect structural similarities between CAT and eukaryotic adenylate cyclases, resulting in immunological tolerance or an evolutionary mechanism to protect key toxin components from neutralizing antibodies (60, 61). Only one of the four mice immunized with the hydrophobic domain reacted with ACT, supporting the SEC data that this construct is poorly folded (Fig. 7B). Antibody responses in the three remaining mice were directed toward the MBP fusion, as determined by ELISA (data not shown).
Next, we wanted to determine which domains induced sera best able to neutralize ACT cAMP intoxication activities in vitro with J774A.1 cells. Here, only immunization with RTX985 elicited sera able to protect cells to a similar extent as sera elicited by full-length ACT (Fig. 7C). Although mechanisms other than receptor blockade may contribute to neutralization, the presence of M2B10- and M1H5-like antibodies in the sera of ACT- or RTX-immunized animals was confirmed by competition ELISA (Fig. 8). Because RTX985 binds antibody M1H5 weakly, this domain may have sufficient conformational similarities as ACT to induce antibodies binding overlapping but nonidentical epitopes as M1H5.
FIGURE 8.

Mice produce antibodies binding the M1H5 and M2B10 neutralizing epitopes whether immunized with ACT (A) or RTX985 (B). Sera at a 1:200 dilution were incubated on ELISA plates coated with ACT, before addition of 0.1 nm M2B10, M1H5, or M1F11 as a competitor. After incubation, immobilized monoclonal antibody was detected with anti-human Fc-HRP, with absorbance normalized to wells without sera. Lower absorbance indicates a higher concentration of epitope-specific murine antibodies.
Finally, to determine whether humans show a similar bias toward RTX recognition, we tested nine serum samples from humans exposed to B. pertussis, selected randomly from a larger collection (62). All nine sera recognized RTX751 at a similar level as full-length ACT, whereas sera from only one individual bound CAT400 (Fig. 9). Taken together, these data provide proof-of-concept that RTX dominates the anti-ACT immune response and that the RTX domain can recapitulate the humoral immune responses induced by ACT.
FIGURE 9.
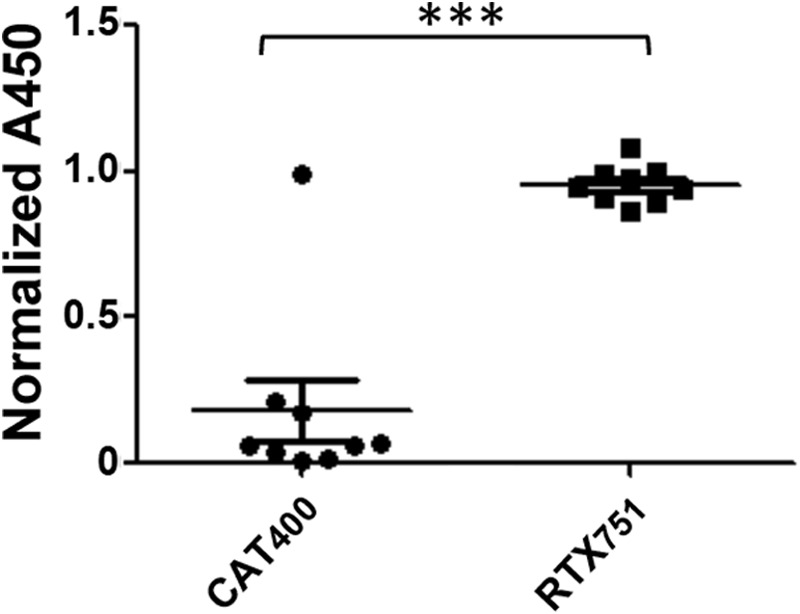
RTX dominates the human immune response to ACT. Nine serum samples from humans exposed to B. pertussis were tested for reactivity to the catalytic domain, RTX751, or intact ACT by ELISA. Absorbance values at a 100-fold dilution of the sera were normalized to that of ACT at the same dilution. A paired t test was used to determine the statistical significance between signals for CAT and RTX binding domains. ***, p ≤ 0.001.
DISCUSSION
The recent surge in pertussis cases, coupled with increasing recognition of the current acellular vaccine's shortcomings, has motivated design of third generation vaccines to prevent pertussis. In humans, even a single dose of whole cell vaccine significantly reduces the risk of illness (63), although in baboons, acellular immunization prevented the severe symptoms of disease but allowed bacterial persistence and transmission to naive animals (5). To design future vaccines that minimize subclinical disease and reduce transmission to susceptible infants, it is crucial to understand the roles played by various protective antigens. ACT has been shown to be protective in animal models and is immunogenic in humans (15, 16, 40, 64, 65). Because ACT activities hinder local anti-bacterial immune responses (66–68), anti-ACT antibodies may protect these cells, indirectly facilitating bacterial elimination. Here, we demonstrated that the RTX domain is able to largely recapitulate the protective humoral immune response induced by ACT in mice and is better expressed and more stable than the intact ACT.
ACT was first discovered based on its ability to increase cAMP levels in neutrophils, inhibiting their anti-bacterial functions, including phagocytosis and respiratory burst, and promoting the early stages of disease establishment (66). At physiologically relevant concentrations (<50 ng/ml) (69), ACT results in cytotoxicity to the murine macrophage cell line J774A.1 after 2 h of exposure; ACT also causes chlorine efflux from polarized epithelial cells and hinders IL-2 secretion and proliferation of T cells (42). More recently, ACT has been shown to suppress development of an IL-17-mediated immune response that appears key for bacterial clearance (21). As a result, passively administered antibodies blocking ACT function may be able to enhance neutrophil-mediated phagocytosis of opsonized bacteria (67). Murine studies have shown that immunization with ACT alone or as a supplement to the acellular vaccine reduces bacterial colonization, an effect that correlated with increased immunoglobulin levels and a Th1/Th2 cytokine phenotype (16, 54). Finally, ACT is a highly conserved antigen, able to induce protective immunity in mouse models against the three dominant Bordetella species (B. pertussis, B. parapertussis, and B. bronchiseptica) (70–72). Although ACT is unlikely to be highly protective as an isolated antigen, it may be a valuable addition to vaccines.
The complex mechanism by which ACT directly translocates its catalytic domain into the host cell cytosol remains incompletely understood. The current working model consists of three steps (48, 73, 74). First, in the presence of millimolar levels of calcium and acylation, the RTX domain forms a β-barrel that binds the αMβ2 receptor on neutrophils or the β2-containing integrin lymphocyte fusion-associated antigen 1 receptor on T cells through N-linked oligosaccharides. Second, this is followed by insertion of two loops (four predicted transmembrane α-helices between residues 502–522 and 565–591) into the host cell membrane, resulting in a translocation intermediate that permeabilizes the membrane to allow an influx of extracellular calcium ions, activating calpain-mediated cleavage of the integrin's talin tether. Third, the ACT-receptor complex is then free to diffuse to cholesterol-rich lipid rafts, which triggers complete translocation of the catalytic domain dependent on residues 375–485 (48). Interestingly, the catalytic domain allows insertion of peptides up to 206 amino acids long for intracellular delivery (75).
This structure-function information provides insight into epitopes required for cellular intoxication and those likely to induce protective responses. For instance, antibodies could block translocation steps, yet only one such antibody has been characterized. Immunization with ACT followed by analysis of the resulting polyclonal serum suggested that antibodies recognizing the RTX domain dominate the response, as 6 of 12 monoclonal antibodies recognized this domain (47, 72). The 3D1 antibody binds a conformational epitope between residues 373 and 399, adjacent to the catalytic domain, trapping a translocation intermediate and preventing complete delivery of the catalytic domain to the host cytosol (48, 73), while the anti-hydrophobic region antibody 2A12 inhibits intoxication and, to a lesser extent, hemolysis, and the anti-RTX antibody 6E1 inhibits only hemolysis (47). Consistent with these prior reports, we observed that the majority of antibodies recovered from phage libraries bind the RTX domain, whereas sera from four mice immunized with the holo-toxin bind RTX only. To the best of our knowledge, no antibodies blocking the ACT-receptor binding, such as M1H5 or M2B10, have been previously described.
A barrier to development of additional technologies based on ACT has been the challenges of recovering monomeric protein from in vitro refolding processes. Standard protocols, including dilution and dialysis from denaturing buffers containing 8 m urea, recover high molecular weight species with variable activity levels. Recently, refolding on a size exclusion column was performed to prevent aggregation of partially folded species and resulted in purification of monomers with very high activity, which depended on the presence of calcium, acylation and molecular confinement (43). Although promising, the yields and scalability of this process are currently unclear. As shown here, the RTX domain retains many structural features, is more readily expressed, and exhibits greater stability than ACT. Furthermore, because RTX lacks the catalytic domain, it has no homology to endogenous proteins and thus poses no potential autoimmune concerns for human use. We evaluated two different RTX constructs, initially RTX985 lacking the acylation sites and thus simpler to express, and then the larger RTX751* retaining the acylation sites. Although both exhibited expected calcium-dependent structural shifts and binding to previously described monoclonal antibodies, RTX751* appears superior to RTX985 in terms of monomericity, stability, and recognition of soluble αMβ2 receptor in vitro.
Because RTX985 is well behaved and binds at least one neutralizing antibody (M2B10), we used this construct for immunization before discovering that RTX751 and RTX751* are better behaved and retain the ability to bind both neutralizing antibodies. Regardless, sera from mice immunized with RTX985 neutralized cAMP intoxication in vitro as efficiently as sera from ACT-immunized mice (Fig. 7C). This may be because these nonoverlapping epitopes induce antibodies neutralizing toxin via similar receptor blocking mechanisms. Supporting this idea, no synergy was observed when the M2B10 and M1H5 antibodies were combined in vitro (data not shown). Thus, it may be possible to induce a strong neutralizing response when only one of the epitopes is structurally intact. We do not have data on RTX751* immunization, but based on the biochemical and biophysical data presented here, we expect it to perform as well or better than RTX985.
Structure-function analyses of antibody-antigen interactions can identify residues forming protective epitopes, key information to guide design of immunogens able to elicit neutralizing antibodies. This approach has been employed for complex antigens with high sequence variability and metastable protective epitopes, such as fHBP from Neisseria meningitidis (76), and the F-protein from respiratory syncytial virus (77). We have demonstrated proof-of-concept that constructs based on the RTX domain are better behaved than ACT, while retaining key epitopes and inducing neutralizing antibodies. Using the structures of homologous RTX toxins (58, 78), RTX variants with enhanced stability, expression level, and reduced immunogenicity of non-neutralizing epitopes could be engineered to evaluate RTX as a vaccine antigen in murine challenge models.
Acknowledgments
We thank Professors Erik Hewlett (University of Virginia) for providing ACT and anti-ACT antibodies, Shelley Payne (University of Texas at Austin) for providing the J774A.1 cell line, and Patrick C. Wilson (University of Chicago) for providing the Igκ-Abvec and IgG-Abvec plasmids. We also thank undergraduate researcher Brennan Hodgson for data collection. The reagent adenylate cyclase from Bordetella pertussis, NR-34572, was obtained through BEI Resources, NIAID, National Institutes of Health.
This work was supported, in whole or in part, by National Institutes of Health Grant GM095638 (to J. A. M.). This work was also supported by Welch Foundation Grant F-1767.
- ACT
- adenylate cyclase toxin
- RTX
- repeat-in-toxin
- MBP
- maltose-binding protein
- scAb
- single-chain antibody fragment
- PE
- phycoerythrin
- IMAC
- immobilized metal affinity chromatography
- IPTG
- isopropyl β-d-thiogalactopyranoside
- SEC
- size exclusion chromatography
- CAT
- catalytic
- HP
- hydrophobic.
REFERENCES
- 1. Witt M. A., Katz P. H., Witt D. J. (2012) Unexpectedly limited durability of immunity following acellular pertussis vaccination in preadolescents in a North American outbreak. Clin. Infect. Dis. 54, 1730–1735 [DOI] [PubMed] [Google Scholar]
- 2. Octavia S., Sintchenko V., Gilbert G. L., Lawrence A., Keil A. D., Hogg G., Lan R. (2012) Newly emerging clones of Bordetella pertussis carrying prn2 and ptxP3 alleles implicated in Australian pertussis epidemic in 2008–2010. J. Infect. Dis. 205, 1220–1224 [DOI] [PubMed] [Google Scholar]
- 3. Higgs R., Higgins S. C., Ross P. J., Mills K. H. (2012) Immunity to the respiratory pathogen Bordetella pertussis. Mucosal. Immunol. 5, 485–500 [DOI] [PubMed] [Google Scholar]
- 4. Sheridan S. L., Ware R. S., Grimwood K., Lambert S. B. (2012) Number and order of whole cell pertussis vaccines in infancy and disease protection. JAMA 308, 454–456 [DOI] [PubMed] [Google Scholar]
- 5. Warfel J. M., Zimmerman L. I., Merkel T. J. (2014) Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a non-human primate model. Proc. Natl. Acad. Sci. U.S.A. 111, 787–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lim A., Ng J. K., Locht C., Alonso S. (2014) Protective role of adenylate cyclase in the context of a live pertussis vaccine candidate. Microbes Infect. 16, 51–60 [DOI] [PubMed] [Google Scholar]
- 7. Polewicz M., Gracia A., Garlapati S., van Kessel J., Strom S., Halperin S. A., Hancock R. E., Potter A. A., Babiuk L. A., Gerdts V. (2013) Novel vaccine formulations against pertussis offer earlier onset of immunity and provide protection in the presence of maternal antibodies. Vaccine 31, 3148–3155 [DOI] [PubMed] [Google Scholar]
- 8. Cherry J. D., Heininger U., Richards D. M., Storsaeter J., Gustafsson L., Ljungman M., Hallander H. O. (2010) Antibody response patterns to Bordetella pertussis antigens in vaccinated (primed) and unvaccinated (unprimed) young children with pertussis. Clin. Vaccine Immunol. 17, 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berbers G. A., de Greeff S. C., Mooi F. R. (2009) Improving pertussis vaccination. Hum. Vaccin. 5, 497–503 [DOI] [PubMed] [Google Scholar]
- 10. Chenal-Francisque V., Caro V., Boursaux-Eude C., Guiso N. (2009) Genomic analysis of the adenylate cyclase-hemolysin C-terminal region of Bordetella pertussis, Bordetella parapertussis, and Bordetella bronchiseptica. Res. Microbiol. 160, 330–336 [DOI] [PubMed] [Google Scholar]
- 11. Parkhill J., Sebaihia M., Preston A., Murphy L. D., Thomson N., Harris D. E., Holden M. T., Churcher C. M., Bentley S. D., Mungall K. L., Cerdeño-Tárraga A. M., Temple L., James K., Harris B., Quail M. A., Achtman M., Atkin R., Baker S., Basham D., Bason N., Cherevach I., Chillingworth T., Collins M., Cronin A., Davis P., Doggett J., Feltwell T., Goble A., Hamlin N., Hauser H., Holroyd S., Jagels K., Leather S., Moule S., Norberczak H., O'Neil S., Ormond D., Price C., Rabbinowitsch E., Rutter S., Sanders M., Saunders D., Seeger K., Sharp S., Simmonds M., Skelton J., Squares R., Squares S., Stevens K., Unwin L., Whitehead S., Barrell B. G., Maskell D. J. (2003) Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35, 32–40 [DOI] [PubMed] [Google Scholar]
- 12. Carbonetti N. H., Artamonova G. V., Andreasen C., Bushar N. (2005) Pertussis toxin and adenylate cyclase toxin provide a one-two punch for establishment of Bordetella pertussis infection of the respiratory tract. Infect. Immun. 73, 2698–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goodwin M. S., Weiss A. A. (1990) Adenylate cyclase toxin is critical for colonization and pertussis toxin is critical for lethal infection by Bordetella pertussis in infant mice. Infect. Immun. 58, 3445–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khelef N., Sakamoto H., Guiso N. (1992) Both adenylate cyclase and hemolytic activities are required by Bordetella pertussis to initiate infection. Microb. Pathog. 12, 227–235 [DOI] [PubMed] [Google Scholar]
- 15. Guiso N., Rocancourt M., Szatanik M., Alonso J.-M. (1989) Bordetella adenylate cyclase is a virulence associated factor and an immunoprotective antigen. Microb. Pathog. 7, 373–380 [DOI] [PubMed] [Google Scholar]
- 16. Guiso N., Szatanik M., Rocancourt M. (1991) Protective activity of Bordetella adenylate cyclase-hemolysin against bacterial colonization. Microb. Pathog. 11, 423–431 [DOI] [PubMed] [Google Scholar]
- 17. Farfel Z., Könen S., Wiertz E., Klapmuts R., Addy P. A., Hanski E. (1990) Antibodies to Bordetella pertussis adenylate cyclase are produced in man during pertussis infection and after vaccination. J. Med. Microbiol. 32, 173–177 [DOI] [PubMed] [Google Scholar]
- 18. Guo Q., Shen Y., Lee Y. S., Gibbs C. S., Mrksich M., Tang W. J. (2005) Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 24, 3190–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raptis A., Knipling L., Wolff J. (1989) Dissociation of catalytic and invasive activities of Bordetella pertussis adenylate cyclase. Infect. Immun. 57, 1725–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paccani S. R., Dal Molin F., Benagiano M., Ladant D., D'Elios M. M., Montecucco C., Baldari C. T. (2008) Suppression of T-lymphocyte activation and chemotaxis by the adenylate cyclase toxin of Bordetella pertussis. Infect. Immun. 76, 2822–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henderson M. W., Inatsuka C. S., Sheets A. J., Williams C. L., Benaron D. J., Donato G. M., Gray M. C., Hewlett E. L., Cotter P. A. (2012) Contribution of Bordetella filamentous hemagglutinin and adenylate cyclase toxin to suppression and evasion of interleukin-17-Mediated Inflammation. Infect. Immun. 80, 2061–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rossi Paccani S., Benagiano M., Capitani N., Zornetta I., Ladant D., Montecucco C., D'Elios M. M., Baldari C. T. (2009) The adenylate cyclase toxins of Bacillus anthracis and Bordetella pertussis promote Th2 cell development by shaping T cell antigen receptor signaling. PLoS Pathog. 5, e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benz R., Maier E., Ladant D., Ullmann A., Sebo P. (1994) Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 269, 27231–27239 [PubMed] [Google Scholar]
- 24. Hackett M., Walker C. B., Guo L., Gray M. C., Van Cuyk S., Ullmann A., Shabanowitz J., Hunt D. F., Hewlett E. L., Sebo P. (1995) Hemolytic, but not cell-invasive activity, of adenylate cyclase toxin is selectively affected by differential fatty-acylation in Escherichia coli. J. Biol. Chem. 270, 20250–20253 [DOI] [PubMed] [Google Scholar]
- 25. Guermonprez P., Khelef N., Blouin E., Rieu P., Ricciardi-Castagnoli P., Guiso N., Ladant D., Leclerc C. (2001) The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the αMβ2 integrin (CD11b/CD18). J. Exp. Med. 193, 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El-Azami-El-Idrissi M., Bauche C., Loucka J., Osicka R., Sebo P., Ladant D., Leclerc C. (2003) Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: Role of toxin acylation and identification of the main integrin interaction domain. J. Biol. Chem. 278, 38514–38521 [DOI] [PubMed] [Google Scholar]
- 27. Basar T., Havlícek V., Bezousková S., Hackett M., Sebo P. (2001) Acylation of lysine 983 is sufficient for toxin activity of Bordetella pertussis adenylate cyclase. Substitutions of alanine 140 modulate acylation site selectivity of the toxin acyltransferase CyaC. J. Biol. Chem. 276, 348–354 [DOI] [PubMed] [Google Scholar]
- 28. Gray M. C., Donato G. M., Jones F. R., Kim T., Hewlett E. L. (2004) Newly secreted adenylate cyclase toxin is responsible for intoxication of target cells by Bordetella pertussis. Mol. Microbiol. 53, 1709–1719 [DOI] [PubMed] [Google Scholar]
- 29. Betsou F., Sebo P., Guiso N. (1993) CyaC-mediated activation is important not only for toxic but also for protective activities of Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 61, 3583–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 31. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krebber A., Bornhauser S., Burmester J., Honegger A., Willuda J., Bosshard H. R., Plückthun A. (1997) Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J. Immunol. Methods 201, 35–55 [DOI] [PubMed] [Google Scholar]
- 33. Hayhurst A., Happe S., Mabry R., Koch Z., Iverson B. L., Georgiou G. (2003) Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. J. Immunol. Methods 276, 185–196 [DOI] [PubMed] [Google Scholar]
- 34. Maynard J., Adams E. J., Krogsgaard M., Petersson K., Liu C. W., Garcia K. C. (2005) High-level bacterial secretion of single-chain αβ T-cell receptors. J. Immunol. Methods 306, 51–67 [DOI] [PubMed] [Google Scholar]
- 35. Smith K., Garman L., Wrammert J., Zheng N. Y., Capra J. D., Ahmed R., Wilson P. C. (2009) Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc. 4, 372–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karimova G., Ladant D. (2007) cAMP assays. CSH Protoc. 2007, pdb.prot4739 [DOI] [PubMed] [Google Scholar]
- 37. Glaser P., Ladant D., Sezer O., Pichot F., Ullmann A., Danchin A. (1988) The calmodulin-sensitive adenylate cyclase of Bordetella pertussis: cloning and expression in Escherichia coli. Mol. Microbiol. 2, 19–30 [PubMed] [Google Scholar]
- 38. Hewlett E. L., Gordon V. M., McCaffery J. D., Sutherland W. M., Gray M. C. (1989) Adenylate cyclase toxin from Bordetella pertussis. Identification and purification of the holotoxin molecule. J. Biol. Chem. 264, 19379–19384 [PubMed] [Google Scholar]
- 39. Rogel A., Schultz J. E., Brownlie R. M., Coote J. G., Parton R., Hanski E. (1989) Bordetella pertussis adenylate cyclase: purification and characterization of the toxic form of the enzyme. EMBO J. 8, 2755–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brézin C., Guiso N., Ladant D., Djavadi-Ohaniance L., Megret F., Onyeocha I., Alonso J.-M. (1987) Protective effects of anti-Bordetella pertussis adenylate cyclase antibodies against lethal respiratory infection of the mouse. FEMS Microbiol. Lett. 42, 75–80 [Google Scholar]
- 41. Rogel A., Farfel Z., Goldschmidt S., Shiloach J., Hanski E. (1988) Bordetella pertussis adenylate cyclase. Identification of multiple forms of the enzyme by antibodies. J. Biol. Chem. 263, 13310–13316 [PubMed] [Google Scholar]
- 42. Eby J. C., Gray M. C., Mangan A. R., Donato G. M., Hewlett E. L. (2012) Role of CD11b/CD18 in the process of intoxication by the adenylate cyclase toxin of Bordetella pertussis. Infect. Immun. 80, 850–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karst J. C., Ntsogo Enguéné V. Y., Cannella S. E., Subrini O., Hessel A., Debard S., Ladant D., Chenal A. (2014) Calcium, acylation and molecular confinement favor folding of Bordetella pertussis adenylate cyclase CyaA toxin into a monomeric and cytotoxic form. J. Biol. Chem. 289, 30702–30716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gallay J., Vincent M., Li de la Sierra I. M., Munier-Lehmann H., Renouard M., Sakamoto H., Bârzu O., Gilles A. M. (2004) Insight into the activation mechanism of Bordetella pertussis adenylate cyclase by calmodulin using fluorescence spectroscopy. Eur. J. Biochem. 271, 821–833 [DOI] [PubMed] [Google Scholar]
- 45. Powthongchin B., Angsuthanasombat C. (2008) High level of soluble expression in Escherichia coli and characterisation of the CyaA pore-forming fragment from a Bordetella pertussis Thai clinical isolate. Arch. Microbiol. 189, 169–174 [DOI] [PubMed] [Google Scholar]
- 46. Pojanapotha P., Thamwiriyasati N., Powthongchin B., Katzenmeier G., Angsuthanasombat C. (2011) Bordetella pertussis CyaA-RTX subdomain requires calcium ions for structural stability against proteolytic degradation. Protein Expr. Purif. 75, 127–132 [DOI] [PubMed] [Google Scholar]
- 47. Lee S. J., Gray M. C., Guo L., Sebo P., Hewlett E. L. (1999) Epitope mapping of monoclonal antibodies against Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 67, 2090–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karst J. C., Barker R., Devi U., Swann M. J., Davi M., Roser S. J., Ladant D., Chenal A. (2012) Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J. Biol. Chem. 287, 9200–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blenner M. A., Shur O., Szilvay G. R., Cropek D. M., Banta S. (2010) Calcium-induced folding of a β roll motif requires C-terminal entropic stabilization. J. Mol. Biol. 400, 244–256 [DOI] [PubMed] [Google Scholar]
- 50. Sotomayor Pérez A. C., Karst J. C., Davi M., Guijarro J. I., Ladant D., Chenal A. (2010) Characterization of the regions involved in the calcium-induced folding of the intrinsically disordered RTX motifs from the Bordetella pertussis adenylate cyclase toxin. J. Mol. Biol. 397, 534–549 [DOI] [PubMed] [Google Scholar]
- 51. Bauche C., Chenal A., Knapp O., Bodenreider C., Benz R., Chaffotte A., Ladant D. (2006) Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 281, 16914–16926 [DOI] [PubMed] [Google Scholar]
- 52. Chenal A., Guijarro J. I., Raynal B., Delepierre M., Ladant D. (2009) RTX calcium binding motifs are intrinsically disordered in the absence of calcium: implication for protein secretion. J. Biol. Chem. 284, 1781–1789 [DOI] [PubMed] [Google Scholar]
- 53. Masin J., Basler M., Knapp O., El-Azami-El-Idrissi M., Maier E., Konopasek I., Benz R., Leclerc C., Sebo P. (2005) Acylation of lysine 860 allows tight binding and cytotoxicity of Bordetella adenylate cyclase on CD11b-expressing cells. Biochemistry 44, 12759–12766 [DOI] [PubMed] [Google Scholar]
- 54. Cheung G. Y., Xing D., Prior S., Corbel M. J., Parton R., Coote J. G. (2006) Effect of different forms of adenylate cyclase toxin of Bordetella pertussis on protection afforded by an acellular pertussis vaccine in a murine model. Infect. Immun. 74, 6797–6805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Basler M., Masin J., Osicka R., Sebo P. (2006) Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect. Immun. 74, 2207–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saggy I., Wine Y., Shefet-Carasso L., Nahary L., Georgiou G., Benhar I. (2012) Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. Protein Eng. Des. Sel. 25, 539–549 [DOI] [PubMed] [Google Scholar]
- 57. Gray M. C., Lee S. J., Gray L. S., Zaretzky F. R., Otero A. S., Szabo G., Hewlett E. L. (2001) Translocation-specific conformation of adenylate cyclase toxin from Bordetella pertussis inhibits toxin-mediated hemolysis. J. Bacteriol. 183, 5904–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lupardus P. J., Shen A., Bogyo M., Garcia K. C. (2008) Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 322, 265–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goldstein D. A. (1990) in Clinical Methods: The History, Physical, and Laboratory Examinations (Walker H. K., Hall W. D., Hurst J. W., eds) 3rd Ed., Chapter 143, Butterworths, Boston: [PubMed] [Google Scholar]
- 60. Goyard S., Orlando C., Sabatier J. M., Labruyere E., d'Alayer J., Fontan G., van Rietschoten J., Mock M., Danchin A., Ullmann A. (1989) Identification of a common domain in calmodulin-activated eukaryotic and bacterial adenylate cyclases. Biochemistry 28, 1964–1967 [DOI] [PubMed] [Google Scholar]
- 61. Monneron A., Ladant D., d'Alayer J., Bellalou J., Bârzu O., Ullmann A. (1988) Immunological relatedness between Bordetella pertussis and rat brain adenylyl cyclases. Biochemistry 27, 536–539 [DOI] [PubMed] [Google Scholar]
- 62. Sutherland J. N., Chang C., Yoder S. M., Rock M. T., Maynard J. A. (2011) Antibodies recognizing protective pertussis toxin epitopes are preferentially elicited by natural infection versus acellular immunization. Clin. Vaccine Immunol. 18, 954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Witt M. A., Arias L., Katz P. H., Truong E. T., Witt D. J. (2013) Reduced risk of pertussis among persons ever vaccinated with whole cell pertussis vaccine compared to recipients of acellular pertussis vaccines in a large US cohort. Clin. Infect. Dis. 56, 1248–1254 [DOI] [PubMed] [Google Scholar]
- 64. Watanabe M., Connelly B., Weiss A. A. (2006) Characterization of serological responses to pertussis. Clin. Vaccine Immunol. 13, 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hormozi K., Parton R., Coote J. (1999) Adjuvant and protective properties of native and recombinant Bordetella pertussis adenylate cyclase toxin preparations in mice. FEMS Immunol. Med. Microbiol. 23, 273–282 [DOI] [PubMed] [Google Scholar]
- 66. Confer D. L., Eaton J. W. (1982) Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 217, 948–950 [DOI] [PubMed] [Google Scholar]
- 67. Weingart C. L., Mobberley-Schuman P. S., Hewlett E. L., Gray M. C., Weiss A. A. (2000) Neutralizing antibodies to adenylate cyclase toxin promote phagocytosis of Bordetella pertussis by human neutrophils. Infect. Immun. 68, 7152–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weingart C. L., Weiss A. A. (2000) Bordetella pertussis virulence factors affect phagocytosis by human neutrophils. Infect. Immun. 68, 1735–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Eby J. C., Gray M. C., Warfel J. M., Paddock C. D., Jones T. F., Day S. R., Bowden J., Poulter M. D., Donato G. M., Merkel T. J., Hewlett E. L. (2013) Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect. Immun. 81, 1390–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gueirard P., Guiso N. (1993) Virulence of Bordetella bronchiseptica: role of adenylate cyclase-hemolysin. Infect. Immun. 61, 4072–4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Khelef N., Danve B., Quentin-Millet M. J., Guiso N. (1993) Bordetella pertussis and Bordetella parapertussis: two immunologically distinct species. Infect. Immun. 61, 486–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Betsou F., Sebo P., Guiso N. (1995) The C-terminal domain is essential for protective activity of the Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 63, 3309–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bumba L., Masin J., Fiser R., Sebo P. (2010) Bordetella adenylate cyclase toxin mobilizes its β2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 6, e1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Basler M., Knapp O., Masin J., Fiser R., Maier E., Benz R., Sebo P., Osicka R. (2007) Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 282, 12419–12429 [DOI] [PubMed] [Google Scholar]
- 75. Gmira S., Karimova G., Ladant D. (2001) Characterization of recombinant Bordetella pertussis adenylate cyclase toxins carrying passenger proteins. Res. Microbiol. 152, 889–900 [DOI] [PubMed] [Google Scholar]
- 76. Scarselli M., Aricò B., Brunelli B., Savino S., Di Marcello F., Palumbo E., Veggi D., Ciucchi L., Cartocci E., Bottomley M. J., Malito E., Lo Surdo P., Comanducci M., Giuliani M. M., Cantini F., Dragonetti S., Colaprico A., Doro F., Giannetti P., Pallaoro M., Brogioni B., Tontini M., Hilleringmann M., Nardi-Dei V., Banci L., Pizza M., Rappuoli R. (2011) Rational design of a meningococcal antigen inducing broad protective immunity. Sci. Transl. Med. 3, 91ra62. [DOI] [PubMed] [Google Scholar]
- 77. McLellan J. S., Chen M., Joyce M. G., Sastry M., Stewart-Jones G. B., Yang Y., Zhang B., Chen L., Srivatsan S., Zheng A., Zhou T., Graepel K. W., Kumar A., Moin S., Boyington J. C., Chuang G. Y., Soto C., Baxa U., Bakker A. Q., Spits H., Beaumont T., Zheng Z., Xia N., Ko S. Y., Todd J. P., Rao S., Graham B. S., Kwong P. D. (2013) Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342, 592–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Angkawidjaja C., You D. J., Matsumura H., Kuwahara K., Koga Y., Takano K., Kanaya S. (2007) Crystal structure of a family I. 3 lipase from Pseudomonas sp. MIS38 in a closed conformation. FEBS Lett. 581, 5060–5064 [DOI] [PubMed] [Google Scholar]