

Background: PPARγ tends to adopt an active conformation that cannot be directly bound by NCoR.
Results: GPS2 binds to active PPARγ, facilitates its repression by NCoR, and is required for the optimal NCoR corepressor function for PPARγ.
Conclusion: GPS2 mediates a novel NCoR repression pathway targeting active PPARγ.
Significance: The GPS2-dependent pathway provides new insights into how NCoR regulates PPARγ function in vivo.
Keywords: Conformational Change, Nuclear Receptor, Peroxisome Proliferator-Activated Receptor (PPAR), Thyroid Hormone, Transcription Coactivator, Transcription Corepressor, GPS2, NCoR, PPARγ, Transcriptional Repression
Abstract
Repression of peroxisome proliferator-activated receptor γ (PPARγ)-dependent transcription by the nuclear receptor corepressor (NCoR) is important for homeostatic expression of PPARγ target genes in vivo. The current model states that NCoR-mediated repression requires its direct interaction with PPARγ in the repressive conformation. Previous studies, however, have shown that DNA-bound PPARγ is incompatible with a direct, high-affinity association with NCoR because of the inherent ability of PPARγ to adopt the active conformation. Here we show that NCoR acquires the ability to repress active PPARγ-mediated transcription via G protein pathway suppressor 2 (GPS2), a component of the NCoR corepressor complex. Unlike NCoR, GPS2 can recognize and bind the active state of PPARγ. In GPS2-deficient mouse embryonic fibroblast cells, loss of GPS2 markedly reduces the corepressor function of NCoR for PPARγ, leading to constitutive activation of PPARγ target genes and spontaneous adipogenesis of the cells. GPS2, however, is dispensable for repression mediated by unliganded thyroid hormone receptor α or a PPARγ mutant unable to adopt the active conformation. This study shows that GPS2, although dispensable for the intrinsic repression function of NCoR, can mediate a novel corepressor repression pathway that allows NCoR to directly repress active PPARγ-mediated transcription, which is important for the optimal corepressor function of NCoR for PPARγ. Interestingly, GPS2-dependent repression specifically targets PPARγ but not PPARα or PPARδ. Therefore, GPS2 may serve as a unique target to manipulate PPARγ signaling in diseases.
Introduction
Nuclear receptors (NRs),3 which comprise the largest superfamily of ligand-inducible transcription factors, play important roles in homeostasis, metabolism, and development. NRs are receptors of various natural and synthetic lipophilic small molecules that can freely enter cells and bind to NRs via their ligand-binding domains (LBDs). Ligand binding induces a conformational change of NRs and alters their ability to recruit corepressors (CoRs) and coactivators (CoAs), leading to transcriptional repression or activation of target genes (1–4). On the basis of sequence homology, NRs can be classified into different categories. One category includes thyroid hormone receptors (TRα and TRβ) and peroxisome proliferator-activated receptors (PPARα, γ, and δ), which share the ability to form heterodimers with the retinoid X receptor. TRs are receptors for thyroid hormone (T3) and play important roles in development and metabolism. PPARs bind to fatty acids and various other ligands. Among the three PPARs, PPARα and PPARγ have been studied extensively. Although both PPARα and PPARγ regulate lipid metabolism and have anti-inflammation functions, PPARα is mainly involved in lipid utilization, whereas PPARγ is associated with lipid storage, adipogenesis, and insulin signaling. In addition, PPARγ serves as the pharmacological target of thiazolidinedione (TZD) antidiabetic drugs.
NRs can adopt two conformational states. Whereas the repressive state binds to CoRs, the active state binds to CoAs. Although it is generally true that unliganded NRs (apo-NRs) exist in the repressive conformation and liganded NRs exist in the active conformation (5), the ligand dependency for the active conformation may vary depending on the specific NRs. For example, the ability of TRα to adopt the active state is strictly dependent on binding to T3. Accordingly, apo-TRα is a constitutive repressor. On the other hand, structural studies have shown that apo-PPARγ can adopt the same active conformation as the agonist-bound PPARγ-CoA complex (6, 7). The intrinsic ability of PPARγ to assume the active state is also supported by its ability to drive activation in the absence of ligands and the reported conformational flexibility of the activation function 2 (AF2) domain (7–9).
The abilities of NRs to adopt the mutually exclusive active and repressive states are governed by the conformation of AF2, a conserved amphipathic sequence located at the C terminus of NRs. Earlier studies have mapped AF2 as a region required for the ability of NRs to activate the transcription of target genes (10). In the active state of NRs, AF2 adopts a conformation that allows it to directly contact CoAs. AF2 has a distinct conformation in the repressive NR state, which allows NRs to bind to CoRs but not CoAs. It has been shown that DNA-bound apo-PPARγ/retinoid X receptor heterodimer cannot directly recruit NCoR or SMRT (11–14). In vivo, the binding of CoAs may further reduce the ability of PPARγ to recruit CoRs. Underscoring the importance of AF2 in regulating PPARγ interactions with CoRs and CoAs, deleting AF2 or mutating its residues involved in CoA interactions allows PPARγ to recruit CoRs and to function as a repressor like unliganded TRα (11, 12, 15).
CoRs and CoAs discriminate repressive and active conformations of NRs via corepressor-nuclear receptor (CoRNR) and NR boxes present in CoRs and CoAs, respectively (16–19). Two CoR proteins, nuclear receptor corepressor (NCoR) (20) and silencing mediator for retinoid and thyroid hormone receptors (SMRT) (21) are present in mammals. These proteins have similar domain structures. In addition to the CoRNR boxes at the C terminus, CoRs also contain two SWI3/ADA2/NCoR/TFIIIB-like domains and three repression domains (RD1, RD2, and RD3) located at the N terminus (Fig. 2A). Both NCoR and SMRT form complexes with histone deacetylase 3 (HDAC3), transducin β-like protein 1 (TBL1)/TBL1-related protein 1 (TBLR1), and G protein pathway suppressor 2 (GPS2) (22–25). HDAC3 interacts with the N-terminal SWI3/ADA2/NCoR/TFIIIB-like domain along with a short upstream sequence. TBL1/TBLR1 and GPS2 simultaneously bind to the RD1 domain to form a heterotrimeric structure (Fig. 2A). Previous studies have shown that both HDAC3 and TBL1/TBLR1 play important roles in mediating downstream repression steps of CoRs (24, 26–29). GPS2 was initially discovered as a protein that can suppress G protein-mediated signal transduction pathways (30, 31). Although numerous studies have shown that GPS2 is an integral corepressor complex component (22, 25, 32), whether it plays a similar role in CoR-mediated repression is largely unknown.
FIGURE 2.

Loss of GPS2 derepresses PPARγ-dependent transcription. A, schematic of NCoR domains. RD1 interacts with GPS2. CoRNR boxes bind to NRs in the unliganded, repressive conformation. B, luciferase activities normalized to Gal4-DBD and assayed in GPS2-WT and GPS2-KO MEFs transfected with Gal4-RD1, Gal4-TRα (LBD), or the empty vector Gal4-DBD, along with a Gal4-UAS-driven luciferase reporter construct. A fold change of >1 denotes activation, whereas a fold change of <1 denotes repression. Inset, Western blot analysis of GPS2 expression in MEFs derived from GPS2-WT and GPS2-KO embryos. C, fold activation relative to Gal4-DBD assayed in GPS2-WT and GPS2-KO MEFs transfected with Gal4-PPARγ (LBD) or Gal4-DBD, along with a Gal4-UAS-driven luciferase reporter construct in the absence and presence of the PPARγ ligand rosiglitazone (Rosi.). *, p < 0.05; **, p < 0.01. D, similar to C, with inclusion of Gal4-PPARγQ286P (LBD). *, p < 0.05; **, p < 0.01. E, similar to C, except that Gal4-PPARγΔAF2 (LBD) was used in place of WT PPARγ. n.s., not significant. F, mammalian two-hybrid assays to measure the in vivo interactions between the NCoR CoRNR box region and PPARγ (LBD) or PPARγΔAF2 (LBD). Interactions derived from PPARγΔAF2 were set at 100%. G, GPS2 specifically inhibited PPARγ but not other PPAR isoforms. The experiments were similar to C, with inclusion of PPARα and PPARδ isoforms. **, p < 0.01. n.s., not significant.
Despite the inability of DNA-bound PPARγ to mediate a strong interaction with CoRNR boxes (11, 12), functional studies have shown that CoRs, in particular, NCoR, can repress PPARγ-dependent transcription in vivo (33–36). In this study, we generated GPS2 KO mice. These mice are embryonically lethal. Using GPS2 KO mouse-derived embryonic fibroblast cells (MEFs), we further studied the role of GPS2 in NCoR- and NR-mediated repression. Our results reveal a novel GPS2-dependent mechanism for PPARγ repression by NCoR. We show that this repression pathway, which targets the active conformation of PPARγ, plays an important role in maintaining NCoR-mediated repression of PPARγ. Loss of GPS2 is sufficient to cause constitutive activation of endogenous PPARγ/NCoR target genes in vivo and to predispose cells to adipogenesis in the absence of ectopic PPARγ. Our results extend the view that NCoR is only capable of regulating NRs in its repressive state and show that the GPS2-enabled strategy to repress active PPARγ is an important gatekeeper mechanism to ensure proper repression of PPARγ target genes, consistent with the susceptibility of PPARγ to adopt the active conformation. Our results also show that the GPS2-mediated regulation is receptor type-, isoform-, and conformation-specific. Loss of GPS2 does not affect the repression mediated by apo-TRα or by an AF2-deleted PPARγ mutant. Neither does it activate PPARα or PPARδ. In addition, given the early lethality of GPS2 KO mice, our work also adds GPS2 to the list of CoR-HDAC3 complex subunits required for embryonic development in mice.
EXPERIMENTAL PROCEDURES
Generation of Global GPS2 KO Mice and MEFs
The targeting vector contained a phosphoglycerate kinase (PGK) promoter-driven neomycin cassette (neo), along with the left and right arms amplified from genomic DNA of 129Sv-derived mouse cells by high-fidelity PCR (Fig. 1A). The left arm contained the GPS2 upstream region, the GPS2 promoter, part of exon 1 lacking the ATG and 3′ section, and a GFP cassette. The right arm contained GPS2 exons 3–10 and 3′ downstream sequences. DNA sequencing confirmed that no mutation existed in either arm. The targeting vector was electroporated into mouse ES cells to generate GPS2+/− ES cell clones via homologous recombination. Upon confirmation by Southern blot analysis (Fig. 1B), two independent ES cell clones were injected into blastocysts to generate chimeric mice from which two independent germ line-transmitted mouse lines were obtained. Both lines showed identical phenotypes.
FIGURE 1.
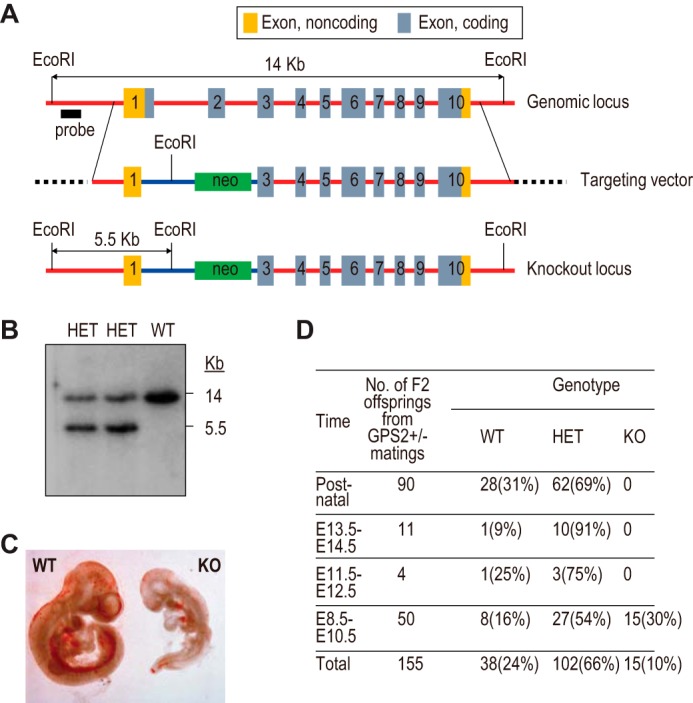
Embryonic lethality of whole-body GPS2 knockout mice. A, schematic of WT and KO GPS2 genomic loci and the design of the targeting vector to generate the whole-body GPS2 knockout mice. B, Southern blot analysis of WT and two independent heterozygous (HET) ES cell colonies. C, microscopic view of WT and KO embryos at E9.5. D, genotyping results of GPS2 WT, heterozygous, and KO mice at various embryonic developmental stages.
To generate MEFs, mouse embryos were isolated under a microscope. After washing once with PBS, the embryos were minced by pipetting up and down 5 times in 50 μl of trypsin in 24-well plates. The embryos were incubated at 37 °C for 30 min and then resuspended in 500 μl of complete DMEM (see below) by pipetting up and down for 20 times using a 1-ml pipette. The cells were passaged every 3 days until they became immortalized (∼3 months).
Adipogenesis
Self-immortalized MEFs were maintained in complete DMEM (DMEM containing 10% FBS) and passaged regularly to prevent overconfluency. For adipogenesis, the cells were allowed to grow to confluency. Two days later (defined as day 0), postconfluent MEFs were treated with rosiglitazone or vehicle. Oil Red O staining was performed on day 14 (37).
GST Pulldown Assays
GST pulldown assays were performed as described previously (38). Briefly, GST fusion proteins were expressed in bacteria and coupled to glutathione-Sepharose beads (GE Healthcare Life Sciences). PPARγ and its mutants were translated and labeled with 35S in vitro using the TnT® coupled reticulocyte lysate system (Promega). A BC200/0.1% Nonidet P-40 buffer containing rosiglitazone (2 μm, Cayman Chemical) or vehicle was used for incubation and washing. CoRNR box peptide (PASNLGLEDIIRKALMGSFD) was dissolved in DMSO (16).
Transfection and Reporter/Mammalian Two-hybrid Assays
These assays were performed as described previously (38). MEFs were transfected with plasmids using Turbofect in vitro transfection reagent (Fisher Scientific). The amounts of transfected plasmids were as follows: full-length PPARγ, 10 ng; Gal4 DNA-binding domain (Gal4-DBD)-derived plasmids, 20 ng; Gal4-upstream activating sequence (Gal4-UAS)-driven luciferase and natural PPARγ response element-driven reporters, 25 ng; GPS2, 50 ng; and NCoR, 40, 100, and 250 ng. Six hours post-transfection, the growth medium was replaced by medium containing hormone-stripped FBS. The next morning, the medium was changed to fresh medium with hormone-stripped FBS containing T3 (100 nm, Sigma), rosiglitazone (1 μm), or vehicles. Luciferase assays were performed 24 h later.
Coimmunoprecipitation Assays
Coimmunoprecipitation assays were performed as described previously (38). 293T cells were transfected with the desired plasmids using Turbofect in vitro transfection reagent. Rosiglitazone (1 μm) was added to the transfected cells the next morning. On the third morning, the cells were lysed in lysis buffer (20 mm Tris (pH 7.9), 1 mm EDTA, 20% glycerol, 1 μm rosiglitazone, 1× protease inhibitor cocktail (Roche), 180 mm NaCl, and 0.5% Nonidet P-40). Cell extracts were incubated with anti-FLAG M2 affinity gel (Sigma) at 4 °C for 3 h, followed by extensive washing with lysis buffer. The same lysis buffer was used for GST-GPS2 and derivatives to pull down endogenous PPARγ from MEFs treated with rosiglitazone (1 μm) for 5 h before lysis of the cells.
Quantitative RT-PCR (RT-qPCR) and RNA Sequencing (RNA-Seq)
RT-qPCR experiments were performed as described previously (39). The primers used in this study are shown below. 18 S rRNA was used as an internal control. RNA-Seq analysis was performed in the DNA Sequencing and Genotyping Core at the Cincinnati Children's Hospital Medical Center. Differential gene expression of GPS2-KO, WT, and GPS2-re-expressed KO cells was analyzed using the DESeq module included in the GeneSpring NGS software (Agilent). The following is a list of primers used for RT-qPCR in MEFs (all sequences are from 5′ to 3′): Fzd1, GTGCTCACGTACCTAGTGGACA and TCCTCCAACAGAAAGCCAGCGA; Socs1, AGTCGCCAACGGAACTGCTTCT and GTAGTGCTCCAGCAGCTCGAAA; Sgk1, AACAGAGAAGGATGGGCCTGAAC and GTTCATAAGCTCCGGCTCCTGAG; Trerf1, AGATGCCTGTGCTCGTGAGGAT and AACTTTGGCGGCGATAGGTGGA; Abca1, GGAGCCTTTGTGGAACTCTTCC and CGCTCTCTTCAGCCACTTTGAG; Idh1, CAGGCTCATAGATGACATGGTGG and CACTGGTCATCATGCCAAGGGA; Adipor2, TCTTCCACACGGTGTACTGCCA and GGTAGATGAAGCAAGGTTGTGGG; adiponectin, AGATGGCACTCCTGGAGAGAAG and ACATAAGCGGCTTCTCCAGGCT; aP2, AACACCGAGATTTCCTT and ACACATTCCACCACCAG; total PPARγ, AGGCCGAGAAGGAGAAGCTGTTG and TGGCCACCTCTTTGCTCTGCTC; PPARγ1, CTGTGAGACCAACAGCCTGACG and AATGTCCTGAATATCAGTGGTTC; and PPARγ2, GAGATTCTCCTGTTGACCCAGAG and AGAGCTGATTCCGAAGTTGGTGG.
Plasmids, Chemicals, and Antibodies for Western Blot Analysis
Mammalian and in vitro expression vectors for PPARγ, PPARα, PPARδ, NCoR, GPS2, and their derivatives have been described previously (9, 22, 40) or were generated by PCR and cloning techniques. The SMRT construct was kindly provided by Dr. Mitchell Lazar (University of Pennsylvania). PPARγ response element- and Gal4-UAS-driven reporters were made by inserting the PPARγ and Gal4 response elements into the polylinker of the pGL2-SV40 plasmid (Promega). Polyclonal rabbit anti-GPS2 antiserum was raised against the C-terminal 300–327 peptide. Anti-Gal4 antibody was obtained from Santa Cruz Biotechnology (catalog no. sc-577). Anti-actin antibody was obtained from Millipore (catalog no. MAB1501). Anti-FLAG antibody was obtained from Sigma. Anti-NCoR antibody was obtained from Thermo Scientific. Anti-PPARγ antibody was obtained from Santa Cruz Biotechnology (catalog no. sc-7196).
Statistical Analysis
Unless otherwise indicated, a two-tailed Student's t test was performed to reveal the significance, as indicated by p values (*, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant, p > 0.05).
RESULTS
Loss of GPS2 Does Not Reduce TRα- or RD1-mediated Repression
To understand the biological function of GPS2, we generated whole-body GPS2 KO mice (Fig. 1). The knockout removed part of exon 1 and the entire exon 2, which encode the ATG start codon and an N-terminal NCoR-interacting region of GPS2. GPS2 KO mice died prenatally around embryonic day 10 (Fig. 1, C and D). A similar embryonic lethality has been reported for the knockout mice of NCoR, SMRT, and HDAC3 (41–44). These results are consistent with an important role of GPS2 as a CoR complex subunit in development.
To study GPS2 function at the cellular level, MEFs from sibling GPS2 KO and WT embryos were isolated and immortalized by continuous passage. Western blot analysis confirmed the lack of expression of GPS2 in KO cells (Fig. 2B, inset). Next we performed Gal4-based reporter assays to test whether GPS2 plays a role in TRα- and NCoR-RD1-dependent transcriptional repression. Both Gal4-TRα and Gal4-RD1 strongly repressed basal transcription in KO cells (Fig. 2B, black columns). The potency of repression was not reduced in KO cells compared with WT cells (Fig. 2B). Because Gal4-TRα did not show a higher level of T3-induced activation in KO cells (Fig. 2B), the strong repression observed in KO cells was probably not caused by higher protein expression in these cells.
PPARγ-dependent Transcription Is Derepressed in GPS2 KO Cells
Because NCoR can repress PPARγ-dependent transcription and because GPS2 binds to NCoR, we asked whether GPS2 plays a role in repressing PPARγ-dependent transcription. Compared with WT cells, the activity of PPARγ observed in KO cells was increased significantly both in the absence and presence of the exogenous ligand rosiglitazone (Fig. 2C). The increase was more dramatic in the absence (3-fold) than in the presence (2-fold) of rosiglitazone, possibly reflecting reduced binding of corepressors in the presence of rosiglitazone.
We excluded the possibility that the increased activity of PPARγ observed in KO cells was due to its differential binding to ligands in these cells (Fig. 2D). PPARγQ286P is a PPARγ mutant defective in binding to ligands (45, 46). Consistent with its inability to bind to ligands, PPARγQ286P failed to activate transcription in WT cells both in the absence and presence of rosiglitazone (Fig. 2D, lanes 2 and 4, white columns). In KO cells, WT PPARγ showed increased activities as observed earlier (Fig. 2D, lanes 1 and 3). Unlike what was observed in WT cells, PPARγQ286P was still able to activate transcription in KO cells, and the fold increase of its activities in KO versus WT cells was similar to that observed with WT PPARγ in the absence of rosiglitazone (Fig. 2D, lanes 2 and 4). These results suggest that GPS2 plays a role in suppressing the constitutive activity of PPARγ that may arise from its ability to adopt the active conformation.
GPS2-dependent Repression Targets the Active Conformation of PPARγ
We next sought to define the conformational state(s) of PPARγ susceptible to GPS2-dependent regulation. Because PPARγ is under equilibrium between repressive and active conformations, loss of GPS2 could possibly increase the activity of PPARγ as a result of derepression of the repressive PPARγ, shifting from the repressive to the active conformation of PPARγ, or failure to repress active PPARγ. Because deleting AF2 will block PPARγ from adopting the active conformation, we first asked whether the ability of PPARγΔAF2 to repress transcription was affected in GPS2 KO cells. Deleting AF2 allowed PPARγΔAF2 to repress basal transcription in WT cells, as expected (Fig. 2E, white columns, lanes 3 and 4 versus lanes 1 and 2). Compared with WT cells, no significant reduction in the ability of PPARγΔAF2 to repress transcription was observed in KO cells (Fig. 2E, black columns, lanes 3 and 4 versus lanes 1 and 2). This result is consistent with the lack of effect of GPS2 KO on TRα-mediated repression, as shown earlier, and indicates that the classical CoRNR box-dependent repression pathway was intact in GPS2 KO cells.
We next asked whether loss of GPS2 would tip the balance toward the active PPARγ conformation. This was tested by mammalian two-hybrid assays to measure the in vivo interaction between PPARγ and an NCoR C-terminal fragment containing all three CoRNR boxes. PPARγΔAF2, which showed comparable repression in WT and KO cells, was used as a control to set the maximal level (100%) of interaction in both cell types (Fig. 2F). As expected, PPARγΔAF2 showed a stronger interaction than WT PPARγ in both cell types (Fig. 2F, white versus black columns). Compared with GPS2-WT cells, the GPS2 KO cells did not show a reduced interaction between WT PPARγ and the NCoR CoRNR boxes (Fig. 2F, cf. white columns). Together, these results showed that loss of GPS2 did not affect either the ability of the repressive PPARγ to repress transcription or the relative abundance of the repressive PPARγ that can directly recruit NCoR, therefore providing support for the idea that GPS2 facilitates the repression of the active form of PPARγ and that loss of its function in this context accounts for the increased PPARγ activity in GPS2 KO cells.
We also examined the effects of GPS2 loss on other PPAR isoforms. As shown in Fig. 2G, loss of GPS2 specifically increased PPARγ-dependent transcription and may slightly repress PPARα- and PPARδ-dependent transcription. This reveals that GPS2-mediated regulation is PPAR isoform-specific.
GPS2 Is Required for NCoR to Repress Active PPARγ
Ectopic expression of GPS2 reduced PPARγ activity in GPS2 KO but not WT cells (Fig. 3A). Interestingly, a weak stimulatory effect was observed in WT cells (see also Fig. 3B). These results indicate that the ability of GPS2 to repress PPARγ should not result from an autonomous function of GPS2. Because a majority of ectopic GPS2 may exist in the free form, we asked whether increasing its complex formation with NCoR would allow GPS2 to manifest its inhibitory function on PPARγ in WT cells. Indeed, although GPS2 alone failed to repress PPARγ, as shown above, it enhanced the ability of ectopic NCoR to repress PPARγ (Fig. 3B, GPS2-WT). A possible explanation for the weak stimulatory effect of GPS2 alone in WT cells may be that the free GPS2 can exert a dominant negative activity against the endogenous GPS2-NCoR complex.
FIGURE 3.

GPS2 is important for PPARγ repression by NCoR. A, ectopic GPS2 restored PPARγ repression in GPS2-KO cells. Luciferase assays were performed in GPS2-WT and GPS-KO MEFs transfected with Gal4-PPARγ or empty vector, along with the Gal4-UAS reporter, in the absence or presence of GPS2. B, similar to A. GPS2-KO and WT MEFs were transfected with NCoR, GPS2, or both, as indicated. *, p < 0.05; n.s., not significant. C and D, NCoR showed defective corepressor function for PPARγ, but not TRα, in GPS2 KO MEFs. Luciferase assays were performed in GPS2 WT and GPS2 KO MEFs transfected with Gal4-PPARγ or Gal4-TRα, different doses of NCoR, or empty vector, along with the Gal4-UAS reporter. The left panels show basal levels of TRα-mediated repression (C) or PPARγ-mediated activation (D). Both were normalized to Gal4-DBD. The right panels show NCoR-elicited potentiation of TRα-dependent repression (C) or inhibition of PPARγ-dependent activation (D). **, p < 0.01. E, fold inhibition by NCoR of PPARγ transcriptional activity in GPS2 WT and GPS2 KO cells in the absence or presence of rosiglitazone (Rosi.). F, fold inhibition of PPARγ by NCoR or SMRT in GPS2 WT and GPS2 KO cells. 250 ng of NCoR and SMRT plasmids was used. The experiment was performed similar to D. ***, p < 0.001.
We also confirmed the cooperative function of GPS2 and NCoR in repressing PPARγ in GPS2 KO cells. In these cells, although both GPS2 and NCoR alone were able to modestly repress PPARγ-dependent transcription, cotransfection of GPS2 and NCoR produced a stronger repression comparable with that observed in cotransfected WT cells (Fig. 3B).
Given that TRα-mediated repression was not affected by GPS2 KO, we next directly compared the ability of NCoR to repress PPARγ and TRα in GPS2 KO and WT cells. As expected, in both cell types, TRα showed similar levels of repression, and NCoR similarly potentiated TRα-dependent repression in a dose-dependent manner (Fig. 3C). In contrast, and consistent with a requirement of GPS2 for the optimal corepressor function of NCoR for PPARγ, a dramatic reduction in the ability of NCoR to mediate dose-dependent repression of PPARγ was observed in KO cells (Fig. 3D, right). This occurred despite the high level of PPARγ activity in KO cells (Fig. 3D, left), further showing that GPS2 is an important rate-limiting factor for NCoR-mediated PPARγ repression.
In KO cells, NCoR was still able to manifest a weak repression on PPARγ (Fig. 3, B and D). Because TRα- and PPARγΔAF2-mediated repression was unaffected in KO cells, we hypothesized that the residual corepressor function of NCoR may reflect its ability to act on a subset of PPARγ in the repressive conformation. To test this, cells were treated with rosiglitazone (1 μm) to saturate its binding to PPARγ. Confirming the hypothesis, rosiglitazone completely abolished the ability of NCoR to repress PPARγ in KO cells (Fig. 3E, lanes 2 and 4). In WT cells, rosiglitazone reduced but did not abolish NCoR-mediated repression (Fig. 3E, lanes 1 and 3). These results demonstrate that NCoR is capable of repressing active PPARγ-mediated transcription via a GPS2-dependent mechanism.
Given the similarity between NCoR and SMRT, we also examined the effect of GPS2 KO on the ability of SMRT to repress PPARγ. SMRT repressed PPARγ-dependent transcription, as expected (Fig. 3F). The repression was similarly reduced in GPS2-KO cells, as observed with NCoR. These results suggest that the GPS2-dependent mechanism is conserved between NCoR and SMRT.
GPS2, but Not NCoR, Is Able to Bind PPARγ in the Liganded Conformation
Because CoRNR boxes cannot bind PPARγ in the active conformation, we hypothesized that GPS2 directly binds to the active PPARγ, thereby allowing its repression by NCoR. Consistent with this idea, GST pulldown assays revealed distinct abilities of NCoR and GPS2 to recognize the repressive and active conformations of PPARγ (Fig. 4A). Compared with GPS2, NCoR bound much more strongly to the repressive conformation of PPARγ (apo-PPARγ and PPARγΔAF2 with or without rosiglitazone). The addition of rosiglitazone essentially abolished NCoR interaction with PPARγ. Rosiglitazone, however, did not reduce, and may slightly increase, the binding of PPARγ to GPS2 (Fig. 4A, densitometry data not shown).
FIGURE 4.

Differential recognition of PPARγ conformations by GPS2 and NCoR. A, GST pulldown assays to detect the in vitro interactions of PPARγ and PPARγΔAF2 with GPS2, NCoR, and TIF2 in the absence or presence of rosiglitazone (Rosi.). B, three-dimensional X-crystallographic structure of PPARγ in complex with rosiglitazone and an NR box peptide from SRC-1 (PDB code 2PRG) (6). It shows that the conserved Thr-325, Lys-329, Leu-339, and Val-343 residues located in the hydrophobic cavity directly contact the NR box peptide. C, mutation of the conserved hydrophobic cavity residues disrupted the PPARγ-NCoR interaction without significantly affecting the PPARγ-GPS2 interaction. D, CoRNR box peptide (100 μm) strongly inhibited the PPARγ-NCoR interaction but not the PPARγ-GPS2 interaction. E and F, coimmunoprecipitation assays performed in 293T cells transfected with FLAG-GPS2 or FLAG-NCoR, along with Gal4-PPARγ (LBD). The cells were cultured in the presence of rosiglitazone. Following anti-FLAG immunoprecipitation (IP), coimmunoprecipitated proteins were detected by Western blot analysis. G, GST pulldown assays were performed using cell lysates from GPS2 WT and GPS2 KO cells pretreated with rosiglitazone (1 μm) for 5 h. The results for WT and KO cells are from the same Western blot analysis but are presented separately.
Unlike the TIF2 NR box, whose binding to PPARγ is strictly ligand-dependent (Fig. 4A), ligand binding is not required for GPS2 to interact with PPARγ. Because both GPS2 and NCoR were able to bind apo-PPARγ, we asked whether their interactions were mutually exclusive. Previous studies have mapped NR residues in the conserved hydrophobic cavity that directly contact CoRNR box and NR box motifs (16–19). In PPARγ, these residues include Thr-325, Lys-329, Leu-339, and Val-343 (Fig. 4B). We confirmed that mutations of T325R-K329A and L339R-V343R abolished the PPARγ/NCoR interaction, as expected. These mutations, however, only slightly affected the PPARγ-GPS2 interactions in the absence or presence of rosiglitazone (Fig. 4C). To test whether the CoRNR box and GPS2 may simultaneously bind to PPARγ, CoRNR box peptide was added to the reaction mixture. Consistent with the mutation results, although the peptide strongly inhibited the NCoR interaction with PPARγ, it did not significantly affect the GPS2 interaction (Fig. 4D). Together, these results suggest that GPS2 targets a separate region (or surface) in PPARγ that is distinct from the classic docking site for CoRNR and NR motifs.
We next confirmed that GPS2, but not NCoR, was able to bind the active form of PPARγ in vivo. 293T cells were transfected with PPARγ (as a fusion to Gal4-DBD) together with FLAG-GPS2 or FLAG-NCoR. Cells were treated with rosiglitazone to ensure that all PPARγ was in the liganded, active conformation. PPARγ was detected in immunoprecipitates derived from FLAG-GPS2 but not in immunoprecipitates derived from FLAG-NCoR (Fig. 4E). No interaction was observed between FLAG-GPS2 and Gal4-DBD (data not shown). Analysis of truncated GPS2 derivatives showed that both the N-terminal region (amino acids 1–155) and C-terminal region (amino acids 105–327) of GPS2 were required for GPS2-PPARγ interaction (Fig. 4F). Although the N-terminal fragment of GPS2 was able to bind endogenous NCoR, as expected (22), it did not associate with PPARγ. This confirmed that the endogenous NCoR, which was also present in FLAG-GPS2 immunoprecipitates, was not responsible for the observed PPARγ association with GPS2.
We also confirmed the ability of GPS2 to interact with endogenous PPARγ in the presence of rosiglitazone. GST-GPS2, but not GST alone, was able to pull down significant levels of endogenous PPARγ from both KO and WT MEF cells (Fig. 4G). Consistent with the coimmunoprecipitation results, the interaction required both N- and C-terminal regions of GPS2. Interestingly, GPS2 was more capable of pulling down PPARγ from KO than from WT cells, consistent with the notion that a subset of PPARγ in WT cells was in complex with endogenous GPS2 and, therefore, unavailable for interaction with GST-GPS2.
Loss of GPS2 Activates Endogenous PPARγ and NCoR Target Genes
Loss of GPS2 also increased the ability of full-length PPARγ to drive transcription from a natural PPARγ response element-dependent reporter both in the absence and presence of rosiglitazone (Fig. 5A). Notably, in vector-transfected cells, rosiglitazone treatment was sufficient to increase the reporter activity in KO but not WT cells (Fig. 5A). This result raised the possibility that subconfluent GPS2-KO cells expressed functional endogenous PPARγ whose activity was also increased as a result of GPS2 depletion. RT-qPCR showed that, compared with WT cells, KO cells expressed higher levels of PPARγ1. The total PPARγ level was also higher in KO cells despite a lower level of PPARγ2 (Fig. 5B). These results are consistent with the notion that PPARγ1 is the predominant PPARγ expressed in subconfluent cells. To explore whether loss of GPS2 activated endogenous PPARγ in KO cells, we asked whether PPARγ target genes were up-regulated in these cells. RNA-Seq studies were performed in KO, WT, and GPS2-transduced KO cells. 362 genes were up-regulated at least 2-fold in GPS2 KO cells but not in GPS2-re-expressed KO cells compared with their expression in WT cells (Fig. 5C). Gene ontology results showed that the 362 genes (also referred here as “GPS2 KO up-regulated” genes) were enriched with features that are characteristic of PPARγ target genes, such as lipid metabolism (gene ontology, 0006629, p = 5.829E-5) and rosiglitazone response (C089730, p = 2.55E-6) (see also Fig. 5F).
FIGURE 5.

Loss of GPS2 was sufficient to activate endogenous PPARγ target genes. A, luciferase assays measuring full-length PPARγ activity. GPS2 WT and GPS2 KO MEFs were transfected with a PPARγ responsive element (PPARE)-driven luciferase reporter along with full-length PPARγ or empty vector control in the absence and presence of rosiglitazone (Rosi.). *, p < 0.05; **, p < 0.01. B, RT-qPCR analysis of endogenous PPARγ1, PPARγ2, and total PPARγ in subconfluent GPS2 KO and GPS2 WT MEFs. C, left panel, overlap of genes up-regulated in GPS2 KO cells and genes physically bound by PPARγ and NCoR at the same site. RNA-Seq analysis was performed in GPS2 WT, GPS2 KO, and GPS2-re-expressed KO MEF cells. The mapped reads were analyzed for differential gene expression using DESeq and GeneSpring NGS software. Compared with GPS2-WT cells, 362 genes showed at least 2-fold higher expression in GPS2 KO but not in GPS2-re-expressed KO cells. The Venn diagram identified 34 overlapping genes between the 362 genes and genes containing overlapping binding sites of PPARγ and NCoR, which were determined by analyzing the ChIP-Seq data sets from mouse macrophages that express PPARγ (47, 48) using Homer (75). Left panel, bottom, de novo motif analysis (75) identified DR1 as the only enriched motif in the PPARγ binding sites on the 34 genes. Right panel, enrichment of PPARγ/NCoR target genes in GPS2 KO up-regulated genes, calculated as the ratio of the overlap with GPS2 KO up-regulated genes (i.e. 34 genes) versus random overlap with RefSeq genes (total of 37,593). 400 randomly generated RefSeq genes were used as a control. The p value was calculated on the basis of a binomial test. D, heatmap of the 34 genes in GPS2 WT, GPS2 KO, and GPS2-transduced KO cells. E, PPARγ and NCoR were present at higher levels on the 34 GPS2 KO up-regulated genes compared with genes not significantly up-regulated by the loss of GPS2. ChIP-Seq tags within a 1-kb region flanking PPARγ binding sites on genes co-occupied by PPARγ/NCoR were quantified using Homer. F, gene list enrichment analysis performed using the ToppGene server (76). GO, gene ontology. G, RT-qPCR analysis of gene expression in subconfluent GPS2 KO, GPS2 WT, and GPS2-re-expressed KO MEFs.
To further demonstrate that GPS2 regulates PPARγ target genes, we analyzed the existing ChIP Sequencing (ChIP-Seq) datasets from mouse macrophages (47, 48). PPARγ and NCoR showed overlapping binding to 1027 genes. Of the 1027 genes, 34 were up-regulated in GPS2 KO cells (Fig. 5, C and D). This overlap between PPARγ/NCoR-co-occupied genes and GPS2 KO up-regulated genes was highly significant compared with the random overlap between PPARγ/NCoR-co-occupied genes and Reference Sequence (RefSeq) genes (Fig. 5C, right panel). Additional evidence further supported that the 34 genes were bona fide GPS2/PPARγ/NCoR target genes. First, de novo motif analysis showed that the PPARγ binding sites on these genes were enriched only with the DR1 PPARγ/retinoid X receptor motif (Fig. 5C, left panel, bottom). Second, the 34 genes recruited more PPARγ and NCoR compared with other genes not significantly up-regulated in GPS2 KO cells (Fig. 5E). Third, gene ontology analysis confirmed that the 34 genes were also enriched with characteristics of PPARγ target genes (Fig. 5F), which essentially recapitulated the results derived from the 362 GPS2 KO up-regulated genes (see above), underscoring PPARγ regulation as a major function of GPS2. Finally, we confirmed the GPS2-dependent regulation of representative genes known to play roles in lipid metabolism (Fig. 5G), including Fzd1 (49), Abca1 (50), Adipor2 (51), Socs1 (52), Sgk1 (53), Trerf1 (54, 55), and Idh1 (56, 57). Among them, Fzd1 and Abca1 are known PPARγ target genes.
Loss of GPS2 Renders Immortalized MEFs Proadipogenic
To functionally demonstrate that loss of GPS2 increases PPARγ activity, we asked whether GPS2-KO MEFs were able to undergo spontaneous adipogenesis in the absence of ectopic PPARγ. Consistent with the notion that self-immortalized MEFs require ectopic expression of PPARγ for adipogenesis (58), WT MEFs were refractory to adipogenesis either in the absence or presence of rosiglitazone (Fig. 6A). A small fraction of GPS2-KO MEFs, however, was able to differentiate into adipocytes spontaneously. The differentiation was greatly enhanced by rosiglitazone treatment, confirming that the process was PPARγ-dependent. The ability of KO cells to undergo adipogenesis was due to the loss of GPS2 because re-expression of GPS2 completely abolished the adipogenic potential of GPS2-KO cells (Fig. 6B). A time-course experiment showed that ectopic GPS2 only slightly reduced the expression of PPARγ1 and the initial level of total PPARγ but was able to completely prevent the induction of PPARγ2, aP2, and adiponectin (Fig. 6C). While the slight reduction of PPARγ1 level by ectopic GPS2 in KO cells was consistent with the difference between WT and KO cells we observed previously, the more dramatic difference observed between KO and WT cells may be related to the use of different cell lines or reflect a long-term cell culture effect on the cells. Nevertheless, the “all-or-none” changes in differentiation potential and the expression of adipocyte-specific PPARγ target genes are consistent with the conclusion that GPS2 regulates adipogenesis by regulating the transcriptional activity of PPARγ. Loss of GPS2 renders MEFs proadipogenic because of impaired repression control of PPARγ.
FIGURE 6.

Loss of GPS2 converts MEFs into a preadipogenic state. A, Oil Red O staining of post-confluent GPS2 KO and GPS2 WT MEFs treated with vehicle or rosiglitazone (Rosi.). B, Oil Red O staining of post-confluent KO and GPS2-transduced KO MEFs treated with vehicle or rosiglitazone. Top panel, Western blot analysis of GPS2 expression in vector- and GPS2-transduced GPS2 KO cells. C, time course analyses of gene expression in post-confluent KO and GPS2-transduced KO cells by RT-qPCR at 0, 2, 4, and 6 days in the absence and presence of rosiglitazone.
DISCUSSION
Since the discovery of GPS2 as a G protein pathway suppressor, GPS2 has emerged as a multifunctional protein. GPS2 can both act as a corepressor and act as a coactivator for various transcription factors, including NRs (25, 59–65), consistent with the presence of distinct repression and activation domains in GPS2 (22). This study focused on the role of GPS2 as an NCoR complex subunit in repression, which is often the first step in signal-dependent transcription activation pathways.
To understand the physiological function of GPS2, we established whole-body GPS2 knockout mice. The mice died during embryonic development between E9.5 and E10.5. A similar lethality has also been reported for NCoR, SMRT, and HDAC3 knockout mice, which die at E15.5, E16.5, and E9.5, respectively (41–43). GPS2+/− mice were fertile and indistinguishable in appearance from their WT counterparts. It is possible that the level of GPS2 in cells may be in excess relative to that of CoRs. Although the precise reason for the death of GPS2 knockout mice remains to be determined, these results are consistent with an essential role of GPS2 as a CoR complex subunit in mouse embryonic development. It is tempting to speculate that the earlier death of GPS2 and HDAC3 KO mice, compared with NCoR and SMRT KO mice, could reflect the fact that GPS2 and HDAC3 are unique subunits of the NCoR/SMRT corepressor complexes, whereas NCoR and SMRT may have redundant functions that allow the embryos to survive longer.
Previous studies have shown that GPS2, TBL1/TBLR1, and RD1 interact with each other to form a stable heterotrimeric complex (22, 32). These results raise the possibility that GPS2 may play a role in complex assembly, which would suggest that GPS2 contributes to RD1- and NCoR-mediated repression. Our results, however, argue against this possibility. Using GPS2-deficient MEFs, we demonstrated that GPS2 was dispensable for RD1-mediated repression. Further supporting the notion that GPS2 is not required for the intrinsic repression function of NCoR, loss of GPS2 did not affect repression mediated by unliganded TRα or TBL1 (Fig. 2B and data not shown). Neither did it affect the corepressor function of NCoR for TRα (Fig. 3C). These results point to fundamental differences between GPS2 and other stoichiometric NCoR-interacting proteins, namely, HDAC3 and TBL1/TBLR1, and suggest that GPS2 is primarily involved in targeting the NCoR complex to transcription factors.
GPS2 plays a critical role in NCoR-mediated repression of PPARγ. Our effort to dissect the conformation requirement of PPARγ for its regulation by GPS2 revealed that GPS2 functions at a step after PPARγ attaining the active conformation (Fig. 7). Our results support the model in which, whereas the CoRNR box allows NCoR to target the repressive state of PPARγ, GPS2 allows NCoR to target the active state of PPARγ. In the absence of the ligand, although GPS2 and the CoRNR box may independently bind to the repressive conformation of PPARγ (and, possibly, apo-TRα), CoRNR box binding should be sufficient to recruit NCoR to mediate repression. This explains why loss of GPS2 did not affect the repression by PPARγΔAF2 and apo-TRα. GPS2 may be the only component in the corepressor complex that can recognize and bind the active PPARγ. Given that a subset of unliganded PPARγ has the active conformation, our model also explains why loss of GPS2 increased PPARγ activities both in the absence and presence of the ligand. Because we have shown that GPS2 and the CoRNR box target different regions of PPARγ, it will be interesting to explore whether GPS2 and the CoRNR box may have cooperative functions in driving the active PPARγ toward a stable inactive complex with NCoR (shown by the reversed arrow line in Fig. 7). Functionally, the GPS2-endowed ability of NCoR to repress active PPARγ is important for cells to maintain proper control of PPARγ target genes, as evidenced by our finding that loss of GPS2 in MEFs was sufficient to cause activation of endogenous PPARγ target genes and to drive adipogenesis of MEFs. Therefore, combined functions of the CoRNR boxes and GPS2 confer on NCoR the abilities to repress diverse NRs, including TRα and PPARγ.
FIGURE 7.
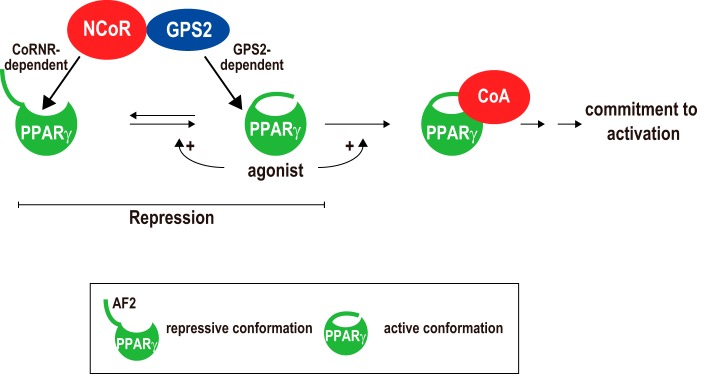
Role of GPS2 in NCoR-mediated repression of PPARγ-dependent transcription. NCoR can directly regulate PPARγ not only in the repressive but also in the active conformation via CoRNR box-dependent and GPS2-dependent mechanisms, respectively. In the absence of agonists, a subset of PPARγ spontaneously adopts the active conformation that requires AF2, explaining why depleting AF2 insensitizes PPARγ to GPS2-dependent regulation. Agonists such as rosiglitazone increase the ability of PPARγ to assume the active state and to bind to CoAs. Unlike thought previously, repression occurs not only to the repressive state but also to the active state of PPARγ. See text for details.
In the presence of agonists such as rosiglitazone, the binding of agonists to PPARγ should abolish the CoRNR box-dependent interaction with PPARγ. Subsequently, the increased association of CoAs with agonist-bound PPARγ further reduces GPS2-dependent repression, leading to commitment of activation. Nevertheless, in the presence of agonists, GPS2 could still impose an inhibition on PPARγ, as evidenced by the increased PPARγ activity in GPS2-KO cells versus WT cells (Figs. 2, C and D, and 3E). These results suggest that GPS2 is not only important for maintaining the repressed state of apo-PPARγ but also plays a role in delimiting the maximal ligand response of PPARγ, which may be important for its biological function.
The GPS2-dependent repression pathway explains why NCoR is still capable of repressing PPARγ-dependent transcription despite the weak interaction between its CoRNR boxes and PPARγ on DNA (11–13). A role of GPS2 for NCoR to repress active PPARγ also provides new mechanistic insights into previous studies showing that NCoR is able to repress PPARγ target genes in the presence of PPARγ agonists. For example, in 3T3-L1 cells cultured in the presence of thiazolidinediones, NCoR depletion dramatically enhances the expression of PPARγ target genes and PPARγ-dependent adipogenesis (33). A recent study also reported that adipocyte-specific knockout of NCoR increases PPARγ activity under high-fat diets (34), a condition that would increase the levels of endogenous PPARγ ligands.
Our finding that GPS2 represses PPARγ activity is in line with previous studies showing that reduced expression of GPS2 is associated with increased obesity in humans (66). It also explains why adipogenesis of immortalized MEFs requires overexpression of ectopic PPARγ (58). On the basis of our data, overexpressing PPARγ may be necessary for PPARγ to escape the interaction with and subsequent inhibition mediated by endogenous GPS2. In a related scenario, the ability of GPS2 to recognize and bind the active state of PPARγ also provides new insights into how GPS2 inhibits inflammation (60, 66). One of the anti-inflammatory pathways involves trans-repression by ligand-associated PPARγ, which inhibits inflammation by preventing the discharge of corepressor complexes from cytokine-inducible genes. Although sumoylation of liganded PPARγ plays a role in its association with the corepressor complex (67), it is not known whether sumoylation is sufficient and how corepressors discriminate different sumoylated proteins. An interaction between the GPS2 subunit of CoR complex and liganded PPARγ may provide the necessary specificity while ensuring the stability of the association between liganded PPARγ and the CoR complex.
Our results show that PPARγ2 expression is strongly induced in post-confluent GPS2-KO cells. Interestingly, the level of PPARγ2 in subconfluent KO cells, as judged by RT-qPCR, was lower than that in WT cells. It should be noted that PPARγ2 expression is already very low in subconfluent cells compared with differentiated cells, consistent with the notion that its expression is adipocyte-specific. In non-differentiated cells, transcription from the PPARγ2 promoter may be epigenetically silenced via H3K9 methylation (68). The further reduction of the PPARγ2 level in KO cells may be related to the increased transcription of PPARγ1 in these cells or reflect an independent “coactivator” function of GPS2 in promoting H3K9 demethylation, as reported recently (61, 69). During differentiation, PPARγ is able to bind to its sites. This allows GPS2 to manifest its corepressor function to prevent the premature activation of PPARγ2, explaining the lack of PPARγ2 induction in WT cells.
In summary, this work identified, for the first time, a novel GPS2-dependent mechanism that allowed NCoR to directly repress active PPARγ-mediated transcription and showed that this repression mechanism was important for the corepressor function of NCoR (and possibly SMRT) for PPARγ. Interestingly, the GPS2-dependent repression appears to affect only PPARγ but not other PPAR isoforms. Therefore, a further understanding of how GPS2 binds and regulates PPARγ may lead to strategies to specifically target PPARγ signaling in various diseases, such as obesity and insulin resistance. In addition to PPARγ, GPS2 also interacts with other NRs, such as liver X receptor (LXR), liver receptor homolog-1 (LRH-1), hepatocyte nuclear factor 4 (HNF4), and farnesoid X receptor (FXR) (61, 63, 64). Moreover, in the case of LXR, GPS2 can similarly bind to the liganded conformation of LXR and facilitate its association with CoR complexes (64). Therefore, the GPS2-dependent regulation, as observed here, may be applicable to these NRs as well. GPS2 appears to belong to a unique class of coregulators different from the classic CoRs and CoAs that only bind to repressive and active conformations of NRs, respectively. Because ligands may not be required for GPS2 to bind NRs, it is possible that GPS2 may recognize a constitutive surface present in both repressive and active forms of NRs, including the constitutively active orphan receptors. We speculate that GPS2 may have evolved to play a general protective role against premature activation (hormone-independent) or hyperactivation (hormone-dependent) of NR target genes. A broad involvement of GPS2-NR interactions may also explain why the CoRNR box region of CoRs (70–74), but not the entire CoR molecule, is dispensable for embryonic development in mice.
Acknowledgment
We thank Dr. Sohaib Khan for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R21CA178513 (to J. Z.) and R01MH093429 (to T. P. B.). This work was also supported by a start-up fund from Saint Louis University (to J. Z.), by a University of Cincinnati Cancer Center Pilot grant (to J. Z.), by the Chinese Scholarship Council (to C. Y.), and by Institute of Medical Biology, Chinese Academy of Medical Science Grant IMB-201 (to H. L.).
- NR
- nuclear receptor
- LBD
- ligand-binding domain
- CoR
- corepressor
- CoA
- coactivator
- TR
- thyroid hormone receptor
- PPAR
- peroxisome proliferator-activated receptor
- NCoR
- nuclear receptor corepressor
- SMRT
- silencing mediator for retinoid and thyroid hormone receptors
- MEF
- mouse embryonic fibroblast
- RT-qPCR
- quantitative RT-PCR
- E9.5
- embryonic day 9.5
- Gal4-DBD
- Gal4 DNA-binding domain
- Gal4-UAS
- Gal4-upstream activating sequence.
REFERENCES
- 1. Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., Evans R. M. (1995) The nuclear receptor superfamily: the second decade. Cell 83, 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glass C. K., Rosenfeld M. G. (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14, 121–141 [PubMed] [Google Scholar]
- 3. Bourguet W., Ruff M., Chambon P., Gronemeyer H., Moras D. (1995) Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-α. Nature 375, 377–382 [DOI] [PubMed] [Google Scholar]
- 4. Wagner R. L., Apriletti J. W., McGrath M. E., West B. L., Baxter J. D., Fletterick R. J. (1995) A structural role for hormone in the thyroid hormone receptor. Nature 378, 690–697 [DOI] [PubMed] [Google Scholar]
- 5. Perissi V., Rosenfeld M. G. (2005) Controlling nuclear receptors: the circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 6, 542–554 [DOI] [PubMed] [Google Scholar]
- 6. Nolte R. T., Wisely G. B., Westin S., Cobb J. E., Lambert M. H., Kurokawa R., Rosenfeld M. G., Willson T. M., Glass C. K., Milburn M. V. (1998) Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-g. Nature 395, 137–143 [DOI] [PubMed] [Google Scholar]
- 7. Uppenberg J., Svensson C., Jaki M., Bertilsson G., Jendeberg L., Berkenstam A. (1998) Crystal structure of the ligand binding domain of the human nuclear receptor PPARγ. J. Biol. Chem. 273, 31108–31112 [DOI] [PubMed] [Google Scholar]
- 8. Hughes T. S., Chalmers M. J., Novick S., Kuruvilla D. S., Chang M. R., Kamenecka T. M., Rance M., Johnson B. A., Burris T. P., Griffin P. R., Kojetin D. J. (2012) Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure 20, 139–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chandra V., Huang P., Hamuro Y., Raghuram S., Wang Y., Burris T. P., Rastinejad F. (2008) Structure of the intact PPAR-γ-RXR- nuclear receptor complex on DNA. Nature 456, 350–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Danielian P. S., White R., Lees J. A., Parker M. G. (1992) Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 11, 1025–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zamir I., Zhang J., Lazar M. A. (1997) Stoichiometric and steric principles governing repression by nuclear hormone receptors. Genes Dev. 11, 835–846 [DOI] [PubMed] [Google Scholar]
- 12. Zhang J., Hu X., Lazar M. A. (1999) A novel role for helix 12 of retinoid X receptor in regulating repression. Mol. Cell. Biol. 19, 6448–6457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi Y., Hon M., Evans R. M. (2002) The peroxisome proliferator-activated receptor δ, an integrator of transcriptional repression and nuclear receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 99, 2613–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dowell P., Ishmael J. E., Avram D., Peterson V. J., Nevrivy D. J., Leid M. (1999) Identification of nuclear receptor corepressor as a peroxisome proliferator-activated receptor α interacting protein. J. Biol. Chem. 274, 15901–15907 [DOI] [PubMed] [Google Scholar]
- 15. Gurnell M., Wentworth J. M., Agostini M., Adams M., Collingwood T. N., Provenzano C., Browne P. O., Rajanayagam O., Burris T. P., Schwabe J. W., Lazar M. A., Chatterjee V. K. (2000) A dominant-negative peroxisome proliferator-activated receptor γ (PPARγ) mutant is a constitutive repressor and inhibits PPARγ-mediated adipogenesis. J. Biol. Chem. 275, 5754–5759 [DOI] [PubMed] [Google Scholar]
- 16. Hu X., Lazar M. A. (1999) The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402, 93–96 [DOI] [PubMed] [Google Scholar]
- 17. Perissi V., Staszewski L. M., McInerney E. M., Kurokawa R., Krones A., Rose D. W., Lambert M. H., Milburn M. V., Glass C. K., Rosenfeld M. G. (1999) Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 13, 3198–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nagy L., Kao H. Y., Love J. D., Li C., Banayo E., Gooch J. T., Krishna V., Chatterjee K., Evans R. M., Schwabe J. W. (1999) Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 13, 3209–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heery D. M., Kalkhoven E., Hoare S., Parker M. G. (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387, 733–736 [DOI] [PubMed] [Google Scholar]
- 20. Hörlein A. J., Näär A. M., Heinzel T., Torchia J., Gloss B., Kurokawa R., Ryan A., Kamei Y., Söderström M., Glass C. K. (1995) Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377, 397–404 [DOI] [PubMed] [Google Scholar]
- 21. Chen J. D., Evans R. M. (1995) A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377, 454–457 [DOI] [PubMed] [Google Scholar]
- 22. Zhang J., Kalkum M., Chait B. T., Roeder R. G. (2002) The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 9, 611–623 [DOI] [PubMed] [Google Scholar]
- 23. Li J., Wang J., Wang J., Nawaz Z., Liu J. M., Qin J., Wong J. (2000) Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 19, 4342–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guenther M. G., Lane W. S., Fischle W., Verdin E., Lazar M. A., Shiekhattar R. (2000) A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 14, 1048–1057 [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng X., Kao H. Y. (2009) G protein pathway suppressor 2 (GPS2) is a transcriptional corepressor important for estrogen receptor α-mediated transcriptional regulation. J. Biol. Chem. 284, 36395–36404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guenther M. G., Barak O., Lazar M. A. (2001) The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 21, 6091–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishizuka T., Lazar M. A. (2003) The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Mol. Cell. Biol. 23, 5122–5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoon H. G., Chan D. W., Huang Z. Q., Li J., Fondell J. D., Qin J., Wong J. (2003) Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 22, 1336–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoon H. G., Choi Y., Cole P. A., Wong J. (2005) Reading and function of a histone code involved in targeting corepressor complexes for repression. Mol. Cell. Biol. 25, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spain B. H., Bowdish K. S., Pacal A. R., Staub S. F., Koo D., Chang C. Y., Xie W., Colicelli J. (1996) Two human cDNAs, including a homolog of Arabidopsis FUS6 (COP11), suppress G-protein- and mitogen-activated protein kinase-mediated signal transduction in yeast and mammalian cells. Mol. Cell. Biol. 16, 6698–6706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jin D. Y., Teramoto H., Giam C. Z., Chun R. F., Gutkind J. S., Jeang K. T. (1997) A human suppressor of c-Jun N-terminal kinase 1 activation by tumor necrosis factor α. J. Biol. Chem. 272, 25816–25823 [DOI] [PubMed] [Google Scholar]
- 32. Oberoi J., Fairall L., Watson P. J., Yang J. C., Czimmerer Z., Kampmann T., Goult B. T., Greenwood J. A., Gooch J. T., Kallenberger B. C., Nagy L., Neuhaus D., Schwabe J. W. (2011) Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 18, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu C., Markan K., Temple K. A., Deplewski D., Brady M. J., Cohen R. N. (2005) The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor γ transcriptional activity and repress 3T3-L1 adipogenesis. J. Biol. Chem. 280, 13600–13605 [DOI] [PubMed] [Google Scholar]
- 34. Li P., Fan W., Xu J., Lu M., Yamamoto H., Auwerx J., Sears D. D., Talukdar S., Oh D., Chen A., Bandyopadhyay G., Scadeng M., Ofrecio J. M., Nalbandian S., Olefsky J. M. (2011) Adipocyte NCoR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell 147, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guan H. P., Ishizuka T., Chui P. C., Lehrke M., Lazar M. A. (2005) Corepressors selectively control the transcriptional activity of PPARγ in adipocytes. Genes Dev. 19, 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mottis A., Mouchiroud L., Auwerx J. (2013) Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 27, 819–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shao D., Lazar M. A. (1997) Peroxisome proliferator activated receptor γ, CCAAT/enhancer-binding protein α, and cell cycle status regulate the commitment to adipocyte differentiation. J. Biol. Chem. 272, 21473–21478 [DOI] [PubMed] [Google Scholar]
- 38. Guo C., Hu Q., Yan C., Zhang J. (2009) Multivalent binding of the ETO corepressor to E proteins facilitates dual repression controls targeting chromatin and the basal transcription machinery. Mol. Cell. Biol. 29, 2644–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gow C. H., Guo C., Wang D., Hu Q., Zhang J. (2014) Differential involvement of E2A-corepressor interactions in distinct leukemogenic pathways. Nucleic Acids Res. 42, 137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo C., Gow C. H., Li Y., Gardner A., Khan S., Zhang J. (2012) Regulated clearance of histone deacetylase 3 protects independent formation of nuclear receptor corepressor complexes. J. Biol. Chem. 287, 12111–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jepsen K., Hermanson O., Onami T. M., Gleiberman A. S., Lunyak V., McEvilly R. J., Kurokawa R., Kumar V., Liu F., Seto E., Hedrick S. M., Mandel G., Glass C. K., Rose D. W., Rosenfeld M. G. (2000) Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 102, 753–763 [DOI] [PubMed] [Google Scholar]
- 42. Jepsen K., Gleiberman A. S., Shi C., Simon D. I., Rosenfeld M. G. (2008) Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 22, 740–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jepsen K., Solum D., Zhou T., McEvilly R. J., Kim H. J., Glass C. K., Hermanson O., Rosenfeld M. G. (2007) SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 450, 415–419 [DOI] [PubMed] [Google Scholar]
- 44. Bhaskara S., Chyla B. J., Amann J. M., Knutson S. K., Cortez D., Sun Z. W., Hiebert S. W. (2008) Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 30, 61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walkey C. J., Spiegelman B. M. (2008) A functional peroxisome proliferator-activated receptor-γ ligand-binding domain is not required for adipogenesis. J. Biol. Chem. 283, 24290–24294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sarraf P., Mueller E., Smith W. M., Wright H. M., Kum J. B., Aaltonen L. A., de la Chapelle A., Spiegelman B. M., Eng C. (1999) Loss-of-function mutations in PPAR γ associated with human colon cancer. Mol. Cell 3, 799–804 [DOI] [PubMed] [Google Scholar]
- 47. Barish G. D., Yu R. T., Karunasiri M. S., Becerra D., Kim J., Tseng T. W., Tai L. J., Leblanc M., Diehl C., Cerchietti L., Miller Y. I., Witztum J. L., Melnick A. M., Dent A. L., Tangirala R. K., Evans R. M. (2012) The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 15, 554–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lefterova M. I., Steger D. J., Zhuo D., Qatanani M., Mullican S. E., Tuteja G., Manduchi E., Grant G. R., Lazar M. A. (2010) Cell-specific determinants of peroxisome proliferator-activated receptor γ function in adipocytes and macrophages. Mol. Cell. Biol. 30, 2078–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oger F., Dubois-Chevalier J., Gheeraert C., Avner S., Durand E., Froguel P., Salbert G., Staels B., Lefebvre P., Eeckhoute J. (2014) Peroxisome proliferator-activated receptor γ regulates genes involved in insulin/insulin-like growth factor signaling and lipid metabolism during adipogenesis through functionally distinct enhancer classes. J. Biol. Chem. 289, 708–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chinetti G., Lestavel S., Bocher V., Remaley A. T., Neve B., Torra I. P., Teissier E., Minnich A., Jaye M., Duverger N., Brewer H. B., Fruchart J. C., Clavey V., Staels B. (2001) PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 7, 53–58 [DOI] [PubMed] [Google Scholar]
- 51. Yamauchi T., Nio Y., Maki T., Kobayashi M., Takazawa T., Iwabu M., Okada-Iwabu M., Kawamoto S., Kubota N., Kubota T., Ito Y., Kamon J., Tsuchida A., Kumagai K., Kozono H., Hada Y., Ogata H., Tokuyama K., Tsunoda M., Ide T., Murakami K., Awazawa M., Takamoto I., Froguel P., Hara K., Tobe K., Nagai R., Ueki K., Kadowaki T. (2007) Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 13, 332–339 [DOI] [PubMed] [Google Scholar]
- 52. Zheng R. D., Liao L. H., Ye J., Wang C. B., Gao J. Z., Ying Y. Q., Ning Q., Luo X. P. (2013) Effects of SOCS 1/3 gene silencing on the expression of C/EBPα and PPARγ during differentiation and maturation of rat preadipocytes. Pediatr. Res. 73, 263–267 [DOI] [PubMed] [Google Scholar]
- 53. Li P., Pan F., Hao Y., Feng W., Song H., Zhu D. (2013) SGK1 is regulated by metabolic-related factors in 3T3-L1 adipocytes and overexpressed in the adipose tissue of subjects with obesity and diabetes. Diabetes Res. Clin. Pract. 102, 35–42 [DOI] [PubMed] [Google Scholar]
- 54. Gizard F., Lavallée B., DeWitte F., Hum D. W. (2001) A novel zinc finger protein TReP-132 interacts with CBP/p300 to regulate human CYP11A1 gene expression. J. Biol. Chem. 276, 33881–33892 [DOI] [PubMed] [Google Scholar]
- 55. Gizard F., Lavallee B., DeWitte F., Teissier E., Staels B., Hum D. W. (2002) The transcriptional regulating protein of 132 kDa (TReP-132) enhances P450scc gene transcription through interaction with steroidogenic factor-1 in human adrenal cells. J. Biol. Chem. 277, 39144–39155 [DOI] [PubMed] [Google Scholar]
- 56. Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., Jewell C. M., Johnson Z. R., Irvine D. J., Guarente L., Kelleher J. K., Vander Heiden M. G., Iliopoulos O., Stephanopoulos G. (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shechter I., Dai P., Huo L., Guan G. (2003) IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: evidence that IDH1 may regulate lipogenesis in hepatic cells. J. Lipid Res. 44, 2169–2180 [DOI] [PubMed] [Google Scholar]
- 58. Rosen E. D., MacDougald O. A. (2006) Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 7, 885–896 [DOI] [PubMed] [Google Scholar]
- 59. Peng Y. C., Breiding D. E., Sverdrup F., Richard J., Androphy E. J. (2000) AMF-1/Gps2 binds p300 and enhances its interaction with papillomavirus E2 proteins. J. Virol. 74, 5872–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cardamone M. D., Krones A., Tanasa B., Taylor H., Ricci L., Ohgi K. A., Glass C. K., Rosenfeld M. G., Perissi V. (2012) A protective strategy against hyperinflammatory responses requiring the nontranscriptional actions of GPS2. Mol. Cell 46, 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jakobsson T., Venteclef N., Toresson G., Damdimopoulos A. E., Ehrlund A., Lou X., Sanyal S., Steffensen K. R., Gustafsson J. A., Treuter E. (2009) GPS2 is required for cholesterol efflux by triggering histone demethylation, LXR recruitment, and coregulator assembly at the ABCG1 locus. Mol. Cell 34, 510–518 [DOI] [PubMed] [Google Scholar]
- 62. Peng Y. C., Kuo F., Breiding D. E., Wang Y. F., Mansur C. P., Androphy E. J. (2001) AMF1 (GPS2) modulates p53 transactivation. Mol. Cell. Biol. 21, 5913–5924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sanyal S., Båvner A., Haroniti A., Nilsson L. M., Lundåsen T., Rehnmark S., Witt M. R., Einarsson C., Talianidis I., Gustafsson J. A., Treuter E. (2007) Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 15665–15670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Venteclef N., Jakobsson T., Ehrlund A., Damdimopoulos A., Mikkonen L., Ellis E., Nilsson L. M., Parini P., Jänne O. A., Gustafsson J. A., Steffensen K. R., Treuter E. (2010) GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRβ in the hepatic acute phase response. Genes Dev. 24, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang D., Harry G. J., Blackshear P. J., Zeldin D. C. (2008) G-protein pathway suppressor 2 (GPS2) interacts with the regulatory factor X4 variant 3 (RFX4_v3) and functions as a transcriptional co-activator. J. Biol. Chem. 283, 8580–8590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Toubal A., Clément K., Fan R., Ancel P., Pelloux V., Rouault C., Veyrie N., Hartemann A., Treuter E., Venteclef N. (2013) SMRT-GPS2 corepressor pathway dysregulation coincides with obesity-linked adipocyte inflammation. J. Clin. Invest. 123, 362–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pascual G., Fong A. L., Ogawa S., Gamliel A., Li A. C., Perissi V., Rose D. W., Willson T. M., Rosenfeld M. G., Glass C. K. (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang L., Xu S., Lee J. E., Baldridge A., Grullon S., Peng W., Ge K. (2013) Histone H3K9 methyltransferase G9a represses PPARγ expression and adipogenesis. EMBO J. 32, 45–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cardamone M. D., Tanasa B., Chan M., Cederquist C. T., Andricovich J., Rosenfeld M. G., Perissi V. (2014) GPS2/KDM4A pioneering activity regulates promoter-specific recruitment of PPARγ. Cell Rep. 8, 163–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nofsinger R. R., Li P., Hong S. H., Jonker J. W., Barish G. D., Ying H., Cheng S. Y., Leblanc M., Xu W., Pei L., Kang Y. J., Nelson M., Downes M., Yu R. T., Olefsky J. M., Lee C. H., Evans R. M. (2008) SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 20021–20026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reilly S. M., Bhargava P., Liu S., Gangl M. R., Gorgun C., Nofsinger R. R., Evans R. M., Qi L., Hu F. B., Lee C. H. (2010) Nuclear receptor corepressor SMRT regulates mitochondrial oxidative metabolism and mediates aging-related metabolic deterioration. Cell Metab. 12, 643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Astapova I., Vella K. R., Ramadoss P., Holtz K. A., Rodwin B. A., Liao X. H., Weiss R. E., Rosenberg M. A., Rosenzweig A., Hollenberg A. N. (2011) The nuclear receptor corepressor (NCoR) controls thyroid hormone sensitivity and the set point of the hypothalamic-pituitary-thyroid axis. Mol. Endocrinol. 25, 212–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fang S., Suh J. M., Atkins A. R., Hong S. H., Leblanc M., Nofsinger R. R., Yu R. T., Downes M., Evans R. M. (2011) Corepressor SMRT promotes oxidative phosphorylation in adipose tissue and protects against diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 3412–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fozzatti L., Lu C., Kim D. W., Park J. W., Astapova I., Gavrilova O., Willingham M. C., Hollenberg A. N., Cheng S. Y. (2011) Resistance to thyroid hormone is modulated in vivo by the nuclear receptor corepressor (NCOR1). Proc. Natl. Acad. Sci. U.S.A. 108, 17462–17467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen J., Bardes E. E., Aronow B. J., Jegga A. G. (2009) ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 [DOI] [PMC free article] [PubMed] [Google Scholar]