

Background: Unconventional secretion of FGF2 occurs by direct translocation across plasma membranes.
Results: The cytoplasmic domain of ATP1A1 directly interacts with FGF2 and is required for FGF2 secretion.
Conclusion: ATP1A1 supports unconventional secretion by recruiting FGF2 to the inner leaflet of plasma membranes.
Significance: A new machinery component required for unconventional secretion of FGF2 was identified and validated.
Keywords: Fibroblast Growth Factor (FGF), Heparan Sulfate, Na+/K+-ATPase, Phosphoinositide, Plasma Membrane, ATP1A1, Membrane Recruitment, Membrane Translocation, Tec Kinase, Unconventional Secretion
Abstract
Previous studies proposed a role for the Na/K-ATPase in unconventional secretion of fibroblast growth factor 2 (FGF2). This conclusion was based upon pharmacological inhibition of FGF2 secretion in the presence of ouabain. However, neither independent experimental evidence nor a potential mechanism was provided. Based upon an unbiased RNAi screen, we now report the identification of ATP1A1, the α1-chain of the Na/K-ATPase, as a factor required for efficient secretion of FGF2. As opposed to ATP1A1, down-regulation of the β1- and β3-chains (ATP1B1 and ATP1B3) of the Na/K-ATPase did not affect FGF2 secretion, suggesting that they are dispensable for this process. These findings indicate that it is not the membrane potential-generating function of the Na/K-ATPase complex but rather a so far unidentified role of potentially unassembled α1-chains that is critical for unconventional secretion of FGF2. Consistently, in the absence of β-chains, we found a direct interaction between the cytoplasmic domain of ATP1A1 and FGF2 with submicromolar affinity. Based upon these observations, we propose that ATP1A1 is a recruitment factor for FGF2 at the inner leaflet of plasma membranes that may control phosphatidylinositol 4,5-bisphosphate-dependent membrane translocation as part of the unconventional secretory pathway of FGF2.
Introduction
Secretory proteins with N-terminal signal peptides qualify for the classical secretory pathway that is based on the membrane systems of the endoplasmic reticulum (ER)3 and the Golgi apparatus (1–3). This process is initiated by a signal peptide recognition particle, resulting in translocation of signal peptide-containing proteins into the lumen of the ER (4). Once secretory proteins have passed quality control in the ER (5, 6), they are transported in vesicular carriers to the Golgi and further to the cell surface, where they are released into the extracellular space (1–3). Although this pathway accounts for the extracellular localization of the majority of secretory proteins, at least two signal peptide recognition particle-independent mechanisms of protein secretion from eukaryotic cells have been identified (7). Intriguingly, unconventional secretory routes are taken by extracellular proteins with fundamental physiological functions, including immune surveillance and tissue organization (8, 9). This is best exemplified by the two most prominent examples of unconventionally secreted proteins, FGF2 and interleukin 1β (IL1β), that act as proangiogenic and proinflammatory molecules, respectively (10, 11).
FGF2 is a classical case of a pathway of unconventional protein secretion that is based upon direct translocation from the cytoplasm across plasma membranes into the extracellular space (12, 13). We have shown that this pathway is initiated by recruitment of FGF2 to the inner leaflet of plasma membranes mediated by the phosphoinositide phosphatidylinositol 4,5-bisphosphate (14, 15). This process causes FGF2 to oligomerize, followed by the formation of a lipidic membrane pore (7, 16, 17). This structure has been interpreted as a translocation intermediate that is resolved by extracellular heparan sulfate proteoglycans (18). Heparan sulfate proteoglycans thus are required to complete FGF2 membrane translocation and trap secreted FGF2 molecules on cell surfaces (19, 20). Further evidence for this mechanism came from the finding that Tec kinase-mediated phosphorylation of FGF2 both facilitates secretion of FGF2 from cells (16, 21) and stimulates membrane pore formation of FGF2 oligomers in reconstitution experiments (17, 22). The role of FGF2 oligomers as translocation intermediates is also supported by findings demonstrating that FGF2 does not undergo unfolding during membrane translocation (22–24).
In the current study, based on an unbiased large scale RNAi screen through which Tec kinase was identified as a positive regulator of FGF2 secretion (21), we report on the identification of another component of the FGF2 secretion machinery, ATP1A1, the α1-chain of the Na/K-ATPase. A functional Na/K-ATPase is composed of a heterotetramer of α- and β-chains in an α2β2 configuration (25). The classical functions of the Na/K ATPase, such as the generation of a general membrane potential or its role in nutrient uptake, depend on its fully assembled form (25). Four isoforms of both α-chains (ATP1A1–4) and β-chains (ATP1B1–4) are known in the human genome. We find that RNAi-mediated down-regulation of ATP1A1 profoundly inhibits FGF2 secretion, whereas the β-chains of the Na/K-ATPase complex appear to be dispensable for this process. These findings suggest that inhibition of FGF2 secretion in the absence of ATP1A1 is not due to an impaired membrane potential because the β-chains are essential for the activity of the Na/K-ATPase (25). Inhibition of FGF2 secretion in the absence of ATP1A1 is a highly specific phenotype because ER/Golgi-dependent protein transport of an integral membrane protein to the cell surface remained unaffected under these conditions. Along with reports suggesting the existence of unassembled α1-chains in plasma membranes (26), our findings indicate a direct role for ATP1A1 in the initiation of FGF2 membrane translocation at the inner leaflet of plasma membranes. This view is supported by biochemical experiments demonstrating a direct interaction with submicromolar affinity between the cytoplasmic domain of ATP1A1 and FGF2. Our data are consistent with previous findings reporting on the inhibition of FGF2 secretion by ouabain (27, 28) and indicate that ATP1A1 is a novel recruitment factor for FGF2 at the inner leaflet of plasma membranes.
EXPERIMENTAL PROCEDURES
Large Scale RNAi Screening to Identify Gene Products Involved in FGF2 Secretion
Stable HeLa cell lines expressing FGF2-GFP in a doxycycline-dependent manner were used to quantify FGF2 secretion as described previously (21). For RNAi screening purposes, the LI-COR infrared imaging platform (LI-COR Biosciences) was employed to analyze FGF2-GFP cell surface expression using the On-Cell-Western procedure. For siRNA transfection under screening conditions, 96-well plates were used. SiRNAs were derived from the Ambion Silencer® human extended druggable genome siRNA library V3, and plates were precoated with mixtures of transfection reagent and siRNAs used for solid-phase transfection (29). The siRNAs used in the experiments shown in Fig. 1 have the following Ambion IDs: 120182, 120183, and 120184 (ATP1A1); 120185, 120186, and 120187 (ATP1A2); 120737, 120840, and 120895 (ATP1A3); 258916, 284615, and 284616 (ATP1A4); 120217, 120218, and 120219 (ATP1B1); 10252, 120221, and 120222 (ATP1B2); 120223, 120224 and 120225 (ATP1B3); and 108263, 108264, and 117446 (ATP1B4). The siRNAs shown in Fig. 1 that were directed against FGF2 have the Ambion IDs 4190, 4286, and 4378. Following RNAi-mediated knockdowns (48 h), expression of FGF2-GFP was induced by the addition of 2 μg/ml doxycycline for 16 h. Cells were then fixed on ice for 20 min with paraformaldehyde (3% in PBS). All of the following steps were carried out at room temperature. Cells were washed (PBS) and treated for 1.5 h with Odyssey blocking solution (LI-COR Biosciences). Fixed cells were incubated for 2 h with affinity-purified anti-GFP antibodies diluted in 1% BSA, PBS. Following washing with PBS, cells were treated for 1 h with both secondary antibody (IRD800 goat anti-rabbit, LI-COR Biosciences) and Syto-60 (Invitrogen) to stain nucleic acids as a relative measure for cell numbers. After extensive washing, plates were imaged on a LI-COR Biosciences Odyssey infrared imaging system. Antibody-derived signals were taken as a relative measure for cell surface-localized FGF2-GFP that was normalized by the Syto-60 signal (cell number). HeLa cells expressing GFP in a doxycycline-dependent manner were used to quantify background signals. RNAi screening data were analyzed as described previously (21). Briefly, four replicates of each experimental condition were expressed as mean scores, a value that describes the deviation of the average of four individual measurements from the corresponding plate medians expressed as the number of S.D. values measured for the corresponding plates. Furthermore, a t test was carried out to analyze whether mean scores for each siRNA differed significantly from zero.
FIGURE 1.
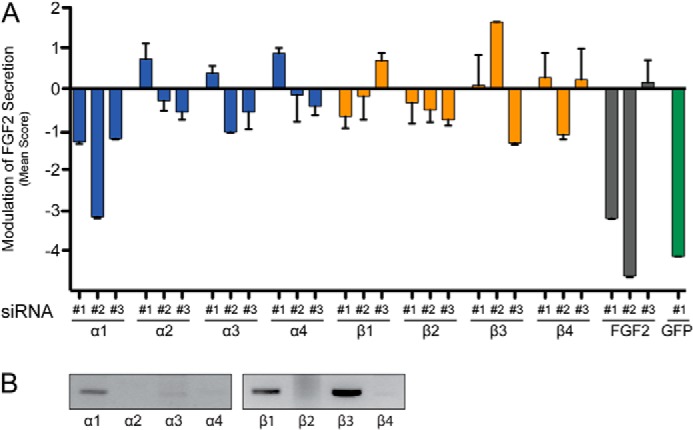
Identification of ATP1A1 as a component of the machinery mediating unconventional secretion of FGF2. A, mean scores of a selected set of siRNAs used in large scale RNAi screening for gene products involved in FGF2 secretion. A stable HeLa cell line expressing FGF2-GFP in a doxycycline-dependent manner was used to quantify FGF2 secretion as described under “Experimental Procedures.” All isoforms of α- and β-chains of the Na/K-ATPase known in the human genome were targeted with three independent siRNAs. In addition, three siRNAs directed against FGF2 are shown. As a control, a validated siRNA against GFP was used to down-regulate the FGF2-GFP reporter itself. Error bars, S.D. (n = 4). B, expression analysis of all known α- and β-chains of the Na/K-ATPase complex in HeLa cells employing RT-PCR.
Quantification of FGF2 Secretion and ER/Golgi-dependent Transport of a Plasma Membrane-resident CD4 Fusion Protein Employing Flow Cytometry
Stable HeLa cell lines either expressing FGF2 and GFP from an internal ribosome entry site construct based on the vector pREVTre2 or expressing a signal peptide-containing GFP-CD4 fusion protein were generated by retroviral transduction. These cell lines were used to quantify unconventional secretion of FGF2 as well as SP-GFP-CD4 transport from the ER to the plasma membrane, respectively. SP-GFP-CD4 contained an N-terminal signal peptide, GFP in its extracellular domain, and the transmembrane span and the cytoplasmic domain of CD4. It was cloned into the retroviral vector pRevTre2, and stable HeLa cell lines were generated as described previously (21, 30). In these cell lines, expression of the reporters was under the control of a doxycycline-dependent promotor (21, 30). To quantify the surface population of FGF2 and SP-GFP-CD4, respectively, a well characterized flow cytometry assay was used (30, 31).
Cells were transfected with Ambion Silencer® Select siRNAs (ATP1A1: siRNA 4 ID s1718; siRNA 5 ID s1719; siRNA 6 ID s1720; ATP1B1: ID s1734; ATP1B3: ID s1741; FGF2 ID 4286 was used as a custom-made Silencer® Select siRNA; GAPDH: ID s5574) at a final concentration of 3 nm using Oligofectamine (Invitrogen) and grown for 48 h. Following induction of protein expression with doxycycline for 16 h, cell surface-localized FGF2 and SP-GFP-CD4 were stained with affinity-purified anti-FGF2 or affinity-purified anti-GFP antibodies (21, 30), followed by incubation with appropriate allophycocyanin-conjugated secondary antibodies. Cells were processed for flow cytometry, and allophycocyanin-derived fluorescence (cell surface population) was determined using a BD Biosciences FACSCalibur instrument. Knockdown efficiencies of the gene products indicated were monitored either by Western analysis or by quantitative RT-PCR (Figs. 2 and 4). Quantitative RT-PCRs were conducted to quantify mRNA levels using the TaqMan gene expression system (Applied Biosystems). The TaqMan primers ID 2597 (ATP1B1) and ID 481 were used to quantify ATP1B1 and ATP1B3, respectively (Fig. 2E). For Western analyses, the following antibodies were used: affinity-purified rabbit anti-FGF2 antibodies (30), anti-GAPDH antibodies (Lifetech catalog no. am3100), anti-tubulin antibodies (Abcam catalog no. 18251), anti-ATP1A1 antibodies (Abcam catalog no. 7671), and anti-ATP1B1 antibodies (Sigma-Aldrich).
FIGURE 2.
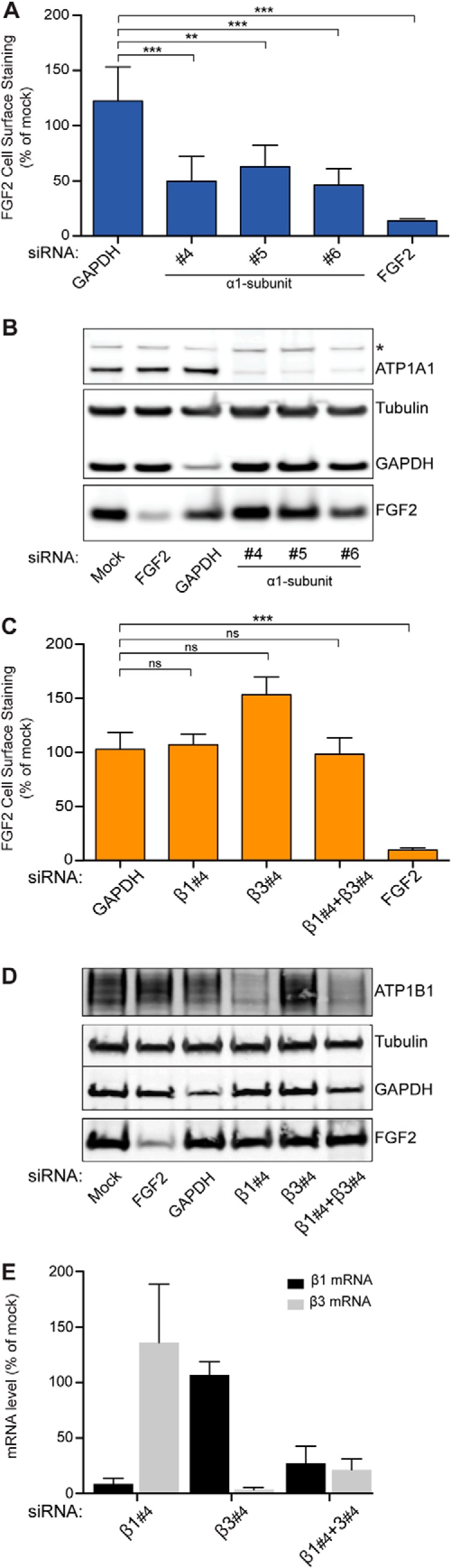
Validation of ATP1A1 as a component of the machinery mediating unconventional secretion of FGF2. A flow cytometry assay was used to quantify unconventional secretion using a stable HeLa cell line expressing FGF2 and GFP in a doxycycline-dependent manner (18, 23, 24, 30, 31). A, FGF2 cell surface expression under control conditions and after down-regulation of ATP1A1. Validated siRNAs directed against GAPDH and FGF2 were used as negative and positive controls, respectively. Error bars, S.D. (n = 4). To test whether observed differences between experimental conditions were statistically significant, an unpaired two-tailed Student's t test was performed (ns, not significant; **, p value ≤ 0.01; ***, p value ≤ 0.001). B, Western blot analysis to test the efficiency of down-regulation by RNAi for the gene products indicated. The asterisk indicates a cross-reactivity of the anti-ATP1A1 antibody used in this analysis. C, FGF2 cell surface expression under control conditions and after down-regulation of ATP1B1 and ATP1B3, the β1- and β3-chains of the Na/K-ATPase expressed in HeLa cells. Validated siRNAs directed against GAPDH and FGF2 were used as negative and positive controls, respectively. Error bars, S.D. (n = 4). To test whether observed differences between experimental conditions were statistically significant, an unpaired two-tailed t test was performed (ns, not significant; ***, p ≤ 0.001). D, Western blot analysis to test the efficiency of down-regulation by RNAi for the gene products indicated. E, quantitative RT-PCR analysis to monitor RNAi-mediated down-regulation of ATP1B1 and ATP1B3, the β1- and β3-chains of the Na/K-ATPase.
FIGURE 4.
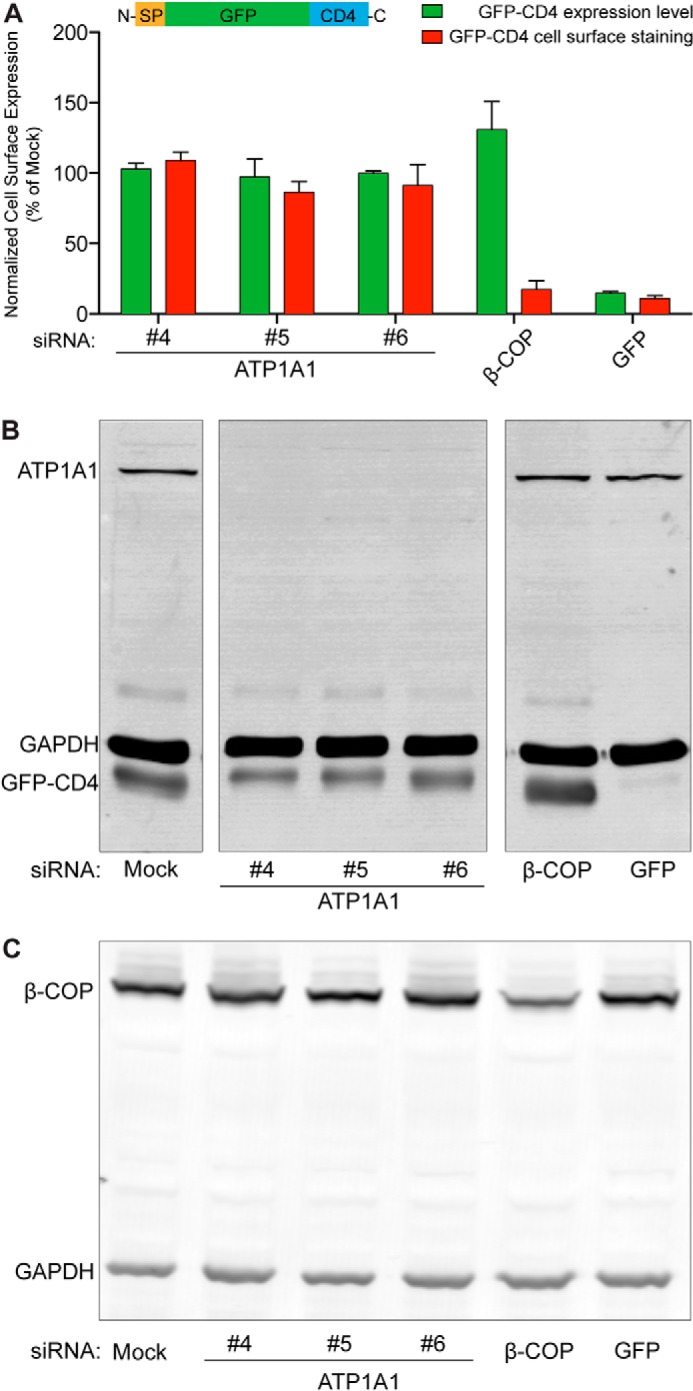
Inhibition of FGF2 secretion caused by down-regulation of ATP1A1 is not due to pleiotropic effects. A stable cell line expressing a GFP-CD4 fusion protein in a doxycycline-dependent manner was generated. This construct carries an N-terminal signal peptide and an extracellular GFP domain, followed by the transmembrane span and the cytoplasmic domain of CD4 (SP-GFP-CD4), and is transported to cell surfaces via the ER/Golgi-dependent secretory pathway. Following incubation of cells in the presence of doxycycline, GFP-CD4 was detected on cell surfaces using anti-GFP antibodies and flow cytometry. A, normalized cell surface expression of GFP-CD4 under control conditions and after down-regulation of ATP1A1 using three independent siRNAs. As a positive control, a validated siRNA directed against β-COP, a component of the coatomer complex that is essential for transport within the ER/Golgi-dependent pathway, was used (2). In addition, a siRNA directed against GFP was used to down-regulate the GFP-CD4 reporter itself. Error bars, S.D. (n = 3). B, Western blot analysis of the efficiency of down-regulation by RNA interference for ATP1A1 and GFP-CD4. C, Western blot analysis of the efficiency of down-regulation by RNA interference for β-COP.
Analysis of Cell Proliferation following RNAi-mediated Down-regulation of ATP1A1, ATP1B1, and ATP1B3
Cell proliferation experiments were conducted employing an Essen BioScience IncuCyte Zoom live cell imaging microscope, providing a kinetic and quantitative analysis of cell proliferation based on confluence measurements (Fig. 3) (32). HeLa cells were cultivated under the conditions used for the experiments shown in Fig. 2 in which FGF2 secretion was quantified following down-regulation of the gene products indicated (see above). Cells were grown in 48-well plates and transfected with the siRNAs indicated. After 24 h, cells were counted, and identical cell numbers from each experimental condition were cultivated in 12-well plates. After 24 h, doxycycline (1 μg/ml) was added to induce expression of FGF2, and cultivation continued for another 24 h. Cell proliferation was monitored for the last 48 h of this protocol (Fig. 3).
FIGURE 3.
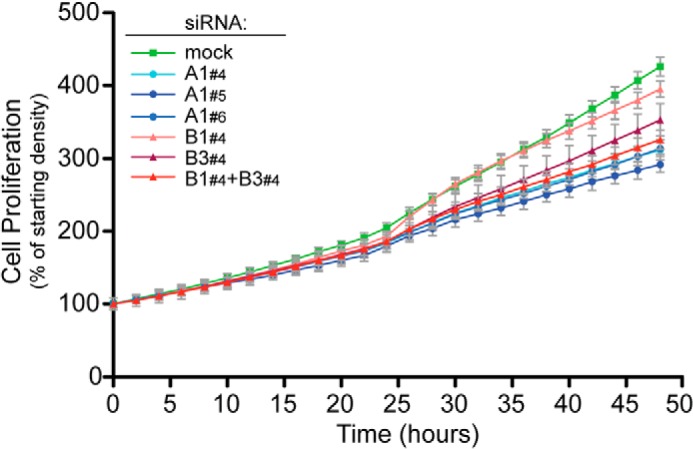
Analysis of cell proliferation following down-regulation of ATP1A1, ATP1B1, and ATP1B3. Cells were cultivated using exactly the conditions described for the FGF2 secretion experiments shown in Fig. 2, including doxycycline-dependent induction of FGF2 expression. Following siRNA-mediated down-regulation of gene products, as indicated, a kinetic analysis of cell proliferation was conducted by quantifying cell confluence using an Essen BioScience IncuCyte Zoom live cell imaging microscope. The starting density measured by cell confluence was set to 100% for each experimental condition. The results shown are representative for four independent biological replicates. Error bars, S.D. derived from three technical replicates.
Recombinant Proteins and Biochemical Protein-Protein Interaction Assays
Recombinant proteins were expressed in Escherichia coli and purified according to standard procedures. The 18-kDa isoform of FGF2 was N-terminally tagged with a His6 epitope (sequence: MRGSHHHHHH-GS-MAAGS with the last 5 amino acids representing the N terminus of FGF2). Additionally, a non-tagged, N-terminally truncated form of FGF2 (NΔ25-FGF2) was used starting from the sequence MGGSMKDPKR. Five variant forms of the cytoplasmic domain of ATP1A1 (ATP1A1-CD) were expressed as N-terminal GST fusion proteins and purified from E. coli according to standard procedures. This included a form that contained all three cytoplasmic loops (GST-ATP1A1-CD1–3): GST-MGRDYEPAAV-loop 1-NALTPPPTTP-GSEFHM-KSSKIMESFK-loop 2-GGQTPIAAEI-GSPGHM-CLTLTAKRMA-loop 3-EEGRLIFDNL with the underlined letters defining the flanking sequences of the three loop regions. The linkers are shown in boldface type. Accordingly, only loops 2 and 3 (GST-ATP1A1-CD2–3) and three variants containing single loops (GST-ATP1A1-CD1, GST-ATP1A1-CD2, and GST-ATP1A1-CD3) were used.
Biochemical Pull-down Experiments to Detect Direct Interactions between FGF2 and the Cytoplasmic Domain of ATP1A1 (ATP1A1-CD)
Pull-down experiments were performed with both possible arrangements of protein immobilization on beads. In one set of experiments, FGF2 was covalently immobilized on epoxy-activated Sepharose beads 6B (GE Healthcare) according to the manufacturer's instructions and incubated with GST fusion proteins containing the various constructs of the cytoplasmic domain of ATP1A1 (Fig. 5A). GST was used as a negative control. FGF2-coupled epoxy beads were blocked with 3% BSA in PBS (supplemented with 0.05% (w/v) Tween 20 and 1 mm benzamidine), and following extensive washing with PBS, the resin was covered with 10 volumes of PBS (supplemented with 0.05% (w/v) Tween 20 and 1 mm benzamidine). Per experimental condition, 100 μl of the epoxy bead suspension (about 60 μg of FGF2/experimental condition) were incubated with 25 μg of each GST-ATP1A1-CD variant form for 2 h at room temperature. Following collection of beads by low speed centrifugation and extensive washing with PBS (supplemented with 0.05% (w/v) Tween 20 and 1 mm benzamidine), bound protein was eluted with SDS sample buffer. Both the bound and the unbound fractions of proteins were subjected to SDS-PAGE, followed by protein staining using Coomassie InstantBlue (Expedeon).
FIGURE 5.

Direct interaction between FGF2 and the cytoplasmic domain of ATP1A1 as demonstrated by biochemical pull-down experiments. A, affinity beads containing FGF2 were incubated with various variant forms of the cytoplasmic domain of ATP1A1, as indicated. The latter were used as GST fusion proteins with GST alone as a negative control (lanes 1–3). The other constructs were GST-ATP1A1-CD1 (lanes 4–6), GST-ATP1A1-CD2 (lanes 7–9), GST-ATP1A1-CD3 (lanes 10–12), GST-ATP1A1-CD2–3 (lanes 13–15), and GST-ATP1A1-CD1–3 (lanes 16–18). Bound (50% of each fraction) and unbound material (5% of each fraction) was analyzed by SDS-PAGE and Coomassie Brilliant Blue protein staining. The results shown are representative for three independent experiments. B, affinity beads containing the various variant forms of the cytoplasmic domain of ATP1A1 were incubated with soluble recombinant FGF2, as indicated. GST alone was used as a negative control (lanes 1 and 2). The other constructs were GST-ATP1A1-CD1 (lanes 3 and 4), GST-ATP1A1-CD2 (lanes 5 and 6), GST-ATP1A1-CD3 (lanes 7 and 8), GST-ATP1A1-CD2–3 (lanes 9 and 10), and GST-ATP1A1-CD1–3 (lanes 11 and 12). Bound (50% of each fraction) and unbound material (5% of each fraction) was analyzed by SDS-PAGE and Coomassie Brilliant Blue protein staining. The results shown are representative for three independent experiments.
In a second set of experiments, GST fusion proteins containing the various forms of cytoplasmic domains of ATP1A1 were immobilized on GSH-Sepharose 4B (GE Healthcare) via a non-covalent interaction with GSH according to the manufacturer's instructions. Following incubation with 3% BSA in PBS (supplemented with 0.05% (w/v) Tween 20 and 1 mm benzamidine) to block nonspecific interactions, the affinity beads were washed extensively with PBS supplemented with 0.05% (w/v) Tween 20 and 1 mm benzamidine. Per experimental condition, 10 μl of affinity beads containing the various forms of the cytoplasmic domain of ATP1A1 (20 μg/experimental condition) were incubated with 25 μg of His-tagged FGF2 and incubated for 2 h at room temperature (Fig. 5B). Following collection of beads by low speed centrifugation and extensive washing with PBS supplemented with 0.05% (w/v) Tween 20 and 1 mm benzamidine, bound protein was eluted with SDS sample buffer. Both the fractions of bound and unbound proteins were subjected to SDS-PAGE, followed by protein staining using Coomassie InstantBlue (Expedeon).
Determination of Binding Affinity for the Interaction between FGF2 and the Cytoplasmic Domain of ATP1A1
To determine binding affinity between FGF2 and ATP1A1-CD (Fig. 6A), we used the AlphaScreen® protein-protein interaction assay.
FIGURE 6.
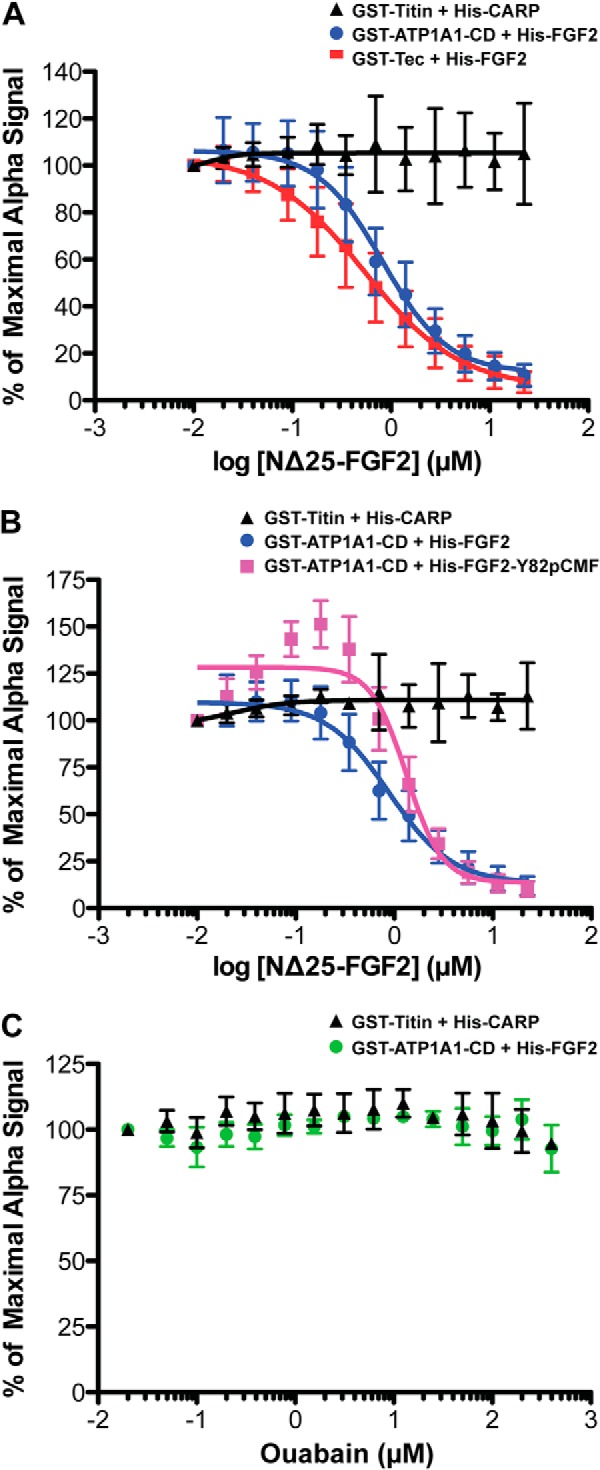
FGF2 binds with submicromolar affinity to the cytoplasmic domain of ATP1A1. A quantitative protein-protein interaction assay based on AlphaScreen® technology (34) was used to determine the affinity of the interaction between FGF2 and ATP1A1-CD. As detailed under “Experimental Procedures,” AlphaScreen® protein-protein interaction signals were recorded for the various pairs of His- and GST-tagged proteins indicated. To determine affinity, an untagged, N-terminally truncated form of FGF2 (NΔ25-FGF2) was titrated into the binding reaction at the concentrations indicated. Error bars, S.D. (n = 3). The experimental data were fitted with a non-linear regression model (log (inhibitor) versus response − variable slope (four parameters)) using GraphPad Prism version 5.0c software to calculate IC50 values. A, His6-tagged FGF2 and GST-tagged ATP1A1-CD (blue spheres), His6-tagged FGF2 and GST-tagged Tec kinase (red squares), and His6-tagged CARP and GST-tagged titin (black triangles). B, comparison between the interactions of WT FGF2 (blue spheres) and FGF2-Y81pCMF (pink squares) with ATP1A1-CD. His6-tagged CARP and GST-tagged titin (black triangles) were used as a negative control. C, analysis of a potential impact of ouabain on the interaction of FGF2 with ATP1A1-CD. Ouabain was titrated at the concentrations indicated. Green spheres, His6-tagged FGF2 and GST-tagged ATP1A1-CD; black triangles, His6-tagged CARP and GST-tagged titin.
In these experiments, WT FGF2 was used with an N-terminal His6 tag (see above) at a final concentration of 60 nm, and the complete cytoplasmic domain of ATP1A1 was used as a GST fusion protein (GST-ATP1A1-CD1–3) at a final concentration of 30 nm. As a positive control, Tec kinase (GST-ΔPH-TH Tec, Tec kinase without the PH domain (21)) was analyzed as a known interaction partner of FGF2 (21) at a final concentration of 30 nm. An unrelated protein-protein interaction was used as a negative control using GST-titin (20 nm) and His6-tagged CARP (20 nm) (33). In the experiments shown in Fig. 6B, the wild-type form of FGF2 was substituted by a phosphomimetic form, FGF2-Y81pCMF (17). In addition, experiments were performed where NΔ25-FGF2 was replaced by ouabain (Fig. 6C). The indicated protein pairs were immobilized on AlphaScreen® nickel acceptor beads and AlphaScreen® glutathione donor beads, respectively. Upon excitation of donor beads at 680 nm, singlet oxygen species are produced that can diffuse within a range of 200 nm. If donor and acceptor beads are in close proximity due to a protein-protein interaction, singlet oxygen species will transfer energy to acceptor beads, which in turn causes emission of light with a wave length of 520 nm that is detected as the AlphaScreen® signal. To conduct competition experiments, a non-tagged, N-terminally truncated form of FGF2 (NΔ25-FGF2) was used to compete for this interaction, which allows the determination of IC50 values. All assays were conducted in PBS supplemented with 0.05% (w/v) Tween 20 and 0.1% (w/v) BSA. The respective pairs of His6- and GST-tagged proteins were mixed (in 5 μl) with competitor molecules (in 5 μl) at the concentrations indicated. These mixtures were preincubated for 60 min at room temperature, followed by the addition of AlphaScreen® nickel acceptor beads and glutathione donor beads at a concentration of 7.5 μg/ml in a final volume of 15 μl. Finally, after 105 min incubation, the AlphaScreen® signal was measured using an EnVision plate reader (PerkinElmer Life Sciences). For each protein pair, the median signal of three technical replicates was calculated, normalized to the signal measured in the absence of the competitor, and plotted against the concentration of the competitor protein (NΔ25-FGF2; Fig. 6). The error estimates shown in the various panels of Fig. 6 are based on three independent biological replicates, each of which consisted of three technical replicates. The competitor concentration promoting half-maximal inhibition (IC50) of the signal was determined by fitting the experimental data with a non-linear regression model (log(inhibitor) versus response − variable slope (four parameters)) using GraphPad Prism version 5.0c software. Under suitable experimental conditions, the apparent IC50 value in this type of competition experiments corresponds to the dissociation constant of the observed protein-protein interaction (34).
Duolink® in Situ Proximity Ligation Immunoassay
HeLa cells were grown on glass bottom culture dishes (MatTek 10-mm microwell), washed with PBS, fixed for 4 min with ice-cold acetone, and blocked with 1% BSA, PBS for 15 min at room temperature. Cells were incubated with the primary antibodies indicated (diluted in 1% BSA, PBS) for 1 h at room temperature. The following primary antibodies were used: mouse anti-ATP1A1 (diluted 1:100; Abcam ab7671), mouse anti-cadherin (diluted 1:100; Abcam ab6528), mouse anti-GM130 (diluted 1:1,000; BD Transduction Laboratories catalog no. 610822), mouse anti-transferrin receptor (diluted 1:500; Invitrogen 13-6800), and rabbit anti-FGF2 (diluted 1:100) (18). Appropriate secondary antibodies conjugated to PLA probes (PLA MINUS anti-mouse (Olink Bioscience 92004-0030) and PLA PLUS anti-rabbit (Olink Bioscience 92002-0030)) were diluted 1:5 in 1% BSA, PBS and incubated for 1 h at 37 °C. Ligation, amplification of DNA, and its detection were conducted according to the manufacturer's manual using the Duolink® detection reagent red (Olink Bioscience 92008-0030). Nuclei were stained with SYTOX green (Invitrogen) prior to imaging on a Zeiss LSM 510 confocal microscope. Signals obtained per cell were quantified using the Duolink® Image Tool software (Olink Bioscience). An unpaired two-tailed Student's t test was performed to analyze whether mean signals per cell obtained from ATP1A1/FGF2 staining significantly differed from those of other pairs (transferrin receptor/FGF2; cadherin/FGF2; GM130/FGF2) or from those under conditions using just one antibody (FGF2 or ATP1A1) as a technical control condition (Fig. 7). In the experiments shown in Fig. 8, in-cell interactions between ATP1A1 and FGF2 were quantified under various knockdown conditions as indicated.
FIGURE 7.

Proximity of ATP1A1 and FGF2 analyzed in cells. To test for proximity of ATP1A1 and FGF2 in cells, the Duolink® in situ proximity ligation immunoassay (PLA®; Sigma-Aldrich) was used. HeLa cells were fixed and permeabilized with acetone. The Duolink® assay was conducted as described under “Experimental Procedures” using pairs of primary antibodies (or single primary antibodies as controls) directed against the antigens indicated. Red dots, interaction/proximity events. To visualize the nuclei of cells, DNA was stained with SYTOX® green (Invitrogen), shown in blue. A, ATP1A1 and FGF2; B, transferrin receptor (TFR) and FGF2; C, cadherin and FGF2; D, GM130 and FGF2; E, control using just one primary antibody against FGF2; F, control using just one primary antibody directed against ATP1A1; G, statistical analysis of proximity events using the Duolink® image tool software (Olink Bioscience). Data are shown as absolute counts of proximity events per cell. Mean values were calculated from four independent experiments with 30–50 cells being analyzed per individual experiment and condition. Error bars, S.D. To test whether observed differences between experimental conditions were statistically significant, an unpaired two-tailed Student's t test was performed (ns, not significant; ***, p ≤ 0.001).
FIGURE 8.

Quantification of in-cell interactions of FGF2 and ATP1A1 following down-regulation of ATP1A1, ATP1B1, and ATP1B3. Following RNAi-mediated down-regulation of the gene products indicated, cells were processed for Duolink® in situ proximity assays as described under “Experimental Procedures” and in the legend to Fig. 7. A, Western analysis to monitor down-regulation of ATP1A1 and ATP1B1 under the experimental conditions indicated. B, Duolink® in situ proximity assay to quantify in-cell interactions between FGF2 and ATP1A1 following down-regulation of the gene products indicated. a, mock; b, siRNA ATP1A1; c, siRNA ATP1B1; d, siRNA ATP1B3; e, siRNA ATP1B1/ATP1B3; f, control 1 (mock; single antibody control) (ATP1A1); g, control 2 (mock; single antibody control) (FGF2); h, control 3 (mock; no primary antibodies against FGF2 and ATP1A1). C, quantification of in-cell interaction signals (proximity events/cell) under the experimental conditions shown in B. The blue dotted line indicates the background signal. Error bars, S.D. (n = 3). To test whether observed differences between experimental conditions were statistically significant, an unpaired two-tailed Student's t test was performed (*, p ≤ 0.05; **, p ≤ 0.01).
RESULTS
Identification of ATP1A1 as a Component of the Machinery Mediating FGF2 Secretion Employing Large Scale RNAi Screening
Based on an unbiased large scale RNAi screen that previously led to the identification of Tec kinase as a positive regulator of FGF2 secretion (21), we have identified ATP1A1 as another plasma membrane-resident factor supporting FGF2 membrane translocation into the extracellular space. This experimental system was based upon stable HeLa cells expressing an FGF2-GFP fusion protein in a doxycycline-dependent manner. Using an on-cell Western protocol, secreted FGF2-GFP present on cell surfaces was quantified using the LI-COR Odyssey imaging platform (21). The screen was conducted in four replicates using a library containing three independent siRNAs against each gene product known in the human genome (21). Mean scores were calculated (as detailed under “Experimental Procedures”), with a negative value indicating a decrease and a positive value indicating an increase in the efficiency of FGF2 secretion. Using this experimental platform, we found that all three siRNAs directed against ATP1A1 that were present in the large scale siRNA library caused inhibition of FGF2 secretion (Fig. 1A). By contrast, none of the siRNAs directed against the other three known α-chains or the four known β-chains had a consistent impact on FGF2 secretion (Fig. 1A). An RT-PCR analysis of all known human α- and β-chains revealed that the α1-chain as well as the β1- and β3-chains are the predominant isoforms expressed in HeLa cells (Fig. 1B). Thus, the data generated by primary screening indicated a specific role of ATP1A1 in unconventional secretion of FGF2.
Validation of ATP1A1 as a Component of the Machinery Mediating FGF2 Secretion Employing a Quantitative Flow Cytometry Assay
To validate the results from RNAi screening (Fig. 1), we used a well established experimental system to quantify FGF2 secretion that is based on flow cytometry (30). Three additional independent siRNAs directed against ATP1A1 were tested, with all of them causing both significant inhibition of FGF2 secretion (Fig. 2A) and a substantial decrease of ATP1A1 levels as determined by Western analysis (Fig. 2B). By contrast, down-regulation of the β1- and β3-chains of the Na/K-ATPase did not result in inhibition of FGF2 secretion (Fig. 2C). Even when both β-chains were down-regulated simultaneously, FGF2 secretion remained unaffected (Fig. 2C). Again, the corresponding knockdown efficiencies were monitored by a Western analysis (Fig. 2D) or by a quantitative RT-PCR analysis (Fig. 2E). These findings validate the results from the RNAi screening procedure and suggest a role for ATP1A1 in unconventional secretion of FGF2.
Impaired FGF2 Secretion after Down-regulation of ATP1A1 Is Not Due to Pleiotropic Effects
To address potential pleiotropic effects causing inhibition of FGF2 secretion following down-regulation of ATP1A1, we conducted two types of experiments. First, using the same experimental conditions described for FGF2 secretion experiments, a kinetic analysis of cell proliferation was conducted (Fig. 3). These experiments revealed that, compared with mock conditions, down-regulation of ATP1A1, ATP1B1, ATP1B3, or a combination of ATP1B1 and ATP1B3 causes a moderate slowdown of cell proliferation. Single knockdowns of either ATP1B1 or ATP1B3 had the mildest phenotypes, whereas a combined knockdown of ATP1B1 and ATP1B3 or a knockdown of ATP1A1 caused a somewhat stronger decrease in cell proliferation. Importantly, under all experimental conditions, cells continued to proliferate (Fig. 3). Because a combined knockdown of ATP1B1 and ATP1B3 does not affect FGF2 secretion (Fig. 2C), we conclude that inhibition of FGF2 secretion after RNAi-mediated down-regulation of ATP1A1 (Fig. 2A) is not caused by pleiotropic effects, such as impaired cell viability.
To challenge this conclusion by an independent approach, we analyzed an unrelated intracellular transport process. Transport of an integral membrane protein from the ER to the plasma membrane was quantified under control conditions and after down-regulation of ATP1A1 (Fig. 4). A stable cell line was constructed expressing a CD4 variant in a doxycycline-dependent manner. In this construct, the luminal domain of CD4 was replaced by GFP. Therefore, upon delivery of the CD4 fusion protein to the plasma membrane, the GFP domain of this construct is exposed on cell surfaces. In this way, using anti-GFP antibodies, a flow cytometry assay could be used to quantify transport of a GFP-CD4 fusion protein that is technically identical to the one used in this study to quantify FGF2 cell surface expression (Fig. 2). Using this system, transport of GFP-CD4 was studied under various experimental conditions, including siRNA-mediated down-regulation of ATP1A1 (Fig. 4, A and B) and β-COP (Fig. 4, A and C), a component of the coatomer complex that is essential for protein transport along the ER/Golgi-dependent secretory pathway (1, 2). These experiments demonstrated that down-regulation of ATP1A1 by three independent siRNAs (Fig. 4B) did not have any impact on the transport of the GFP-CD4 fusion protein to the plasma membrane (Fig. 4A). By contrast, down-regulation of β-COP (Fig. 4C) resulted in an almost complete block of transport of the GFP-CD4 fusion protein to the plasma membrane (Fig. 4A). Similar to the experiments shown in Fig. 3 but using an independent approach, these findings exclude pleiotropic effects as the cause of impaired FGF2 secretion after down-regulation of ATP1A1. In conclusion, the combined findings shown in Figs. 1–4 establish a specific role of ATP1A1 in unconventional secretion of FGF2.
A Direct Interaction between FGF2 and the Cytoplasmic Domain of ATP1A1
In order to obtain initial insight into the mechanism of how ATP1A1 affects unconventional secretion of FGF2, we analyzed whether they interact with each other in a direct manner. Despite small loops connecting the transmembrane segments, ATP1A1 does not contain an extracellular domain. Therefore, we reasoned that FGF2 may interact with the cytoplasmic domain of ATP1A1. A construct was generated containing all three cytoplasmic domains of ATP1A1 that were connected by short linkers and expressed as a soluble recombinant GST fusion protein (ATP1A1-CD1–3). In addition, we generated versions containing only one loop (ATP1A1-CD1, ATP1A1-CD2, and ATP1A1-CD3) or a combination of loops 2 and 3 (ATP1A1-CD2–3; Fig. 5). In a first set of experiments, purified recombinant FGF2 was covalently coupled to epoxy-Sepharose beads, and a potential binding of the various GST-ATP1A1-CD fusion proteins was assayed by classical pull-down experiments (Fig. 5A). These experiments revealed substantial binding of FGF2 to GST-ATP1A1-CD fusion proteins containing either loop 2 or loop 3. In addition, efficient binding of FGF2 to GST-ATP1A1-CD constructs was observed containing both loop 2 and loop 3 or all three loops of the cytoplasmic domain of ATP1A1. By contrast, FGF2 did not bind to GST-ATP1A1-CD1 containing only loop 1 or GST, establishing the specificity of the observed interaction (Fig. 5A).
In a second set of experiments, the various GST-ATP1A1-CD fusion proteins were non-covalently immobilized on glutathione beads, and binding of soluble FGF2 was tested again by pull-down experiments (Fig. 5B). Compared with the analysis shown in Fig. 5A, very similar binding properties were observed. Although FGF2 did not bind to loop 1 of ATP1A1-CD, it did bind efficiently to versions of ATP1A1-CD containing either loop 2 or loop 3. In addition, FGF2 did bind efficiently to versions of ATP1A1-CD containing both loop 2 and loop 3 or containing all three loops of the cytoplasmic domain of ATP1A1 (Fig. 5B). The combined findings from the experiments shown in Fig. 5 establish a direct interaction of FGF2 with the cytoplasmic domain of ATP1A1 mediated by loops 2 and 3.
FGF2 Binds to the Cytoplasmic Domain of ATP1A1 with Submicromolar Affinity
To further characterize the interaction between the cytoplasmic domain of ATP1A1 and FGF2, we aimed to determine binding affinity using the AlphaScreen® protein-protein interaction assay (34, 35). To mimic the situation in cells as closely as possible, we used a GST fusion protein of ATP1A1-CD containing all three loops of the cytoplasmic domain of ATP1A1 and a His-tagged version of FGF2 (Fig. 6). Two additional protein-protein interaction assays were analyzed, with the interaction of FGF2 with Tec kinase used as a positive control (21) and an unrelated protein-protein interaction between titin and CARP (33) serving as a negative control. A non-tagged and N-terminally truncated form of FGF2 (NΔ25-FGF2) was used as a competitor to determine binding affinity between ATP1A1-CD and FGF2. NΔ25-FGF2 efficiently competed for the interaction between ATP1A1-CD and FGF2 with an IC50 of 0.826 ± 0.33 μm (Fig. 6A). As expected, NΔ25-FGF2 also competed for the interaction between Tec kinase and FGF2 with an IC50 of 0.542 ± 0.29 μm. By contrast, the interaction between titin and CARP remained unaffected in the presence of NΔ25-FGF2 (Fig. 6A). Under the experimental conditions used, the determined IC50 values are good estimates for the dissociation constants of the observed interactions of FGF2 with the cytoplasmic domain of ATP1A1 and Tec kinase, respectively.
In a second set of experiments, we tested whether a phosphomimetic form of FGF2, FGF2-Y81pCMF (17), behaves differently from WT FGF2 in terms of affinity toward the cytoplasmic domain of ATP1A1. As shown in Fig. 6B, the IC50 value obtained by again using NΔ25-FGF2 as competitor was determined to be 1.33 ± 0.3 μm, indicating that affinity between FGF2-Y81pCMF and ATP1A1-CD is not significantly different from the corresponding interaction between WT FGF2 and ATP1A1-CD. Furthermore, we tested whether ouabain affects the interaction between FGF2 and the cytoplasmic domain of ATP1A1. As shown in Fig. 6C, a titration of ouabain at the concentrations indicated did not provide any evidence for an inhibitory effect. This result was expected because the ouabain binding site lies within a small extracellular loop of ATP1A1 (25).
Proximity of ATP1A1 and FGF2 in Cells
To obtain evidence for an interaction of ATP1A1 and FGF2 in cells, we used the Duolink® in situ proximity assay (see “Experimental Procedures” for details). In brief, HeLa cells were fixed and permeabilized, followed by incubation with different pairs of primary antibodies directed against the antigens indicated (Fig. 7). Using appropriate secondary antibodies coupled to PLA® probes (see “Experimental Procedures”), proximity of the two antigens recognized by the primary antibodies can be detected and quantified in a cellular context. Positive signals indicate either a direct interaction or proximity of two antigens mediated by indirect interactions. As shown in Fig. 7A, almost 100 proximity events per cell could be detected for ATP1A1 and FGF2. Two other plasma membrane-resident proteins were tested as a pair with FGF2, the transferrin receptor (Fig. 7B) and cadherin (Fig. 7C). Whereas the transferrin receptor showed proximity to FGF2, with about 70 events/cell (Fig. 7, B and G), no proximity of cadherin with FGF2 could be detected (Fig. 7, C and G). Similarly, the Golgi-resident integral membrane protein GM-130 did not display proximity to FGF2 (Fig. 7, D and G). When compared with background controls using just one primary antibody, no significant interaction of FGF2 with either cadherin or GM-130 was observed (Fig. 7, E–G). These observations are consistent with immunoprecipitation studies from other laboratories (28) and the biochemical reconstitution experiments presented in this study (Figs. 5 and 6) demonstrating a direct interaction of ATP1A1 and FGF2.
In a second series of Duolink proximity experiments shown in Fig. 8, we tested in-cell interactions between ATP1A1 and FGF2 under conditions that correspond to the secretion experiments shown in Fig. 2. This included down-regulation of ATP1A1, ATP1B1, and ATP1B3 as well as an ATP1B1/ATP1B3 double knockdown. Knockdown efficiencies were monitored by Western analysis of ATP1A1 and ATP1B1 for all experimental conditions (Fig. 8A). Whereas single knockdowns of ATP1B1 and ATP1B3 did not affect ATP1A1 protein levels to a significant extent, a double knockdown of both ATP1B1 and ATP1B3 resulted in reduced amounts of ATP1A1 (Fig. 8A). However, the levels of ATP1A1 under these conditions were significantly higher compared with a direct knockdown of ATP1A1 (Fig. 8A). These findings correlated well with the results from in-cell interactions between FGF2 and ATP1A1 (Fig. 8, B and C). Down-regulation of ATP1A1 caused a drop of proximity events between FGF2 and ATP1A1 down to almost background levels (Fig. 8, B and C). By contrast, following an ATP1B1/ATP1B3 double knockdown, in-cell interactions between ATP1A1 and FGF2 continued to be present and, therefore, were significantly more abundant compared with under conditions of a direct knockdown of ATP1A1. Thus, the differential phenotypes observed in FGF2 secretion assays comparing ATP1A1 knockdowns (inhibition of FGF2 secretion; Fig. 2A) versus ATP1B1/ATP1B3 double knockdowns (normal FGF2 secretion; Fig. 2C) directly correlated with the absence (ATP1A1 knockdown) or presence (ATP1B1/ATP1B3 double knockdown) of functional in-cell interactions between ATP1A1 and FGF2. These findings therefore explain the differential FGF2 secretion phenotypes observed for ATP1A1 versus ATP1B1/ATP1B3 knockdown conditions (Fig. 2).
DISCUSSION
In the current study, based on an unbiased large scale RNAi screen, we provide direct evidence for a specific role of ATP1A1, the α1-chain of the Na/K-ATPase, in unconventional secretion of FGF2. Furthermore, we identify the cytoplasmic domain of ATP1A1 as directly interacting with FGF2 with submicromolar affinity and provide direct evidence for in-cell interactions of FGF2 and ATP1A1. We propose that ATP1A1 represents a novel high affinity recruitment factor at the inner leaflet of plasma membranes that is involved in the translocation of FGF2 to cell surfaces.
In previous studies (27, 28, 30), a potential role for the Na/K-ATPase in unconventional secretion of FGF2 was discussed; however, these studies were limited to pharmacological inhibition using ouabain, an inhibitor of the enzymatic activity of the Na/K-ATPase (36). Because independent reagents compromising the membrane potential did not affect FGF2 secretion, a so far unidentified role of the Na/K-ATPase in FGF2 secretion was proposed (27, 28). This view was supported by experiments showing that ouabain-resistant mutants of the Na/K-ATPase restore FGF2 secretion in the presence of ouabain (27). Finally, evidence was presented for an interaction between FGF2 and ATP1A1 based on immunoprecipitation experiments from cell lysates. Shortcomings of these studies were that all cell-based experiments were entirely based on pharmacological inhibition along with protein-protein interaction data that did not provide insight into the question of direct contact, the strength of the interaction in terms of affinity, or the domains of ATP1A1 involved in the interaction with FGF2.
The current study now provides conclusive experimental evidence for a role of ATP1A1 in unconventional secretion of FGF2. Following down-regulation of ATP1A1 using a total of six independent siRNAs, substantial inhibition of FGF2 secretion was observed. Beyond the primary screening data, this phenotype could be confirmed using a well established flow cytometry assay used to quantify FGF2 secretion. Furthermore, a specific role for the α1-chain (ATP1A1) of the Na/K-ATPase could be established because down-regulation of the two forms of β-chains expressed in HeLa cells (ATP1B1 and ATP1B3) did not inhibit FGF2 secretion. Furthermore, inhibition of FGF2 secretion by down-regulation of ATP1A1 was not based on potential pleiotropic effects, such as impaired cell viability, because transport of an integral membrane protein from the ER to the plasma membrane remained unaffected under these conditions. These findings establish a specific role for ATP1A1 in unconventional secretion of FGF2 that is independent of the classical, membrane potential-generating function of the assembled α2β2 hetero-oligomeric complex that makes up the functional Na/K-ATPase.
To obtain insight into potential physical interactions between ATP1A1 and FGF2, we sought to conduct biochemical experiments suitable to address a potential direct interaction and, in the event that one exists, to obtain information on affinity and the domains of ATP1A1 that are involved. Because the cell-based experiments presented in this study indicated a role for ATP1A1 in FGF2 secretion and because ATP1A1 does not contain extracellular domains other than small loops that connect its transmembrane segments, we hypothesized that the cytoplasmic domain of ATP1A1 represents a potential binding site for FGF2. We generated a soluble recombinant protein that contains the three cytoplasmic domains of ATP1A1 (ATP1A1-CD). Using purified recombinant proteins, a direct physical interaction between ATP1A1-CD and FGF2 could be established employing classical pull-down experiments. Two loops in the cytoplasmic domain of ATP1A1 were found to mediate binding to FGF2, whereas the N-terminal part of ATP1A1-CD did not contribute to FGF2 binding. Furthermore, a construct containing all three cytoplasmic loops of ATP1A1 was expressed and purified as a soluble recombinant protein and shown to bind to FGF2 with submicromolar affinity, indicated by an IC50 value of 0.826 μm in competition experiments. Beyond the biochemical data, using the Duolink® in situ proximity assay, we also provide evidence for an interaction of FGF2 with ATP1A1 in cells.
How do the findings on ATP1A1 reported in this study fit into our general model of how FGF2 is secreted from cells? As illustrated in Fig. 9, FGF2 has been shown to translocate across plasma membranes (12, 13) based on phosphatidylinositol 4,5-bisphosphate-dependent membrane recruitment and oligomerization (14, 15), membrane insertion (16, 17), and extracellular trapping mediated by cell surface heparan sulfate proteoglycans (18, 19). This process is regulated by Tec kinase (21, 37) that itself is recruited to the inner leaflet through the phosphoinositide phosphatidylinositol 3,4,5-trisphosphate. This, in turn, activates the enzymatic activity of Tec through the interaction with plasma membrane-resident upstream and downstream effectors (38, 39). Tec kinase phosphorylates FGF2 at tyrosine 81, a modification that stimulates membrane-insertion of FGF2 oligomers (16, 17). The basic principle of membrane-inserted FGF2 oligomers as translocation intermediates in FGF2 secretion is also reflected by observations demonstrating a requirement for proper folding of FGF2 to qualify for membrane translocation (20, 23, 24, 40). Beyond the phosphoinositides phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate as well as Tec kinase at the inner leaflet and heparan sulfates at the outer leaflet, ATP1A1 is the fifth factor being identified as part of the secretory machinery mediating unconventional secretion of FGF2. Thus, all known molecular components of this pathway are physically associated with the plasma membrane. As illustrated in Fig. 9, we propose ATP1A1 to be part of a chain of sequential interactions at the inner leaflet resulting in FGF2 oligomerization and membrane insertion. Intriguingly, unassembled α-chains have been proposed to exist in plasma membranes based on significantly faster turnover rates of β- versus α-chains (26). It is therefore an interesting working hypothesis that unassembled α1-chains (ATP1A1) play a role in unconventional secretion of FGF2 that is unrelated to their participation in the α2β2 hetero-oligomeric complex that makes up a functional Na/K-ATPase.
FIGURE 9.
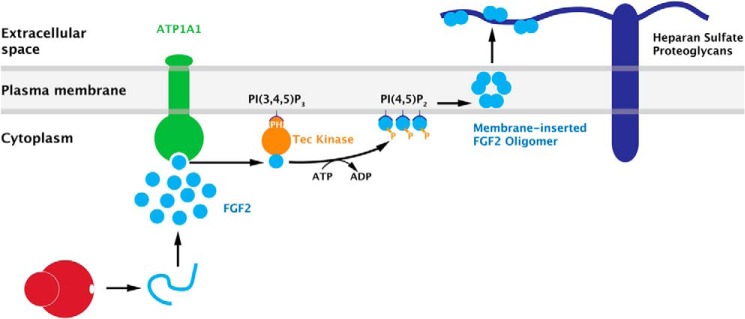
A current model of FGF2 membrane translocation with ATP1A1 as a high affinity recruitment factor for FGF2 at the inner leaflet of plasma membranes. For details, see “Discussion.”
Acknowledgment
We thank Julia P. Steringer (Heidelberg University Biochemistry Center) for reagents used in this study.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grants SFB 638, SFB/TRR 83, and GRK1188; the AID-NET program of the Federal Ministry for Education and Research of Germany (to W. N.); the Boehringer Ingelheim Fonds (to S. Z.); and the DFG cluster of excellence CellNetworks.
- ER
- endoplasmic reticulum
- PH
- pleckstrin homology.
REFERENCES
- 1. Rothman J. E. (1994) Mechanisms of intracellular protein transport. Nature 372, 55–63 [DOI] [PubMed] [Google Scholar]
- 2. Rothman J. E., Wieland F. T. (1996) Protein sorting by transport vesicles. Science 272, 227–234 [DOI] [PubMed] [Google Scholar]
- 3. Schekman R., Orci L. (1996) Coat proteins and vesicle budding. Science 271, 1526–1533 [DOI] [PubMed] [Google Scholar]
- 4. Rapoport T. A. (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 [DOI] [PubMed] [Google Scholar]
- 5. Hegde R. S., Ploegh H. L. (2010) Quality and quantity control at the endoplasmic reticulum. Curr. Opin. Cell Biol. 22, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sitia R., Braakman I. (2003) Quality control in the endoplasmic reticulum protein factory. Nature 426, 891–894 [DOI] [PubMed] [Google Scholar]
- 7. Rabouille C., Malhotra V., Nickel W. (2012) Diversity in unconventional protein secretion. J. Cell Sci. 125, 5251–5255 [DOI] [PubMed] [Google Scholar]
- 8. Nickel W. (2003) The mystery of nonclassical protein secretion: a current view on cargo proteins and potential export routes. Eur. J. Biochem. 270, 2109–2119 [DOI] [PubMed] [Google Scholar]
- 9. Prudovsky I., Tarantini F., Landriscina M., Neivandt D., Soldi R., Kirov A., Small D., Kathir K. M., Rajalingam D., Kumar T. K. (2008) Secretion without Golgi. J. Cell Biochem. 103, 1327–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dinarello C. A. (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 [DOI] [PubMed] [Google Scholar]
- 11. Presta M., Dell'Era P., Mitola S., Moroni E., Ronca R., Rusnati M. (2005) Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 16, 159–178 [DOI] [PubMed] [Google Scholar]
- 12. Nickel W. (2005) Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic 6, 607–614 [DOI] [PubMed] [Google Scholar]
- 13. Schäfer T., Zentgraf H., Zehe C., Brügger B., Bernhagen J., Nickel W. (2004) Unconventional secretion of fibroblast growth factor 2 is mediated by direct translocation across the plasma membrane of mammalian cells. J. Biol. Chem. 279, 6244–6251 [DOI] [PubMed] [Google Scholar]
- 14. Temmerman K., Ebert A. D., Müller H. M., Sinning I., Tews I., Nickel W. (2008) A direct role for phosphatidylinositol-4,5-bisphosphate in unconventional secretion of fibroblast growth factor 2. Traffic 9, 1204–1217 [DOI] [PubMed] [Google Scholar]
- 15. Temmerman K., Nickel W. (2009) A novel flow cytometric assay to quantify interactions between proteins and membrane lipids. J. Lipid Res. 50, 1245–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nickel W. (2011) The unconventional secretory machinery of fibroblast growth factor 2. Traffic 12, 799–805 [DOI] [PubMed] [Google Scholar]
- 17. Steringer J. P., Bleicken S., Andreas H., Zacherl S., Laussmann M., Temmerman K., Contreras F. X., Bharat T. A., Lechner J., Müller H. M., Briggs J. A., García-Sáez A. J., Nickel W. (2012) Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J. Biol. Chem. 287, 27659–27669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zehe C., Engling A., Wegehingel S., Schäfer T., Nickel W. (2006) Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc. Natl. Acad. Sci. U.S.A. 103, 15479–15484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nickel W. (2007) Unconventional secretion: an extracellular trap for export of fibroblast growth factor 2. J. Cell Sci. 120, 2295–2299 [DOI] [PubMed] [Google Scholar]
- 20. Nickel W., Seedorf M. (2008) Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annu. Rev. Cell Dev. Biol. 24, 287–308 [DOI] [PubMed] [Google Scholar]
- 21. Ebert A. D., Laussmann M., Wegehingel S., Kaderali L., Erfle H., Reichert J., Lechner J., Beer H. D., Pepperkok R., Nickel W. (2010) Tec-kinase-mediated phosphorylation of fibroblast growth factor 2 is essential for unconventional secretion. Traffic 11, 813–826 [DOI] [PubMed] [Google Scholar]
- 22. Steringer J. P., Muller H. M., Nickel W. (2014) Unconventional secretion of fibroblast growth factor 2: a novel type of protein translocation across membranes? J. Mol. Biol. 10.1016/j.jmb.2014.07.012 [DOI] [PubMed] [Google Scholar]
- 23. Backhaus R., Zehe C., Wegehingel S., Kehlenbach A., Schwappach B., Nickel W. (2004) Unconventional protein secretion: membrane translocation of FGF-2 does not require protein unfolding. J. Cell Sci. 117, 1727–1736 [DOI] [PubMed] [Google Scholar]
- 24. Torrado L. C., Temmerman K., Müller H. M., Mayer M. P., Seelenmeyer C., Backhaus R., Nickel W. (2009) An intrinsic quality-control mechanism ensures unconventional secretion of fibroblast growth factor 2 in a folded conformation. J. Cell Sci. 122, 3322–3329 [DOI] [PubMed] [Google Scholar]
- 25. Kaplan J. H. (2002) Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535 [DOI] [PubMed] [Google Scholar]
- 26. Yoshimura S. H., Iwasaka S., Schwarz W., Takeyasu K. (2008) Fast degradation of the auxiliary subunit of Na+/K+-ATPase in the plasma membrane of HeLa cells. J. Cell Sci. 121, 2159–2168 [DOI] [PubMed] [Google Scholar]
- 27. Dahl J. P., Binda A., Canfield V. A., Levenson R. (2000) Participation of Na,K-ATPase in FGF-2 secretion: rescue of ouabain-inhibitable FGF-2 secretion by ouabain-resistant Na,K-ATPase α subunits. Biochemistry 39, 14877–14883 [DOI] [PubMed] [Google Scholar]
- 28. Florkiewicz R. Z., Anchin J., Baird A. (1998) The inhibition of fibroblast growth factor-2 export by cardenolides implies a novel function for the catalytic subunit of Na+,K+-ATPase. J. Biol. Chem. 273, 544–551 [DOI] [PubMed] [Google Scholar]
- 29. Erfle H., Neumann B., Rogers P., Bulkescher J., Ellenberg J., Pepperkok R. (2008) Work flow for multiplexing siRNA assays by solid-phase reverse transfection in multiwell plates. J. Biomol. Screen. 13, 575–580 [DOI] [PubMed] [Google Scholar]
- 30. Engling A., Backhaus R., Stegmayer C., Zehe C., Seelenmeyer C., Kehlenbach A., Schwappach B., Wegehingel S., Nickel W. (2002) Biosynthetic FGF-2 is targeted to non-lipid raft microdomains following translocation to the extracellular surface of CHO cells. J. Cell Sci. 115, 3619–3631 [DOI] [PubMed] [Google Scholar]
- 31. Stegmayer C., Kehlenbach A., Tournaviti S., Wegehingel S., Zehe C., Denny P., Smith D. F., Schwappach B., Nickel W. (2005) Direct transport across the plasma membrane of mammalian cells of Leishmania HASPB as revealed by a CHO export mutant. J. Cell Sci. 118, 517–527 [DOI] [PubMed] [Google Scholar]
- 32. Aftab O., Nazir M., Fryknäs M., Hammerling U., Larsson R., Gustafsson M. G. (2014) Label free high throughput screening for apoptosis inducing chemicals using time-lapse microscopy signal processing. Apoptosis 19, 1411–1418 [DOI] [PubMed] [Google Scholar]
- 33. Miller M. K., Bang M. L., Witt C. C., Labeit D., Trombitas C., Watanabe K., Granzier H., McElhinny A. S., Gregorio C. C., Labeit S. (2003) The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J. Mol. Biol. 333, 951–964 [DOI] [PubMed] [Google Scholar]
- 34. Taouji S., Dahan S., Bossé R., Chevet E. (2009) Current screens based on the AlphaScreen technology for deciphering cell signalling pathways. Curr. Genomics 10, 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lazar G. A., Dang W., Karki S., Vafa O., Peng J. S., Hyun L., Chan C., Chung H. S., Eivazi A., Yoder S. C., Vielmetter J., Carmichael D. F., Hayes R. J., Dahiyat B. I. (2006) Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A. 103, 4005–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lingrel J. B., Kuntzweiler T. (1994) Na+,K+-ATPase. J. Biol. Chem. 269, 19659–19662 [PubMed] [Google Scholar]
- 37. Nickel W. (2010) Pathways of unconventional protein secretion. Curr. Opin. Biotechnol. 21, 621–626 [DOI] [PubMed] [Google Scholar]
- 38. Mano H. (1999) Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 10, 267–280 [DOI] [PubMed] [Google Scholar]
- 39. Schaeffer E. M., Schwartzberg P. L. (2000) Tec family kinases in lymphocyte signaling and function. Curr. Opin. Immunol. 12, 282–288 [DOI] [PubMed] [Google Scholar]
- 40. Nickel W., Rabouille C. (2009) Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 10, 148–155 [DOI] [PubMed] [Google Scholar]