

Background: Although knockdown of heat shock protein 47 (Hsp47) attenuates liver fibrosis, the underlying molecular mechanism is unknown.
Results: Deletion of Hsp47 caused activated hepatic stellate cells (HSCs) to undergo ER stress-mediated apoptosis when autophagy was inhibited.
Conclusion: ER stress-induced apoptosis may underlie the clearance of collagen-producing HSCs.
Significance: Hsp47 could be an attractive therapeutic target for fibrosis treatment.
Keywords: Apoptosis, Autophagy, Collagen, Endoplasmic Reticulum Stress (ER Stress), Hepatic Stellate Cell (HSC), Hsp47
Abstract
Chronic liver injury, often caused by alcoholism and viral hepatitis, causes liver fibrosis via the induction of collagen production. In liver fibrosis, hepatic stellate cells (HSCs) are activated and transform into myofibroblasts, which actively produce and secrete collagen into the extracellular matrix. Hsp47 (heat shock protein 47) is a collagen-specific molecular chaperone that is essential for the maturation and secretion of collagen. Here, we used the Cre-LoxP system to disrupt the Hsp47 gene in isolated HSCs from Hsp47 floxed mice. Immature type I procollagen accumulated and partially aggregated in Hsp47-KO HSCs. This accumulation was augmented when autophagy was inhibited, which induced expression of the endoplasmic reticulum (ER) stress-inducible proteins BiP (immunoglobulin heavy chain-binding protein) and Grp94 (94-kDa glucose-regulated protein). The inhibition of autophagy in Hsp47-KO HSCs also induced CHOP (CCAAT/enhancer-binding protein homologous protein), which is an ER stress-induced transcription factor responsible for apoptosis. These data suggest that apoptosis is induced through ER stress by procollagen accumulation in Hsp47-KO HSCs when autophagy is inhibited. Thus, Hsp47 could be a promising therapeutic target in liver fibrosis.
Introduction
Chronic liver injury causes fibrosis and cirrhosis, resulting in organ failure through excess accumulation of extracellular matrix (1). Hepatic stellate cells (HSCs)3 store vitamin A as lipid droplets and encircle sinusoids in normal liver (2). During liver fibrosis, HSCs differentiate into myofibroblast-like cells in response to inflammatory cytokines such as TGF-β. This process is characterized by the loss of lipid droplets, accelerated production and accumulation of type I collagen, expression of smooth muscle α-actin, and an increased proliferation rate (3). Transient activation of HSCs is thought to be a protective acute response against liver injury, whereas persistent HSCs activation is responsible for fibrosis (1, 3).
Procollagen requires several molecular chaperones and enzymes for it to generate a trimer consisting of three α-chains and to fold correctly in the endoplasmic reticulum (ER). Such molecular chaperones include BiP/Grp78, Grp94 (94-kDa glucose-regulated protein), protein-disulfide isomerase, and Hsp47 (heat shock protein 47) (4–8). The enzymes prolyl 4-hydroxylase, prolyl 3-hydroxylase, and cyclophilin B are required for the stable triple-helix formation of procollagen (9–12). We previously reported that Hsp47 is a collagen-specific molecular chaperone in the ER (13–16). Disruption of the Hsp47 gene causes embryonic lethality by 11.5 days post coitus because of the disruption of basement membranes and the misformation of collagen fibrils (17–19). When the Hsp47 gene was disrupted in chondrocytes using the Cre-LoxP system, where Hsp47 floxed mice were crossed with mice carrying a chondrocyte-specific Col2α1-Cre transgene, mice died just before or shortly after birth displaying severe chondrodysplasia and bone deformities with reduced type II collagen production (20). In Hsp47-null fibroblasts, the triple-helix formation and secretion of type I collagen are severely perturbed (19, 21). Type I procollagen α-chains that are misfolded and fail to form a trimer in the ER are degraded via ER-associated degradation after retrograde transport from the ER to the cytosol, whereas misfolded α-chains that form a trimer are eliminated through autophagy, a process termed ER phagy (22, 23). In Hsp47-null fibroblasts, type I procollagen is misfolded and forms trimers, which accumulate as detergent-insoluble aggregates in the ER that are eliminated via the autophagic lysosome pathway (22).
Although expression of Hsp47 is up-regulated by various cytosolic stresses, including heat shock, constitutive expression of Hsp47 correlates with that of collagen in various organs and collagen-related pathophysiological conditions, including liver fibrosis, connective tissue diseases, and dermal fibrotic diseases (24, 25). A recent study showed that TGF-β, a pro-fibrotic cytokine, regulates Hsp47 production in fibroblast cell lines (26). Fibrotic progression is attenuated by down-regulation of Hsp47 expression (27–31). Because of the pivotal role of collagen-producing cells in fibrosis, clearance of activated HSCs through cell death might be expected to alleviate and recover from liver fibrosis. Therefore, the induction of the death of activated HSCs by suppression of Hsp47 expression may be an attractive therapeutic strategy for liver fibrosis. Actually, suppression of Hsp47 expression was reported to attenuate liver fibrosis via inducing the apoptosis of HSCs (28, 30); however, the reason why apoptosis is induced upon suppression of Hsp47 expression remains unknown. We previously reported that ER stress causes apoptotic cell death with the induction of CHOP expression in Hsp47-disrupted mice and cells (18, 20, 22). Accumulation of misfolded proteins in the ER causes ER stress and triggers activation of the unfolded protein response (32). At least three ER transmembrane sensor proteins are activated in the unfolded protein response: PERK (PKR-like endoplasmic reticulum kinase) (33), ATF6 (activating transcription factor 6) (34), and IRE1 (inositol requiring 1) (35, 36). Under ER stress, the unfolded protein response can operate as a pro-survival process through the transient termination of translation, the induction of a series of proteins that coordinately function to renature misfolded proteins, and/or the elimination of misfolded proteins. However, under conditions of persistent ER stress, pro-apoptotic signals are induced through the activation of several transcription factors, such as ATF4 (activating transcription factor 4) and CHOP, downstream of the PERK pathway (37).
In the present study, we investigated the mechanisms underlying the apoptosis of Hsp47-deficient HSCs and the involvement of autophagy in the clearance of misfolded type I procollagen in the absence of Hsp47. We hypothesized that misfolded type I procollagen in the ER might cause ER stress, resulting in the apoptosis of Hsp47-disrupted HSCs when autophagy is inhibited. To this end, we isolated HSCs from the livers of Hsp47 floxed mice, disrupted the Hsp47 gene by infecting cells with an adenovirus harboring the Cre gene, and performed biochemical and cell biological analyses.
EXPERIMENTAL PROCEDURES
Reagents
Collagenase type I and ascorbic acid were purchased from WAKO (Osaka, Japan). Pronase E was purchased from Roche, DNase I was purchased from Merck, and TGF-β was purchased from PeproTech Inc. (Rocky Hill, NJ). Chloroquine (CQ), 3-methyladenine (3-MA), leupeptin, and pepstatin A were purchased from Sigma Aldrich (St. Louis, MO). Anti-Hsp47 and anti-Grp94 antibodies were purchased from ENZO Life Sciences (Plymouth Meeting, PA), an anti-BiP/Grp78 antibody was purchased from BD Transduction Laboratories, an anti-CHOP antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and an anti-GAPDH antibody was purchased from HyTest (Turku, Finland). Antibodies against mouse type I collagen and β-actin were purchased from Millipore Corporation (formerly NYSE: MIL), and those against LC3 and p62 were purchased from MBL (Nagoya, Japan).
Mice
Hsp47 floxed mice were previously generated (20). All experiments using animals were approved by the Kyoto Sangyo University Committee for Animal Care and Welfare.
Isolation and Culture of HSCs
HSCs were isolated from Hsp47flox/flox mice as described by Radaeva et al. (38). The isolated HSCs were cultured on a plastic dish in low glucose DMEM supplemented with 10% FBS and antibiotic-antimycotic solution (Sigma-Aldrich) and maintained at 37 °C in a humidified atmosphere with 5% CO2.
Adenovirus Infection
Adenoviruses harboring the Cre recombinase gene, or the neo gene as the control, under the control of the CAG promoter were kindly provided by Dr. Ikeda (Osaka City University, Osaka, Japan) (39). Adenoviruses were purified using the Adeno-X maxi purification kit (Takara Bio Inc., Shiga, Japan), and the titer of the purified virus was determined using the Adeno-X rapid titer kit (Takara Bio Inc., Shiga, Japan). At days 18–31 after their isolation, HSCs were infected with adenovirus at a multiplicity of infection of 17, 20, or 25 and cultured with 136 μg/ml ascorbic acid.
Immunoblot Analysis
HSCs were trypsinized and seeded onto multiwell plate on day 8 after adenovirus infection with 136 μg/ml ascorbic acid and 1 ng/ml TGF-β. At day 4 after seeding, HSCs were treated with or without 20 μm CQ or 10 mm 3-MA for 24 h. HSCs were lysed with buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 5.0 mm EDTA, 1% (v/v) octylphenoxypolyethoxyethanol, and protease inhibitors (1 μg/ml of leupeptin and pepstatin A) at 4 °C for 20 min. After centrifugation, supernatants and pellets were collected and electrophoresed on a 7, 10, or 15% SDS-PAGE gel (40). Thereafter, the gels were transferred to PVDF membranes with a pore size of 0.45 or 0.22 μm. The membranes were blocked with Blocking One (Nacalai Tesque, Kyoto, Japan) and then stained with specific antibodies. The targeted proteins were detected by a chemiluminescence method using an ECL Western blotting detection reagent or an ECL prime Western blotting detection reagent (GE Healthcare Bio-Sciences) followed by visualization using the LAS-3000 system (Fujifilm, Tokyo, Japan).
RT-PCR and Quantitative Real Time PCR
Total RNA was extracted from HSCs using the RNeasy mini kit (Qiagen), and then first strand cDNA was synthesized using the SuperScript III first strand synthesis SuperMix for RT-PCR and quantitative real time PCR. For RT-PCR, the PCR primers used to amplify mouse Hsp47 were 5′-AAGATGCAGAAGAAGGCTGTCG-3′ (forward) and 5′-CTGTGACACCCCTGAATTTGGT-3′ (reverse), and those used to amplify mouse XBP-1 were 5′-TGAGAACCAGGAGTTAAGAACACGC-3′ (forward) and 5′-TTCTGGGTAGACCTCTGGGAGTTCC-3′ (reverse). For real time PCR, the primers and probes were designed using the Assay Design Center (Roche). The sequences of the primers and the probe used to detect mouse Hsp47 were as follows: 5′-GAAGGCTGTCGCCATCTC-3′ (forward), and 5′-TCCTGCCAGATGTTTCTGC-3′ (reverse), and 5′-TGGTGGAG-3′ (probe). The Universal Probe Library Mouse GAPD Gene Assay (Roche) was used to detect GAPDH as the control. Real time PCR was performed using the Applied Biosystems StepOnePlus real time PCR System (Applied Biosystems, Foster, CA). Expression of Hsp47 was normalized against that of GAPDH.
Immunostaining
HSCs were trypsinized and seeded onto poly-l-lysine-coated cover glasses with ascorbic acid on day 8 after adenovirus infection. At day 4 after seeding, Immunofluorescence was performed as previously described (21). Briefly, HSCs were fixed with 4% paraformaldehyde for 20 min at 37 °C, treated with 0.25% collagenase type I for 3 min at 37 °C, and then treated with 0.1% Triton X-100 for 5 min at room temperature to permeabilize the cells. For staining of extracellular matrix components, HSCs were not treated with collagenase or Triton X-100. HSCs were treated with blocking buffer containing 2% goat serum and 20% glycerol for 30 min at room temperature and then incubated with specific antibodies. Thereafter, HSCs were stained with Alexa Fluor 488-conjugated anti-rabbit IgG or Alexa Fluor 546-conjugated anti-mouse IgG. Immunofluorescence was detected using a LSM-700 microscope (Zeiss, Jena, Germany). The same exposure time was used for the acquisition of all images.
Detection of Caspase-3 Activity
Caspase-3 activity was detected using the NucView 488 caspase-3 assay kit (Biotium, Hayward, CA) according to the manufacturer's instructions. Signals were observed using a BZ-710 microscope (Keyence, Osaka, Japan). The same exposure time was used for the acquisition of all images.
Statistical Analysis
Quantitative data are described as means ± S.E., and the difference between the two groups was statistically analyzed using the two-tailed unpaired Student's t test. All quantitative data were obtained from at least three independent experiments. Densitometric analysis of immunoblots was performed using Multi Gauge version 3.0 software (Fujifilm, Tokyo, Japan), and the caspase-3 activity signal was calculated using the BZ-X analysis application (Keyence).
RESULTS
Depletion of the Hsp47 Gene in Activated HSCs
To reveal the role of Hsp47 in HSCs, we established Hsp47-KO HSCs using the Cre-loxP system. We previously established cartilage-specific Hsp47-KO mice by crossing Hsp47flox/flox mice with mice expressing the Cre recombinase gene under the control of the type II collagen promoter (Col2-cre). In the current study, HSCs were isolated from Hsp47flox/flox mice and activated by culture on a noncoated plastic dish. Hsp47 was depleted in the activated HSCs by infection with an adenovirus encoding Cre recombinase (AdCre). At day 4 after infection, expression of Hsp47 mRNA was decreased in HSCs infected with AdCre to less than 10% of the level in HSCs infected with the control adenovirus (AdControl), so-called control HSCs (Fig. 1, A and B). This suggests that Hsp47 was almost completely knocked out. At days 4 and 8 after infection, the protein level of Hsp47 in Hsp47-KO HSCs was decreased to ∼40% and less than 20% of the level in control HSCs, respectively (Fig. 1, C and D). At day 12 after infection, the mRNA and protein levels of Hsp47 in Hsp47-KO HSCs were less than 10 and 20% of those in control HSCs, respectively. These results indicate that the Hsp47 gene was disrupted in activated HSCs by infection with AdCre.
FIGURE 1.
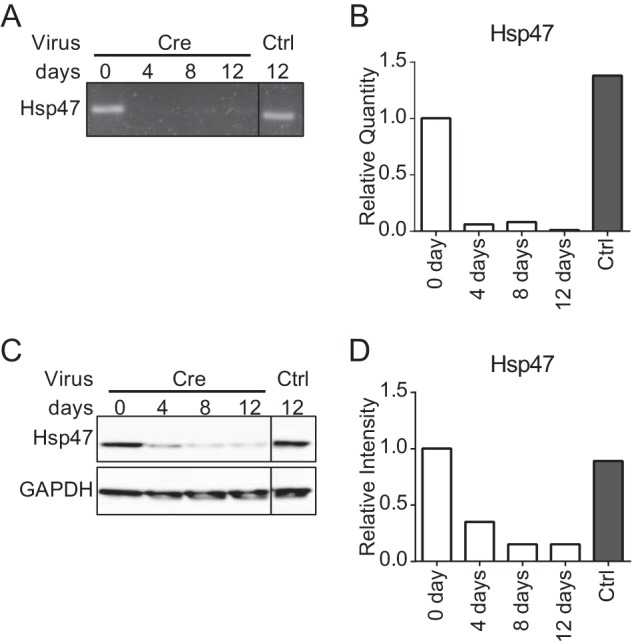
Depletion of Hsp47 in activated HSCs. A–C, the mRNA and protein levels of Hsp47 at the indicated number of days after infection with a control adenovirus (Ctrl) or an adenovirus encoding Cre recombinase (Cre) at a multiplicity of infection of 17 in activated HSCs were analyzed by semiquantitative RT-PCR (A), real time PCR (B), and Western blotting (C). D, the protein level of Hsp47 in C is shown relative to that of GAPDH.
Accumulation of Type I Procollagen in the ER of Hsp47-KO HSCs
The effect of Hsp47 on the maturation of type I collagen in activated HSCs was examined using Hsp47-KO HSCs. The accumulation of extracellular type I collagen was analyzed by immunostaining with an anti-collagen antibody without permeabilizing cells. The level of extracellular collagen was dramatically decreased in Hsp47-KO HSCs, in comparison with control HSCs (Fig. 2A). The amount of type I procollagen that accumulated inside cells, as determined by immunoblot analysis, was clearly higher in Hsp47-KO HSCs than in control HSCs (Fig. 2, B and C). Accumulation of type I procollagen in the detergent-insoluble fraction was hardly observed either in control or Hsp47-KO HSCs. It might be noted that procollagen was clearly detected in the detergent-insoluble fraction in Hsp47-KO MEFs (22). The localization of procollagen within cells was next examined by immunostaining HSCs after they had been permeabilized using Triton X-100. Type I procollagen localized primarily in the Golgi apparatus in control HSCs, as shown by its colocalization with GM130 (cis-Golgi marker) (41) but not with protein-disulfide isomerase (ER marker) (42). However, in Hsp47-KO HSCs, the level of type I procollagen in the ER was markedly increased, and this procollagen was also clearly observed in the Golgi apparatus (Fig. 2D). These results suggest that in the absence of Hsp47, type I procollagen failed to form the mature form and therefore could not be deposited in the extracellular matrix. Consequently, a portion of procollagen remained in the ER as a detergent-insoluble form in Hsp47-KO HSCs.
FIGURE 2.
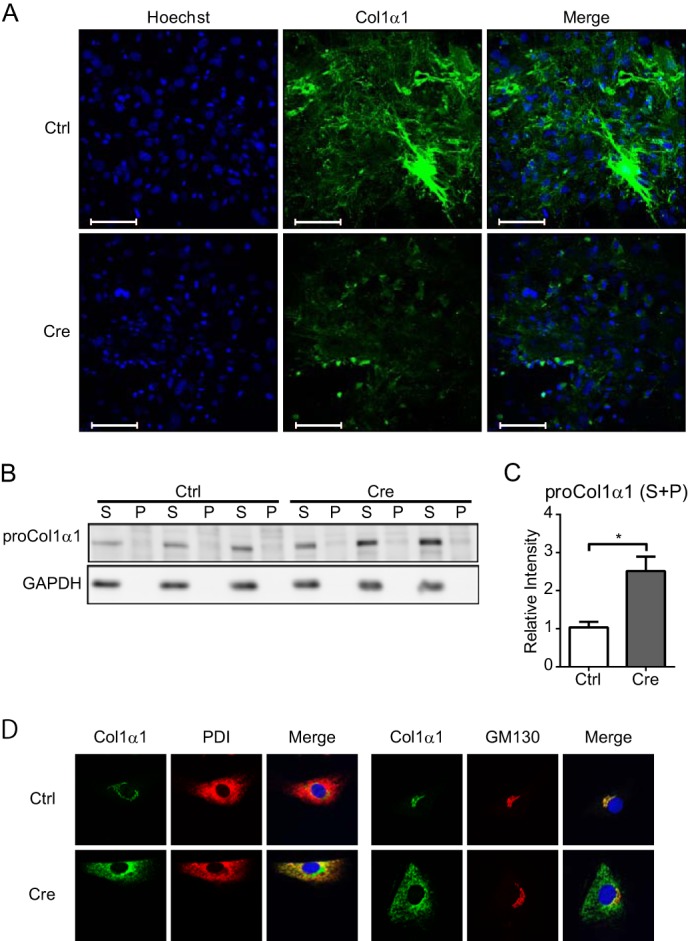
Maturation of type I collagen in Hsp47-KO HSCs. A, deposition of type I collagen in the extracellular matrix after infection of activated HSCs with AdControl or AdCre at a MOI of 17 was detected by staining with an anti-type I collagen (green) antibody and Hoechst 33342 (blue) without permeabilizing cells. Scale bars, 100 μm. B, Western blot analyses of type I procollagen α1 in activated HSCs infected with AdControl or AdCre at a MOI of 25. Cell lysates were separated by centrifugation to generate detergent-soluble (S) and detergent-insoluble (P) fractions. C, the protein level of type I procollagen α1 in B is shown relative to that of GAPDH. Experiments were performed four times independently, and values are means ± S.E. *, p ≤ 0.05. D, the localization of type I collagen in activated HSCs infected with AdControl or AdCre at a MOI of 20 was determined by staining with anti-type I collagen (Col1α1; green), anti-protein-disulfide isomerase (PDI; left, red), and anti-GM130 (right, red) antibodies and Hoechst 33342 (blue). Ctrl, control.
Induction of ER Stress and Apoptosis in AdCre-infected HSCs
The accumulation of misfolded proteins and/or detergent-insoluble protein aggregates in the ER reportedly causes ER stress and consequently apoptosis via the induction of CHOP expression, which is up-regulated by PERK in the unfolded protein response pathways (32, 37). The amount of type I procollagen in the ER was greatly increased in Hsp47-KO HSCs (Fig. 2, B and C); therefore, the induction of ER stress and/or apoptosis was examined in these cells. First, the induction of several stress proteins in the ER, including BiP and GRP94, was examined to determine whether ER stress was induced in AdCre-infected HSCs. Although Hsp47 expression was dramatically decreased in Hsp47-KO HSCs, expression of BiP or Grp94 was not clearly changed (Fig. 3, A and B). The splicing of XBP-1 by activated IRE1 is known to be induced during ER stress (32). XBP-1 splicing analyzed by RT-PCR was not observed in Hsp47-KO HSCs (Fig. 3C). The induction of CHOP (Fig. 3, A and B) and caspase-3 (data not shown) expression, which are apoptosis makers, was also not detected in Hsp47-KO HSCs. These results suggest that neither ER stress nor apoptosis were induced in Hsp47-KO HSCs. We previously reported that ER stress induces apoptosis in MEFs in which Hsp47 is disrupted by homologous recombination (22). However, aggregates of misfolded procollagen in the ER of these cells are degraded by autophagy (22). Therefore, the induction of apoptosis in these cells was considered to be accentuate under the inhibition of autophagy. Thus, the effect of autophagy inhibition on the induction of ER stress and apoptosis in Hsp47-KO HSCs was next examined by treating these cells with inhibitors of autophagy.
FIGURE 3.

Endoplasmic reticulum stress and apoptosis are not observed in Hsp47-KO HSCs. A, Western blot analyses of activated HSCs infected with AdControl or AdCre at a multiplicity of infection of 25 were performed with the indicated antibodies. HSCs were treated with tunicamycin (TM) as a positive control for CHOP detection. B, the protein levels of Hsp47, BiP, Grp94, and CHOP in A are shown relative to that of GAPDH. Experiments were performed four times independently, and values are means ± S.E. **, p ≤ 0.01. NS, not significant. C, RT-PCR for XBP-1 in activated HSCs infected with AdControl or AdCre at a MOI of 25. HSCs were treated with thapsigargin (TG) as a positive control for detecting of XBP-1 spliced form. Ctrl, control.
HSCs were treated with CQ, which inhibits autophagy by preventing acidification of lysosomes (43). First, the accumulation of type I procollagen was examined by immunoblot analysis of CQ-treated cells. Type I procollagen accumulated in CQ-treated control HSCs (Fig. 4, A and B). The level of procollagen was significantly higher in Hsp47-KO HSCs than in control HSCs in the absence of CQ treatment, and the level of procollagen in Hsp47-KO HSCs was greatly increased following CQ treatment. Furthermore, the amount of procollagen was modestly higher in the detergent-insoluble fraction of CQ-treated Hsp47-KO HSCs than in that of untreated Hsp47-KO HSCs (Fig. 4A). The increase of procollagen in Hsp47-KO HSCs was also observed when autophagy was inhibited with 3-MA, another autophagy inhibitor that inhibits type III phosphatidylinositol 3-kinases (44) (Fig. 4C). After treatment with CQ for 24 h on day 12 after infection with AdControl or AdCre, the levels of LC3 and p62, which are autophagy markers, were determined by immunoblot analysis. During activation of autophagy, LC3, a membrane component of the autophagosome, is converted from LC3-I to LC3-II by the conjugation of phosphatidylethanolamine (45). In control HSCs, CQ treatment did not significantly affect the levels of p62 and LC3-II (Fig. 4, D and E). By contrast, in Hsp47-KO HSCs, the levels of LC3-II and p62 were significantly increased by CQ treatment. These results clearly indicated that autophagy was up-regulated in Hsp47-KO HSCs (Fig. 4, D and E), which is consistent with our previous work (22). The levels of BiP and Grp94 were significantly increased in Hsp47-KO HSCs when autophagy was inhibited, whereas this was not observed in control HSCs (Fig. 5, A–C). CHOP was also up-regulated in Hsp47-KO HSCs, but not in control HSCs, when autophagy was inhibited (Fig. 5, A and B). The activity of caspase-3 was examined using a fluorescent marker-conjugated caspase-3 substrate (Fig. 5, D and E). The number of Hsp47-KO HSCs with caspase-3 activity was increased even in the absence of CQ (Fig. 5E), whereas the CHOP induction was not changed (Fig. 5B). CQ treatment increased the numbers of control and Hsp47-KO HSCs that exhibited caspase-3 activity (Fig. 5E). These results suggest that ER stress was induced in Hsp47-KO HSCs through the accumulation of type I procollagen in the ER when autophagy was inhibited and caused the apoptosis in activated HSCs.
FIGURE 4.

Accumulation of type I procollagen in Hsp47-KO HSCs when autophagy is inhibited. A, the protein levels of type I procollagen α1 and GAPDH in activated HSCs infected with AdControl or AdCre at a MOI of 25 and treated with or without CQ were detected by immunoblotting with anti-type I collagen (proCol1α1) and anti-GAPDH antibodies. Cell lysates were separated by centrifugation to generate detergent-soluble (S) and detergent-insoluble (P) fractions. B, the protein level of type I procollagen α1 in A is shown relative to that of GAPDH. Experiments were performed eight times independently. C, the protein levels of type I procollagen α1 and GAPDH in activated HSCs infected with AdControl or AdCre at a MOI of 25 and treated with or without 3-MA were detected by immunoblotting with anti-type I collagen (proCol1a1) and anti-GAPDH antibodies. Cell lysates were separated by centrifugation to generate detergent-soluble (S) and detergent-insoluble (P) fractions. The samples representing the detergent-insoluble fraction are 5-fold concentrated in comparison with the detergent-soluble fraction. D, Western blot analyses of LC3, p62, and GAPDH in activated HSCs infected with AdControl or AdCre at a MOI of 25 and treated with or without CQ were performed with the indicated antibodies. Cell lysates were separated by centrifugation to generate detergent-soluble (supernatant) and detergent-insoluble (pellet) fractions. E, the protein levels of LC3-II and p62 in supernatant fraction in D are shown relative to that of GAPDH. Experiments were performed nine times independently. All values are means ± S.E. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. NS, not significant; NT, not treated; Ctrl, control.
FIGURE 5.

Induction of endoplasmic reticulum stress and apoptosis in Hsp47-KO HSCs when autophagy is inhibited. A, the protein levels of Hsp47, BiP, Grp94, CHOP, and GAPDH in activated HSCs infected with AdControl or AdCre at a MOI of 25 and treated with or without CQ were detected by Western blot analyses using the indicated antibodies. HSCs were treated with tunicamycin (TM) as positive control for CHOP detection. B, the protein levels of Hsp47, BiP, Grp94, and CHOP in A are shown relative to that of GAPDH. Experiments were performed nine times independently. C, the protein levels of Hsp47, BiP, Grp94, and GAPDH in activated HSCs infected with AdControl or AdCre at a MOI of 25 and treated with or without CQ and 3-MA were detected by Western blot analyses using the indicated antibodies. D, activation of caspase-3 in activated HSCs infected with AdCre or AdControl at a MOI of 25 and treated with or without CQ was analyzed using the Nucview488 caspase-3/7 substrate (green) and Hoechst 33342 (blue). E, the ratio of caspase-3 activity-positive cells to Hoechst 33342-positive cells in D is shown. HSCs were treated with staurosporine (ST) as positive control for apoptosis. Experiments were performed three times independently. All values are means ± S.E. *, p ≤ 0.05; **, p ≤ 0.01. NS, not significant; NT, not treated; Ctrl, control.
DISCUSSION
Since we first identified Hsp47 as a collagen-specific molecular chaperone residing in the ER of mammalian cells (13, 15), correlational expression analysis of Hsp47 and various types of collagens has been reported in numerous tissues (15) and collagen-related diseases including fibrosis (24, 25). This correlational expression of Hsp47 and collagens is important for the potential treatment of fibrotic disorders because down-regulation of Hsp47 expression by RNAi or antisense RNA methods markedly delays the progression of fibrosis and concomitantly reduces the level of collagen in the extracellular matrix (27–31). Recently, knockdown of Hsp47 in the HSCs of rat, which was achieved using a drug delivery system to target RNAi specifically to these cells, was shown to markedly prevent the progression of experimental liver fibrosis (28, 30). In this report, apoptotic death of HSCs in rats treated with Hsp47-targeting RNAi was suggested to be a major reason underlying the dramatic therapeutic effects on liver fibrosis.
We previously reported that disruption of the Hsp47 gene in mice causes improper molecular maturation of type I and/or IV procollagens and the accumulation of these molecules in the ER as detergent-insoluble aggregates that are not secreted (17–19). In Hsp47-KO mouse embryos, ER stress-mediated apoptotic cell death is observed in various tissues and MEFs (18, 20, 22). Thus, we hypothesized that the abnormal accumulation of misfolded and/or aggregated procollagens in the ER of mice treated with Hsp47-targeting RNAi might cause ER stress, leading to the expression of CHOP and other apoptosis-inducing genes, and that this ER stress might be the major reason underlying the apoptosis of HSCs.
To address this issue, we established an inducible knock-out system for the Hsp47 gene by infecting activated HSCs from Hsp47flox/flox mouse liver with AdCre. ER stress was not observed in activated control or Hsp47-KO HSCs (Figs. 3 and 5), which is inconsistent with our previous results in Hsp47-KO MEFs (22). This apparent discrepancy may be due to the difference in the basal expression level of procollagen; it is higher in MEFs compared with that in HSCs. In addition, the autophagy was reported to be activated in HSCs (48, 49), which attenuates the induction of ER stress in HSCs, because the misfolded proteins in the ER could be eliminated by autophagy (ER-phagy). We previously reported that misfolded procollagen trimers in the ER are degraded by autophagy, whereas misfolded single procollagen α-chains are degraded by ER-associated degradation (22, 46, 47). Thus, we treated activated HSCs with inhibitors of autophagy. Upon treatment with autophagy inhibitor, the level of procollagens in the ER was increased in both control and Hsp47-KO HSCs (Fig. 4, A–C). It suggests that a part of procollagens was degraded through autophagic pathway. As mentioned above, it was reported recently that the up-regulation of autophagy is required for the activation of hepatic stellate cell (48, 49). It is conceivable that this higher activity of autophagy may be responsible for degrading procollagen even in control HSCs. The increase in the accumulation of procollagens in the ER of CQ-treated cells was reflected by the induction of ER stress, which was detected by the increased levels of BiP and Grp94 (Fig. 5, A–C). It should be noted that the increased accumulation of procollagens caused ER stress in Hsp47-KO HSCs, but not in control HSCs (Fig. 5, A–C), which suggests that the molecular conformation of accumulated procollagens differs according to whether Hsp47 is present or absent. This is supported by our observation that autophagy was up-regulated much more in Hsp47-KO HSCs than in control HSCs (Fig. 4, D and E). ER stress in Hsp47-KO HSCs induced apoptosis, which was detected by increases in the level of CHOP and the activity of caspase-3 (Fig. 5).
Thus, we clearly showed that procollagens that accumulate in the ER of activated HSCs because of the absence of Hsp47 can be degraded by autophagy. However, when autophagy is blocked or overwhelmed by the excess accumulation of misfolded procollagens, activated HSCs undergo apoptosis.
During regression of fibrosis, half of the activated HSCs undergo apoptosis, and the other half revert back to inactivated HSCs (50). Reinactivated HSCs are more sensitive to inflammatory cytokines and are more easily activated than quiescent HSCs. Therefore, reinactivated HSCs are regarded as a risk factor for fibrosis. Thus, immediate and sufficient elimination of activated HSCs is important for the treatment of fibrosis. We showed that apoptosis was induced in activated HSCs by a combination of Hsp47 KO and inhibition of autophagy.
The observations of this study are important with regards to the use of Hsp47 down-regulation as a therapeutic strategy for various fibroses, including liver cirrhosis. Down-regulation of Hsp47 blocks the deposition of procollagens in the extracellular matrix by activated HSCs, which decreases the level of collagens in fibrotic tissues. In addition, prevention of the secretion of procollagens from the ER causes these proteins to accumulate in the ER, which leads to apoptosis of HSCs, the major collagen-producing cells in the liver. Thus, down-regulation of Hsp47 can alleviate fibrosis in two concomitant ways: by inhibiting the secretion of collagen and by inducing apoptosis in collagen-producing cells. This sheds light on a new strategy to treat fibrosis.
Acknowledgments
We thank Dr. Yoshiya Kawaguchi and Dr. Kenichiro Furuyama for providing professional instructions with isolation of HSCs.
This work was supported by Grant-in-Aid for Scientific Research (S) 24227009 from the Japan Society for the Promotion of Science (to K. N.), Grants-in-Aid for Scientific Research on Innovative Areas 24121725 and 26111521 and for Scientific Research for Young Scientists (B) 25840079 from the Japan Society for the Promotion of Science (to R. U.), and Grant-in-Aid for Japan Society for the Promotion of Science Fellows 11J05697 (to S. I.).
- HSC
- hepatic stellate cell
- ER
- endoplasmic reticulum
- MOI
- multiplicity of infection
- CQ
- chloroquine
- 3-MA
- 3-methyladenine
- MEF
- mouse embryonic fibroblast
- AdControl
- control adenovirus
- AdCre
- adenovirus encoding Cre recombinase.
REFERENCES
- 1. Friedman S. L. (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134, 1655–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wake K. (1980) Perisinusoidal stellate cells (fat-storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A-storing cells in extrahepatic organs. Int. Rev. Cytol. 66, 303–353 [DOI] [PubMed] [Google Scholar]
- 3. Friedman S. L. (2008) Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 88, 125–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chessler S. D., Byers P. H. (1993) BiP binds type I procollagen pro alpha chains with mutations in the carboxyl-terminal propeptide synthesized by cells from patients with osteogenesis imperfecta. J. Biol. Chem. 268, 18226–18233 [PubMed] [Google Scholar]
- 5. Ferreira L. R., Norris K., Smith T., Hebert C., Sauk J. J. (1994) Association of Hsp47, Grp78, and Grp94 with procollagen supports the successive or coupled action of molecular chaperones. J. Cell. Biochem. 56, 518–526 [DOI] [PubMed] [Google Scholar]
- 6. Lamandé S. R., Chessler S. D., Golub S. B., Byers P. H., Chan D., Cole W. G., Sillence D. O., Bateman J. F. (1995) Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro alpha 1 (I) chain carboxyl-terminal propeptide which impair subunit assembly. J. Biol. Chem. 270, 8642–8649 [DOI] [PubMed] [Google Scholar]
- 7. Wilson R., Lees J. F., Bulleid N. J. (1998) Protein disulfide isomerase acts as a molecular chaperone during the assembly of procollagen. J. Biol. Chem. 273, 9637–9643 [DOI] [PubMed] [Google Scholar]
- 8. Nagata K. (2003) HSP47 as a collagen-specific molecular chaperone: function and expression in normal mouse development. Semin. Cell Dev. Biol. 14, 275–282 [DOI] [PubMed] [Google Scholar]
- 9. Walmsley A. R., Batten M. R., Lad U., Bulleid N. J. (1999) Intracellular retention of procollagen within the endoplasmic reticulum is mediated by prolyl 4-hydroxylase. J. Biol. Chem. 274, 14884–14892 [DOI] [PubMed] [Google Scholar]
- 10. Smith T., Ferreira L. R., Hebert C., Norris K., Sauk J. J. (1995) Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles: a role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 270, 18323–18328 [DOI] [PubMed] [Google Scholar]
- 11. Vranka J. A., Sakai L. Y., Bächinger H. P. (2004) Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 279, 23615–23621 [DOI] [PubMed] [Google Scholar]
- 12. Ishikawa Y., Wirz J., Vranka J. A., Nagata K., Bächinger H. P. (2009) Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein·cyclophilin B complex. J. Biol. Chem. 284, 17641–17647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagata K., Saga S., Yamada K. M. (1986) A major collagen-binding protein of chick embryo fibroblasts is a novel heat shock protein. J. Cell Biol. 103, 223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saga S., Nagata K., Chen W. T., Yamada K. M. (1987) pH-dependent function, purification, and intracellular location of a major collagen-binding glycoprotein. J. Cell Biol. 105, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagata K. (1996) Hsp47: a collagen-specific molecular chaperone. Trends Biochem. Sci. 21, 22–26 [DOI] [PubMed] [Google Scholar]
- 16. Tasab M., Batten M. R., Bulleid N. J. (2000) Hsp47: a molecular chaperone that interacts with and stabilizes correctly-folded procollagen. EMBO J. 19, 2204–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagai N., Hosokawa M., Itohara S., Adachi E., Matsushita T., Hosokawa N., Nagata K. (2000) Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J. Cell Biol. 150, 1499–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marutani T., Yamamoto A., Nagai N., Kubota H., Nagata K. (2004) Accumulation of type IV collagen in dilated ER leads to apoptosis in Hsp47-knockout mouse embryos via induction of CHOP. J. Cell Sci. 117, 5913–5922 [DOI] [PubMed] [Google Scholar]
- 19. Matsuoka Y., Kubota H., Adachi E., Nagai N., Marutani T., Hosokawa N., Nagata K. (2004) Insufficient folding of type IV collagen and formation of abnormal basement membrane-like structure in embryoid bodies derived from Hsp47-null embryonic stem cells. Mol. Biol. Cell 15, 4467–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masago Y., Hosoya A., Kawasaki K., Kawano S., Nasu A., Toguchida J., Fujita K., Nakamura H., Kondoh G., Nagata K. (2012) The molecular chaperone Hsp47 is essential for cartilage and endochondral bone formation. J. Cell Sci. 125, 1118–1128 [DOI] [PubMed] [Google Scholar]
- 21. Ishida Y., Kubota H., Yamamoto A., Kitamura A., Bächinger H. P., Nagata K. (2006) Type I collagen in Hsp47-null cells is aggregated in endoplasmic reticulum and deficient in N-propeptide processing and fibrillogenesis. Mol. Biol. Cell 17, 2346–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishida Y., Yamamoto A., Kitamura A., Lamandé S. R., Yoshimori T., Bateman J. F., Kubota H., Nagata K. (2009) Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol. Biol. Cell 20, 2744–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanida I. (2011) Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox. Signal. 14, 2201–2214 [DOI] [PubMed] [Google Scholar]
- 24. Masuda H., Fukumoto M., Hirayoshi K., Nagata K. (1994) Coexpression of the collagen-binding stress protein HSP47 gene and the alpha 1(I) and alpha 1(III) collagen genes in carbon tetrachloride-induced rat liver fibrosis. J. Clin. Invest. 94, 2481–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naitoh M., Hosokawa N., Kubota H., Tanaka T., Shirane H., Sawada M., Nishimura Y., Nagata K. (2001) Upregulation of HSP47 and collagen type III in the dermal fibrotic disease, keloid. Biochem. Biophys. Res. Commun. 280, 1316–1322 [DOI] [PubMed] [Google Scholar]
- 26. Nakai A., Satoh M., Hirayoshi K., Nagata K. (1992) Involvement of the stress protein HSP47 in procollagen processing in the endoplasmic reticulum. J. Cell Biol. 117, 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sunamoto M., Kuze K., Tsuji H., Ohishi N., Yagi K., Nagata K., Kita T., Doi T. (1998) Antisense oligonucleotides against collagen-binding stress protein HSP47 suppress collagen accumulation in experimental glomerulonephritis. Lab. Invest. 78, 967–972 [PubMed] [Google Scholar]
- 28. Sato Y., Murase K., Kato J., Kobune M., Sato T., Kawano Y., Takimoto R., Takada K., Miyanishi K., Matsunaga T., Takayama T., Niitsu Y. (2008) Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 26, 431–442 [DOI] [PubMed] [Google Scholar]
- 29. Kitamura H., Yamamoto S., Nakase H., Matsuura M., Honzawa Y., Matsumura K., Takeda Y., Uza N., Nagata K., Chiba T. (2011) Role of heat shock protein 47 in intestinal fibrosis of experimental colitis. Biochem. Biophys. Res. Commun. 404, 599–604 [DOI] [PubMed] [Google Scholar]
- 30. Ishiwatari H., Sato Y., Murase K., Yoneda A., Fujita R., Nishita H., Birukawa N. K., Hayashi T., Sato T., Miyanishi K., Takimoto R., Kobune M., Ota S., Kimura Y., Hirata K., Kato J., Niitsu Y. (2013) Treatment of pancreatic fibrosis with siRNA against a collagen-specific chaperone in vitamin A-coupled liposomes. Gut 62, 1328–1339 [DOI] [PubMed] [Google Scholar]
- 31. Honzawa Y., Nakase H., Shiokawa M., Yoshino T., Imaeda H., Matsuura M., Kodama Y., Ikeuchi H., Andoh A., Sakai Y., Nagata K., Chiba T. (2014) Involvement of interleukin-17A-induced expression of heat shock protein 47 in intestinal fibrosis in Crohn's disease. Gut 63, 1902–1912 [DOI] [PubMed] [Google Scholar]
- 32. Walter P., Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 33. Harding H. P., Zhang Y., Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 34. Haze K., Yoshida H., Yanagi H., Yura T., Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox J. S., Shamu C. E., Walter P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 [DOI] [PubMed] [Google Scholar]
- 36. Mori K., Ma W., Gething M. J., Sambrook J. (1993) A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756 [DOI] [PubMed] [Google Scholar]
- 37. Gotoh T., Mori M. (2006) Nitric oxide and endoplasmic reticulum stress. Arterioscler. Thromb. Vasc. Biol. 26, 1439–1446 [DOI] [PubMed] [Google Scholar]
- 38. Radaeva S., Wang L., Radaev S., Jeong W. I., Park O., Gao B. (2007) Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G809–G816 [DOI] [PubMed] [Google Scholar]
- 39. Kinoshita K., Iimuro Y., Fujimoto J., Inagaki Y., Namikawa K., Kiyama H., Nakajima Y., Otogawa K., Kawada N., Friedman S. L., Ikeda K. (2007) Targeted and regulable expression of transgenes in hepatic stellate cells and myofibroblasts in culture and in vivo using an adenoviral Cre/loxP system to antagonise hepatic fibrosis. Gut 56, 396–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 41. Perez F., Pernet-Gallay K., Nizak C., Goodson H. V., Kreis T. E., Goud B. (2002) CLIPR-59, a new trans-Golgi/TGN cytoplasmic linker protein belonging to the CLIP-170 family. J. Cell Biol. 156, 631–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mehtani S., Gong Q., Panella J., Subbiah S., Peffley D. M., Frankfater A. (1998) In vivo expression of an alternatively spliced human tumor message that encodes a truncated form of cathepsin B. Subcellular distribution of the truncated enzyme in COS cells. J. Biol. Chem. 273, 13236–13244 [DOI] [PubMed] [Google Scholar]
- 43. Steinman R. M., Mellman I. S., Muller W. A., Cohn Z. A. (1983) Endocytosis and the recycling of plasma membrane. J. Cell Biol. 96, 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seglen P. O., Gordon P. B. (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 79, 1889–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fitzgerald J., Lamandé S. R., Bateman J. F. (1999) Proteasomal degradation of unassembled mutant type I collagen pro-alpha1(I) chains. J. Biol. Chem. 274, 27392–27398 [DOI] [PubMed] [Google Scholar]
- 47. Lamandé S. R., Bateman J. F. (1993) A type I collagen reporter gene construct for protein engineering studies. Functional equivalence of transfected reporter COL1A1 and endogenous gene products during biosynthesis and in vitro extracellular matrix accumulation. Biochem. J. 293, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thoen L. F., Guimarães E. L., Dollé L., Mannaerts I., Najimi M., Sokal E., van Grunsven L. A. (2011) A role for autophagy during hepatic stellate cell activation. J. Hepatol. 55, 1353–1360 [DOI] [PubMed] [Google Scholar]
- 49. Hernández-Gea V., Ghiassi-Nejad Z., Rozenfeld R., Gordon R., Fiel M. I., Yue Z., Czaja M. J., Friedman S. L. (2012) Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142, 938–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kisseleva T., Cong M., Paik Y., Scholten D., Jiang C., Benner C., Iwaisako K., Moore-Morris T., Scott B., Tsukamoto H., Evans S. M., Dillmann W., Glass C. K., Brenner D. A. (2012) Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. U.S.A. 109, 9448–9453 [DOI] [PMC free article] [PubMed] [Google Scholar]