

Background: We tested whether insulin cells expressing an inducible RIP-Cre transgene display a normal phenotype.
Results: RipCre insulin cells die when the stimulation of insulin synthesis is protracted.
Conclusion: Beta cells expressing an inducible RIP-Cre transgene are functionally deficient.
Significance: The use of an inducible RIPCre system to examine beta cell gene function/development could distort experimental results.
Keywords: Apoptosis, Gene Knockout, Insulin Synthesis, Pancreatic Islet, Transgenic Mice, Beta Cells
Abstract
We used cre-lox technology to test whether the inducible expression of Cre minimize the deleterious effect of the enzyme on beta cell function. We studied mice in which Cre is linked to a modified estrogen receptor (ER), and its expression is controlled by the rat insulin promoter (RIP). Following the injection of tamoxifen (TM), CreER- migrates to the nucleus and promotes the appearance of a reporter protein, enhanced yellow fluorescent protein (EYFP), in cells. Immunocytochemical analysis indicated that 46.6 ± 2.1% insulin cells of adult RIPCreER- EYFP expressed EYFP. RIPCreER-EYFP (+TM) mice were normoglycemic throughout the study, and their glucose tolerance test results were similar to control CD-1 mice. However, an extended exposure to reagents that stimulate insulin synthesis was detrimental to the survival of IN+EYFP+cells. The administration of an inhibitor of the enzyme dipeptidyl-peptidase (DPP4i), which prevents the cleavage of glucagon-like peptide (GLP-1), to adult RIPCreER-EYFP mice lead to a decrease in the percentage of IN+EYFP+ to 17.5 ± 1.73 and a significant increase in apoptotic cells in islets. Similarly, a 2-week administration of the GLP-1 analog exendin 4 (ex-4) induced an almost complete ablation of IN+ expressing a different reporter protein and a significant decrease in the beta cell mass and rate of beta cell proliferation. Since normal beta cells do not die when induced to increase insulin synthesis, our observations indicate that insulin cells expressing an inducible RIPCre transgene are functionally deficient. Studies employing these mice should carefully consider the pitfalls of the Cre-Lox technique.
Introduction
The Cre-lox system has become an essential tool in biomedical research since it allows cell-type specific modifications to test cell lineage derivation and gene function. The technique involves the generation of transgenic mice with constitutive expression of Cre recombinase and of mice harboring a gene of interest flanked by loxP sites. Crossing these two lines results in DNA recombination in the cell type of interest, with the elimination of expression of a specific gene (knock-out). Alternatively, the loxP sites may have prevented the expression of a particular gene, such as a reporter protein, which is expressed upon removal of the stop sign. In pancreas, one of the commonly used transgenic lines employ Ins2 promoter sequences to drive Cre expression in beta cells (reviewed in Ref. 1), and the lines in use were initially selected because of their high level of Cre expression.
There is now evidence that mice constitutively expressing Cre-recombinase in beta cells are glucose intolerant (1) and that the Cre transgene driven by the rat insulin promoter (RIP)2 is expressed in the hypothalamus (2), raising concern regarding the evaluation of studies exploring the effect of genes on metabolic function. It has been proposed that the physiological abnormalities of transgenic RIP-Cre mice may be avoided using an inducible form of Cre (1, 3, 4). Although the efficiency of recombination is lower than in lines with constitutive Cre expression, the possibility was raised that the appearance of metabolic abnormalities would be evaded with the lower levels of recombinase.
In the present study, we tested whether the inducible expression of Cre affects beta cell function. We used a line, termed RIPCreER-EYFP, generated by crossing mice (RIP-CreER) harboring a transgene comprised of the RIP linked to Cre recombinase-estrogen receptor with a strain containing a floxed reporter gene encoding for Enhanced Yellow Fluorescent Protein (EYFP). Injection of tamoxifen (TM) into bigenic mice results in a rapid translocation of the Cre protein to the nucleus, which permits Cre-mediated recombination in a subset of beta cells and the expression of EYFP. Since it has been suggested that results obtained using RIPCre transgenes vary with the type of floxed reporter protein (3), we also examined RIPCreER-PLAP mice, generated by crossing mice (RIP-CreER) with a strain containing a floxed reporter gene encoding for human Placental Alkaline Phosphatase (PLAP) (12).
We show that RIPCreER mice expressing a reporter protein in a subset of beta cells are glucose tolerant, indicating that their beta cells increased insulin synthesis to reduce the rise in circulating glucose levels. However, since the measurement of glucose responsiveness evaluates the response of all the beta cell population to the transient induction of insulin synthesis and secretion by glucose, we reasoned that defects in the beta cells that underwent Cre-mediated recombination could be masked by the normal response of the insulin cells that never expressed the recombinase in the nucleus. Therefore, we examined islets of RIPCReER-EYFP and RIPCreER-PLAP normoglycemic mice following the administration of insulinotropic agents. These agents were either exendin-4 (ex-4), a mimetic of glucagon like peptide-1(GLP-1) (5) or an inhibitor of the enzyme DPP4 (DPP4i) (6). DPP4i prevents the cleavage of GLP-1, maintaining the intact levels of GLP-1 in the circulation (7). Our findings show that these agents result in the preferential death of the beta cells expressing the reporter gene. Since normal beta cells of normoglycemic mice do not die when induced to increase insulin synthesis, our observations indicate that insulin cells expressing an inducible RIPCre transgene are functionally deficient. These results raise important questions regarding the validity of observations obtained using these mice in developmental, genetic, and metabolic studies.
EXPERIMENTAL PROCEDURES
Animals
RIPCreER and PLAP (Z/AP) reporter mice were kind gifts from D. A. Melton (Harvard University, Boston, MA). RIPCreER-EYFP mice were generated by crossing RIPCreER mice with a line containing a floxed EYFP gene (R26R YFP; Jackson Laboratories stock 6148). RIPCreER-PLAP double-transgenic mice were generated by crossing single heterozygous transgenics.
Two-month-old bigenic mice were injected with tamoxifen (TM, Sigma, 5 mg ip/day/5days) dissolved in oil. This dose of TM induces Cre-mediated recombination in 30% of IN cells (8) and were fed a regular diet (rodent chow no. 5015; Purina). Three mice were sacrificed one month later. Starting at 4 months, 4 bigenic mice were switched to a diet (rodent chow no. 5015, Purina) containing a DPP4i (MK0626,Research Diets, New Brunswick, NJ), an analog of sitagliptin, for an additional two months while other 4 mice remained on the regular Purina diet. The dose of MK 0626 used (4 mg/kg/day) significantly reduces circulating DDP-4 levels and stabilizes postprandial levels of GLP-1 in mice (9, 10). Body weight and fed blood glucose levels were determined bi-weekly. To test the effect of exendin-4 (ex-4), five 4–5-month-old RIPCreER-PLAP mice were injected with the GLP-1 mimetic (10 nmol/kg bodyweight (9), Bachem, Torrance, CA) dissolved in PBS plus 1% bovine serum albumin (BSA) twice a day for 2 weeks. Five RIPCreER-PLAP received vehicle (PBS plus 1% BSA) for the same length of time. RIPCreER-EYFP/PLAP mice receiving the DPP4i or ex-4 were normoglycemic throughout the length of the study.
Mice were perfused through the heart with 4% paraformaldehyde buffered to pH 7.4 with 0.1 m phosphate-buffered saline (PBS). The fixed tissues were infiltrated overnight in 30% sucrose, mounted in embedding matrix (Lipshaw Co., Pittsburgh, PA) and 20-μm cryostat sections were collected onto charged microscope slides. All animal protocols were approved by the institutional Animal Care and Use Committee.
Mouse Genotyping
DNA was extracted from the tail using DNeasy kit (Qiagen, Valencia, CA). For Cre, the following oligonucleotides were used: forward AACCTGGATAGTGAAACAGGGGC, reverse TTCCATGGAGCGAACGACGAGACC, which amplified a 400-bp product. Primers were obtained from Eurofino MWG Operon (Huntsville, AL). Primers used for EYFP were those designed by Charles River Inc. PCR conditions were: 95 °C for 15 min followed by 35 cycles at 94 °C for 30 s, 58 °C for 1 min, 72 °C for 1 min and a final extension step at 72 °C for 5 min. Lac-Z activity was determined in tail biopsies using the biochemical detection method described by Jackson Laboratories (Bar Harbor, ME).
Immunostaining
Sections were incubated sequentially in an empirically derived optimal dilution of control serum or primary antibody raised in species “X” overnight and with a 1:200 dilution of the secondary antibodies. For activated caspase 3 and PLAP immunostaining, sections were processed for antigen retrieval (Fisher Scientific, Fair Lawn, NJ; Cat. BP2473) before incubation with primary antibody. For multiple label experiments, antibodies produced in different hosts were used. Antibodies were diluted in 0.1 m PBS containing 0.05% Tween.
Source of Antibodies and Purified Peptides
Primary Antiserum
Rabbit antiserum to activated Caspase-3 and to Ki67: Cell Signaling, Danvers, MA and EMD Millipore, Billerica, MA, respectively. Rabbit antisera to EYFP (Millipore). Mouse antiserum to IN (Sigma). Guinea pig antibodies to bovine IN (Linco Research, Inc., Eureka, MO). Rabbit antiserum to glucagon (Calbiochem, San Diego, CA). Mouse antiserum to PLAP and BrdU (DAKO Inc., Carpinteria, CA). Antibodies were diluted in 0.1 m PBS containing 0.05% Tween and were used at the following dilutions: antisera to PLAP 1: 1:1000; to insulin 1:400; to glucagon; 1:12,000; to BrdU 1:500; to Ki67: 1:400, and to EYFP: 1:250, respectively.
Secondary Antibodies
Alexa Fluor 488 antimouse and antirabbit IgG, Alexa Fluor 594 antiguinea pig, antirabbit, and antimouse IgG were purchased from Invitrogen (Carlsbad, CA). All secondary IgG's were used at 1:200 dilutions in 0.1 m PBS containing 0.05% Tween buffer.
BrdU Labeling
Mice were given BrdU (Sigma) dissolved in water at a concentration of 0.8 mg/ml for 6 days. The solution was changed every other day. Mice were perfused, and the pancreas sectioned as described above. To visualize BrdU, sections were incubated with 2N HCl for 20 min and with 0.1N HCl containing 0.5% pepsin (Sigma-Aldrich Inc.) for 20 min at 37 °C and processed for immunostaining. Results are expressed as the mean ± S.E. Number of scored cells is indicated in the individual experiments.
Morphometry
The relative number of beta cells per section was determined in sections immunostained for insulin by the point sampling method (11) using a 300 point ocular grid at a total magnification of ×400. The relative IN+ cell mass per section was calculated by dividing the number of points over immunostained IN+ cells over the number of points scored for that section. Similar sections through the pancreatic duct were selected for counting. Results were expressed as mean values of all animals at each time point ± S.E.
The number of IN+ PLAP/EYFP+ cells was determined using the point selection icon of Image J (imagej.nih.gov/ij). The percentage of IN+PLAP+/EYFP+ cells was calculated by dividing the number of IN+PLAP+(EYFP+) cells by the total number of insulin cells scored x 100. The number of insulin cells scored is indicated in the individual experiments. To determine the density of apoptotic cells per islet, islet surface area was measured using ImageJ, and the data for each islet, in arbitrary units of numeric value × 10−3, were used to normalize the number of apoptotic cells in that islet.
Confocal Microscopy
A Leica TCS SP-5 laser scanning confocal microscope was used with a 63× 1.4 NA pan Apochromat objective (Carl Zeiss). Typically, 0.7-μm vertical steps were used with a vertical optical resolution of <1.0 μm.
Intraperitoneal Glucose Tolerance Test (IPGTT)
Mice were deprived of food for 6 h before the administration of glucose (intraperitoneal,1.5 mg/g body weight). Blood was collected immediately before and at 30, 60, 90, and 120 min following administration. Blood glucose levels were determined using a Precision QID (MediSense, Abbott Labs, Bedford, MA) monitor.
Statistical Analysis
All values are shown as mean ± S.E. For comparison between two groups, the unpaired t test (two tail) was used. A p value < 0.05 was considered significant.
RESULTS
Since transgenic mice expressing a constitutive form of RIPCre are glucose impaired, we tested whether lower levels of Cre expression in islets would avoid the development of this metabolic dysfunction. RIPCreER-EYFP mice were normoglycemic throughout the study, and the IPGTT of 5-month-old bigenic mice revealed that their response to glucose was normal, indicating that they were glucose tolerant (Fig. 1A).
FIGURE 1.

DPP4i induce an increase in apoptotic cells. A, IPGTT-Blood glucose levels after administration of glucose (ip) to 5-month-old CD-1 mice(dashed lines) or RIPCreER-EYFP (solid line) mice. n = 5 animals/group. No statistical difference was found between the results from the two different lines. B, body weight of bigenic mice fed control diet (dashed lines) or pellets containing the DPP4i (solid lines) through an 12-week-period; n = 4 animals/group. C, beta cell mass is similar in bigenic mice receiving control diet or pellets containing MK0626. At least 25,000 points were scored per group (n = 4). D, percentage of apoptotic cells per islet area is significantly higher in bigenic mice receiving a diet containing a DPP4i than in those fed a control diet. At least 20 islets were scored/group (n = 4 mice/group). E, islet of a DPP4i treated RIPCreER-EYFP mice immunostained for visualization of activated caspase-3 (ac-3; green). Arrow indicates a cell expressing cytoplasmic activated caspase 3. This cell is shown magnified in F. Asterisk is located between two apoptotic bodies; these bodies are shown magnified in G. H, islet of a bigenic mouse injected with saline. Note the absence of apoptotic figures.
To test whether an increase in the beta cell functional activity leads to beta cell failure, mice were fed for two months with pellets containing a DPP4 inhibitor or control diet. While bigenic mice fed with either diet had comparable beta cell mass (Fig. 1, C and D, respectively), the administration of the DPP4i induced beta cell death since islets of 6-month-old bigenic fed with a DPP4i diet from months 4–6 contained cells expressing activated caspase 3 (Fig. 1, E--G). The percentage of beta cells undergoing programmed cell death, illustrated in Fig. 1D, documents a significant increase in the percentage of apoptotic cells following the administration of DPP4i.
Then, we sought to determine whether the administration of the DPP4i affects the survival of the beta cells expressing EYFP. In islets of six-month-old RIPCreER-EYFP mice, the number of nuclei with positive histofluorescence per islet was lower in mice receiving the DPP4i (Fig. 2B) that in those receiving the control diet (Fig. 2A). Double immunohistochemistry analysis indicated that the percentage of IN+ cells expressing EYFP in islets of 3-month-old bigenic mice was 29%± 1.8 (4 mice and a total of 7531IN+ cells scored), and increased to 46.6 ± 2.1 (4 mice and a total of 8681IN+ cells scored) in 6-month-old mice fed throughout the study with control diet (Fig. 2E). This finding confirms previous results indicating a temporal increase in the percentage of beta cells expressing the RIP-transgene (8). In contrast to mice fed with control diet, the percentage of IN+EYFP+ cells decreased from 29% at three months to 17.5 ± 1.73 (4 mice and a total of 9474 IN+ cells scored) in 6-month-old RIPCreER-EYFP mice that received DPP4i therapy during months 4–6 (Fig. 2E). Representative images of islets of RIPCreER-EYFP mice fed control or a diet containing DPP4i are illustrated in Fig. 2, C and D, respectively.
FIGURE 2.
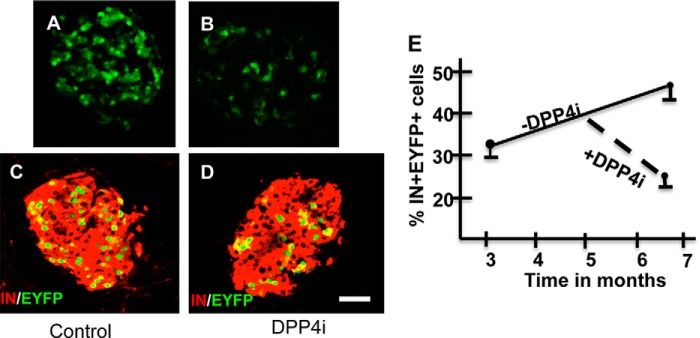
IN+EYFP+ cells are ablated by the DPP4i. A and B, EYFP histofluorescence (green) in pancreatic islets of RIPCreER-EYFP mice (+TM) fed with control (A) or DPP4i (B) diet; C, D, pancreatic islets of RIPCreER-EYFP mice (+TM) fed with control (C) or DPP4i (D) diet immunostained for insulin (red) and EYFP (green). E, percentage of EYFP+ cells in RIPCreER-EYFP mice receiving control or DPP4i diet. n = 3/line. Note that the percentage of IN+EYFP+ cells increase with time in mice fed with control diet. In contrast, the percentage of IN+EYFP+ cells decrease in bigenic mice receiving DPP4i. *, p < 0.005
To determine whether the decrease in the percentage of IN+EYFP+ cells in mice exposed to the enzyme inhibitor was due to selective proliferation of IN+EYFP- cells, 6-month old RIPCreER-EYFP mice fed with DPP4i diet received BrdU (80 mg/100 ml) in the water for 6 days prior to being euthanized. Tissue sections were processed for visualization of BrdU, EYFP and insulin by immunostaining. Confocal images revealed the presence of IN+EYFP+ cells expressing BrdU (Fig. 3, A–C). The rate of turnover of IN+EYFP+ cells and of IN+EYFP-cells was 1.1% ± 0.1 (943 IN+EYFP+ cells scored) and 1.46% ± 0.1(7338 IN+EYFP- cells scored), respectively (Fig. 3D) indicating that both cell types had similar rates of proliferation. Since results obtained with Cre lines varies when different reporter lines are used (3, 12), we tested the effect of the induction of insulin synthesis in RIPCreER mice crossed with mice expressing PLAP and fed with the DPP4i diet for two months. The DPP4i diet induced a 28% reduction in the percentage of insulin cells expressing PLAP (not shown), which is similar to that obtained with the bigenic line expressing EYFP.
FIGURE 3.
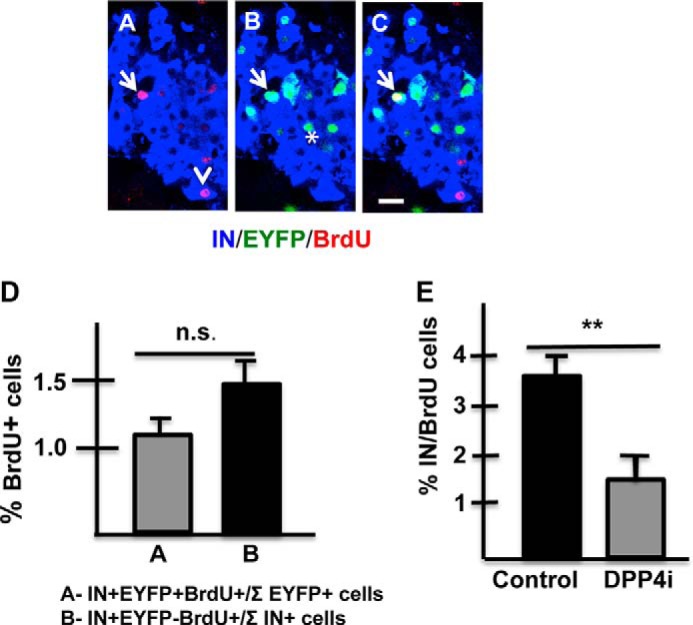
The decrease in the percentage of EYFP+IN+ cells is not due to a reduction in their rate of proliferation. A–C, islet of RIPCreER-EYFP (+TM) mouse immunostained for visualization of insulin (blue), BrdU(red), and EYFP(green). A, insulin +BrdU; B, IN + EYFP, and C, IN + EYFP+ BrdU. Arrow indicates an IN+ EYFP+BrdU+ cell; arrowhead indicates an IN+EYFP-BrdU+ cell and asterisk in B indicates two IN+EYFP+BrdU- cells. D, percentage of cells expressing BrdU is similar in the EYFP+IN+ and EYFP-IN+ cell populations. 7338 IN+EYFP cells and 943 IN+EYFP+ cells were scored/mouse (n = 3). E, the percentage of IN+ BrdU+ cells is lower in mice receiving a diet containing MK0626 than in mice fed a control diet. *, p < 0.05. At least 5000 IN cells were scored/group (n = 3 mice/group).
Administration of the DPP4i also resulted in a decreased rate of beta cell proliferation. Pancreas of RIPCreER-EYFP mice fed with control or DPP4i diet that received BrdU in water (80 mg/100 mls) for 6 days before being euthanized were examined. This analysis indicated that the percentage of cycling IN+ cells (Fig. 3E) was lower in DPP4i mice than in controls.
Exendin-4 Drastically Reduces the Number of IN Cells Expressing a Transgene
Next, we sought to determine whether ex-4 also reduce the number of insulin cells expressing the RIP-transgene. RIPCreER-PLAP mice were injected with vehicle (5 mice) or with ex-4 (5 mice) for 2 weeks as indicated under “Experimental Procedures,” and the pancreas was examined at the end of the treatment. While islets of 18-week-old bigenic mice injected with vehicle contained 41.3 ± 2.45 IN+PLAP+ cells (Fig. 4A, 7894 IN+ cells scored), the percentage of IN+PLAP+ cells in islets of bigenic mice injected with ex-4 decreased to 2.70 ± 0.6 (Fig. 4A, 7752 IN+ cells scored). Photomicrographs of representative islets of RIPCreER-PLAP mice injected with vehicle or with exendin-4 are shown in Fig. 4 (B, C, and D, respectively).
FIGURE 4.

Exendin-4 ablates IN+ cells expressing a RIPCre transgene. A, percentage of IN+PLAP+ cells is significantly reduced in RIPCreER-PLAP mice receiving exendin-4. At least 20 islets were scored/mice/3 mice/group. ***, p < 0.001. B, immunohistochemical visualization of insulin(red) and PLAP(green) in islets of RIPCreER-PLAP mice injected with saline (B) or with exendin-4 (C and D). Note that islets of mice receiving the incretin analog contain few (C) or almost no (D) PLAP+ cells. Bars, B and C: 20 um; D: 40 μm. E, Exendin-4 induced a 50% reduction in the beta cell mass compared with controls. Over 15,000 points were scored in sections (5 mice/group); ***, p < 0.001. F, percentage of insulin-positive cells expressing Ki67 is dramatically lower in RIPCreER-PLAP mice receiving exendin-4 than in mice injected with saline (over 3000 cells were scored per mice/5 mice per group). No statistical error was calculated for results from RIPCreER-PLAP mice injected with exendin-4 because most islets did not contain Ki67+ cells insulin cells.
The administration of exendin-4 to RIPCreER-PLAP mice induced a near 50% decrease in the beta cell mass when compared with mice injected with saline (exendin-4: 2.14 ± 0.71, a total of 17,323 points were scored; vehicle: 5.4 ± 0.74, a total of 15,155 points were scored, p < 0.006; Fig. 4E). Although this observation suggests that the decrease in beta cell mass is due to cell death, rare cells expressing activated caspase-3 were found in very few islets of mice receiving exendin-4 (Fig. 1J). Analysis of the rate of beta cell growth using the proliferation marker Ki67 indicated a sharp decrease in the percentage of IN+KI67+ cells in islets of RIPCreER-PLAP mice injected with exendin-4 (Fig. 4F), indicating that the GLP-1 mimetic reduced the rate of beta cell proliferation.
DISCUSSION
The experiments reported here were designed to test whether beta cells of mice expressing an inducible RIPCre transgene display a normal physiological phenotype. The GTT was performed in 5-month-old RIPCreEYFP mice, when ∼45% of the beta cells expressed the reporter protein. Bigenic mice fed with control diet displayed a normal pattern of regulation of glucose levels during the test, suggesting either that all beta cells responded to the increased glucose levels by secreting insulin or that the EYFP+ beta cells were unresponsive while those that had not undergone recombination and were EYFP- secreted more insulin and compensated for their silent companions.
Our results suggest that beta cells expressing a reporter protein are responsive to high glucose levels because they responded to the action of the DPP4i. Hence, the protracted increase in secretory activity induced by this reagent lead to the death of the beta cells expressing a transgene. Thus, the percentage of IN+EYFP+ cells in RIPCreER-EYFP mice fed with control diet increased from 29% at three months of age to 46% at 6 months. In contrast to mice fed with control diet, the percentage of IN+EYFP+ cells decreased from 29% at three months to ∼18% two months later in mice fed a diet containing the DPP4i MK0626. Since the turnover rate of EYFP+ and EYFP- insulin positive cells was similar, the decrease in the percentage of IN+EYFP+ cells was not due to increased proliferation of the IN+EYFP- cell population but, rather, to the death of the insulin cells expressing EYFP. This possibility is supported by the finding of apoptotic cells in islets of RIPCreER-EYFP receiving DPP4i treatment but not in islets of mice fed with control diet. Therefore, our findings indicate that the extended activation of insulin synthesis by the DPP4i was detrimental to the survival of the IN+EYFP+ cells.
The ablation of cells expressing the RIP-transgene occurs at a faster rate with ex-4, which eliminated almost all insulin cells expressing the RIPCreER transgene cells after only 2 weeks of treatment, than with the DPP4i, where islets contained insulin cells expressing a reporter protein after two months of exposure. This observation implies that the rate of elimination of insulin cells expressing the RIP-transgene is dependent on the activity of the incretin in the circulation. Indeed, it is well established that ex-4 has a higher potency of GLP-1r activation and of induction of insulin synthesis than GLP-1 (5, 13). Mice treated with DPP4i have a high number of cells expressing activated caspase-3 in islets, but the DPP4i-induced apoptosis did not lead to a decrease in beta cell mass, suggesting that dying cells still expressed insulin and probably were included in the morphometric analysis. It is possible that differences in beta cell mass between groups fed with DPP4i or control diet will appear in mice after a longer treatment period with the DPP4i. In contrast, islets of mice receiving exendin-4 had few apoptotic figures but they showed significant decrease in the beta cell mass, implying that beta cell death occurred soon after the injection of the GLP-1 mimetic. Presumably, the period of apoptosis ended before the mice were euthanized.
The decrease in the number of cells expressing the reporter gene following ex-4 treatment was correlated with a 50% reduction in the beta cell mass and a significant fall in the rate of beta cell proliferation. The reduction in the beta cell mass in the RIPCreEr-EYFP mice probably reflects the elimination of almost all the IN+ cells expressing the transgene. It has been reported that normoglycemic CD-1 mice treated with liraglutide, a GLP-1r agonist, for 6 weeks had a decreased beta cell mass and in proliferation (14). In view of our present findings with bigenic mice, the observations in normoglycemic CD-1 mice suggest that the stimuli to secrete more insulin eliminated suboptimal beta cells, and that normal glucose levels was maintained by the cells still left in the islets.
The finding that cells expressing an inducible Cre are dysfunctional raises important questions regarding the validity of results obtained using these lines. In some of these studies, RipCreER mice have been used to examine the effect of the deletion of the transcription factors PDX-1 and FoxO1 in beta cell function (15, 16). Although RipCreER mice were tested as controls, the effect of the mutation was evaluated on beta cells that underwent Cre-mediated recombination and were already suboptimal. Similar concerns are raised with studies in which RIPCreER mice were used for cell lineage analysis (17), clonal studies of beta cell development (18), insulin resistance (19), glucose sensing by beta cells (20); effects of hyperglycemia on islet morphology (21), changes in islet composition during pregnancy (22), beta cell proliferation (23, 24), generation of new beta cells during regeneration (25), beta cell apoptosis (26), and for the analysis of the role of NeuroD, a transactivator of the insulin gene, in beta cell development (27). Moreover, results from classical studies using RIPCre mice for cell lineage analysis of alpha and beta cells during development (28) should also be reexamined, since the fate of abnormal RIPCre cells may be different from that of insulin cells that do not express the transgene.
A new transgenic mouse line in which the expression of an inducible Cre-fused estrogen receptor is targeted to β -cells by the mouse insulin 1 gene promoter (termed MIP1-CreERT) was generated by Tamarina et al. (29). This mice lack ectopic expression of the transgene in brain and other tissues and displayed a normal response to glucose in a glucose tolerance test (2, 29). It will be important to determine whether the expression of Cre in beta cells of MIP1-CreERT mice affects the survival of cells expressing the transgene in different metabolic paradigms.
In summary, this study demonstrates that beta cells of RIPCReER mice that underwent Cre-mediated recombination are eliminated by exendin-4. The effect of the GLP-1 mimetic on islet cell composition is reproduced by the DPP4i, albeit at a slower pace indicating that beta cell ablation is correlated with the concentration of the incretin in the circulation. These observations add to previously raised concerns regarding the toxic effects of Cre on cell function (1, 3, 4, 30) and highlight the need for caution in the interpretation of results using the Cre-lox technology.
Footnotes
- RIP
- rat insulin promoter
- TM
- tamoxifen
- EYFP
- enhanced yellow fluorescent protein
- PLAP
- placental alkaline phosphatase.
REFERENCES
- 1. Lee J. Y., Ristow M., Lin X., White M. F., Magnuson M. A., Hennighausen L. (2006) RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J. Biol. Chem. 281, 2649–2653 [DOI] [PubMed] [Google Scholar]
- 2. Wicksteed B., Brissova M., Yan W., Opland D. M., Plank J. L., Reinert R. B., Dickson L. M., Tamarina N. A., Philipson L. H., Shostak A., Bernal-Mizrachi E., Elghazi L., Roe M. W., Labosky P. A., Myers M. G., Jr., Gannon M., Powers A. C., Dempsey P. J. (2010) Conditional Gene Targeting in Mouse Pancreatic {beta}-Cells: Analysis of Ectopic Cre Transgene Expression in the Brain. Diabetes 59, 3090–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harno E., Cottrell E. C., White A. (2013) Metabolic pitfalls of CNS Cre-based technology. Cell Metab. 18, 21–28 [DOI] [PubMed] [Google Scholar]
- 4. Schmidt-Supprian M., Rajewsky K. (2007) Vagaries of conditional gene targeting. Nat. Immunol. 8, 665–668 [DOI] [PubMed] [Google Scholar]
- 5. Göke R., Fehmann H. C., Linn T., Schmidt H., Krause M., Eng J., Göke B. (1993) Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin-secreting beta-cells. J. Biol. Chem. 268, 19650–19655 [PubMed] [Google Scholar]
- 6. Drucker D. J. (2006) The biology of incretin hormones. Cell Metab. 3, 153–165 [DOI] [PubMed] [Google Scholar]
- 7. Holst J. J., Deacon C. F. (2005) Glucagon-like peptide-1 mediates the therapeutic actions of DPP-IV inhibitors. Diabetologia 48, 612–615 [DOI] [PubMed] [Google Scholar]
- 8. Liu H., Guz Y., Kedees M. H., Winkler J., Teitelman G. (2010) Precursor cells in mouse islets generate new beta-cells in vivo during aging and after islet injury. Endocrinology 151, 520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grigoryan M., Kedees M. H., Charron M. J., Guz Y., Teitelman G. (2012) Regulation of mouse intestinal L cell progenitors proliferation by the glucagon family of peptides. Endocrinology 153, 3076–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mu J., Woods J., Zhou Y. P., Roy R. S., Li Z., Zycband E., Feng Y., Zhu L., Li C., Howard A. D., Moller D. E., Thornberry N. A., Zhang B. B. (2006) Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 55, 1695–1704 [DOI] [PubMed] [Google Scholar]
- 11. Hellestrom C., Swenne I. (1985) Growth pattern of pancreatic islet cells in animals in The Diabetic Pancreas. 2nd Ed (Volk B., Arquilla E., eds), pp. 53–80, Plenum Medical Book Co, New York and London [Google Scholar]
- 12. Lee K. Y., Russell S. J., Ussar S., Boucher J., Vernochet C., Mori M. A., Smyth G., Rourk M., Cederquist C., Rosen E. D., Kahn B. B., Kahn C. R. (2013) Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62, 864–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Young A. A., Gedulin B. R., Bhavsar S., Bodkin N., Jodka C., Hansen B., Denaro M. (1999) Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 48, 1026–1034 [DOI] [PubMed] [Google Scholar]
- 14. Ellenbroek J. H., Töns H. A., Westerouen van Meeteren M. J., de Graaf N., Hanegraaf M. A., Rabelink T. J., Carlotti F., de Koning E. J. (2013) Glucagon-like peptide-1 receptor agonist treatment reduces beta cell mass in normoglycaemic mice. Diabetologia 56, 1980–1986 [DOI] [PubMed] [Google Scholar]
- 15. Gao T., McKenna B., Li C., Reichert M., Nguyen J., Singh T., Yang C., Pannikar A., Doliba N., Zhang T., Stoffers D. A., Edlund H., Matschinsky F., Stein R., Stanger B. Z. (2014) Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 19, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Talchai C., Xuan S., Lin H. V., Sussel L., Accili D. (2012) Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150, 1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weinberg N., Ouziel-Yahalom L., Knoller S., Efrat S., Dor Y. (2007) Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes 56, 1299–1304 [DOI] [PubMed] [Google Scholar]
- 18. Brennand K., Huangfu D., Melton D. (2007) All beta Cells Contribute Equally to Islet Growth and Maintenance. PLoS Biol. 5, e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang T., Abbott M. J., Ahmadian M., Lopes A. B., Wang Y., Sul H. S. (2013) Desnutrin/ATGL activates PPARδ to promote mitochondrial function for insulin secretion in islet beta cells. Cell Metab. 18, 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beall C., Piipari K., Al-Qassab H., Smith M. A., Parker N., Carling D., Viollet B., Withers D. J., Ashford M. L. (2010) Loss of AMP-activated protein kinase alpha2 subunit in mouse beta-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia. Biochem. J. 429, 323–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brereton M. F., Iberl M., Shimomura K., Zhang Q., Adriaenssens A. E., Proks P., Spiliotis II, Dace W., Mattis K. K., Ramracheya R., Gribble F. M., Reimann F., Clark A., Rorsman P., Ashcroft F. M. (2014) Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat. Commun. 5, 4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abouna S., Old R. W., Pelengaris S., Epstein D., Ifandi V., Sweeney I., Khan M. (2010) Non-beta-cell progenitors of beta-cells in pregnant mice. Organogenesis 6, 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeng N., Yang K. T., Bayan J. A., He L., Aggarwal R., Stiles J. W., Hou X., Medina V., Abad D., Palian B. M., Al-Abdullah I., Kandeel F., Johnson D. L., Stiles B. L. (2013) PTEN controls beta-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell 12, 1000–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang K. T., Bayan J. A., Zeng N., Aggarwal R., He L., Peng Z., Kassa A., Kim M., Luo Z., Shi Z., Medina V., Boddupally K., Stiles B. L. (2014) Adult-onset deletion of Pten increases islet mass and beta cell proliferation in mice. Diabetologia 57, 352–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rankin M. M., Wilbur C. J., Rak K., Shields E. J., Granger A., Kushner J. A. (2013) beta-Cells are not generated in pancreatic duct ligation-induced injury in adult mice. Diabetes 62, 1634–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carrington E. M., McKenzie M. D., Jansen E., Myers M., Fynch S., Kos C., Strasser A., Kay T. W., Scott C. L., Allison J. (2009) Islet beta-cells deficient in Bcl-xL develop but are abnormally sensitive to apoptotic stimuli. Diabetes 58, 2316–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gu C., Stein G. H., Pan N., Goebbels S., Hörnberg H., Nave K. A., Herrera P., White P., Kaestner K. H., Sussel L., Lee J. E. (2010) Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 11, 298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Herrera P. L. (2000) Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127, 2317–2322 [DOI] [PubMed] [Google Scholar]
- 29. Tamarina N. A., Roe M. W., Philipson L. (2014) Characterization of mice expressing Ins1 gene promoter driven CreERT recombinase for conditional gene deletion in pancreatic beta-cells. Islets 6, 27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Magnuson M. A., Osipovich A. B. (2013) Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab. 18, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]