

Background: The Na+,K+-ATPase discriminates between similar and abundant ions.
Results: The K+ congener acetamidinium interacts with the outward facing sites of Na+,K+-ATPase, but does not interact with the inward facing sites.
Conclusion: Water in the ion binding cavity regulates ion selectivity of the Na+,K+-ATPase.
Significance: This study identifies new determinants of ion selectivity of K+-transporting P-type pumps.
Keywords: ATPase, Membrane Enzyme, Membrane Protein, Na+/K+-ATPase, Potassium Transport
Abstract
The Na+,K+-ATPase is essential for ionic homeostasis in animal cells. The dephosphoenzyme contains Na+ selective inward facing sites, whereas the phosphoenzyme contains K+ selective outward facing sites. Under normal physiological conditions, K+ inhibits cytoplasmic Na+ activation of the enzyme. Acetamidinium (Acet+) and formamidinium (Form+) have been shown to permeate the pump through the outward facing sites. Here, we show that these cations, unlike K+, are unable to enter the inward facing sites in the dephosphorylated enzyme. Consistently, the organic cations exhibited little to no antagonism to cytoplasmic Na+ activation. Na+,K+-ATPase structures revealed a previously undescribed rotamer transition of the hydroxymethyl side chain of the absolutely conserved Thr772 of the α-subunit. The side chain contributes its hydroxyl to Na+ in site I in the E1 form and rotates to contribute its methyl group toward K+ in the E2 form. Molecular dynamics simulations to the E1·AlF4−·ADP·3Na+ structure indicated that 1) bound organic cations differentially distorted the ion binding sites, 2) the hydroxymethyl of Thr772 rotates to stabilize bound Form+ through water molecules, and 3) the rotamer transition is mediated by water traffic into the ion binding cavity. Accordingly, dehydration induced by osmotic stress enhanced the interaction of the congeners with the outward facing sites and profoundly modified the organization of membrane domains of the α-subunit. These results assign a catalytic role for water in pump function, and shed light on a backbone-independent but a conformation-dependent switch between H-bond and dispersion contact as part of the catalytic mechanism of the Na+,K+-ATPase.
Introduction
The Na+,K+-ATPase (sodium pump) belongs to P-type cation transporting ATPases, which use energy from ATP hydrolysis to establish electrochemical gradients for different ions across cellular membranes. Gastric H+,K+-ATPase and sarcoplasmic reticulum Ca2+-ATPase are closely related members (1). The Na+,K+-ATPase exchanges intracellular Na+ for extracellular K+, at a stoichiometry of 3Na+/2K+ for each ATP molecule hydrolyzed (2). The functional pump consists of two subunits; α is the catalytic subunit that couples ATP hydrolysis to the uphill transport of ions, and β is a glycoprotein important for targeting and stability of the enzyme complex (3). Under steady state conditions, the pump is challenged by numerous passive Na+ influx events associated with physiological functions such as nutrient uptake, nerve conduction, and osmoregulation.
The mechanism of the Na+,K+-ATPase is described by the Post-Albers scheme; three Na+ ions bind to cytoplasmic sites in the E1 form, providing the trigger for phosphoryl transfer and formation of the Na+-occluding E1P(3Na+) state. The Na+ ions are released to the extracellular side in concomitance with the E1P(3Na+) → E2P·3Na+ transition. Following Na+ release, two K+ ions bind to extracellular sites in the E2P form, inducing dephosphorylation and formation of the K+-occluding E2(2K+) state. ATP facilitates K+ release to the cytoplasm and shift to the E1 form.
During catalysis, Na+,K+-ATPase alternates between two major conformations, exposing ion binding sites to either side of the membrane. Inward facing sites in the dephosphorylated enzyme bind Na+ with high affinity and K+ with low affinity, whereas the outward facing sites in the phosphorylated enzyme bind K+ with high affinity and Na+ with low affinity. Mutational studies have identified several residues in membrane spans M4, M5, and M6 of the α-subunit as essential to ion binding. In particular, comparison between the different P-type pumps indicated that the proximal part of M5 is a determinant of ion selectivity (4). Although disposition of the membrane spans are considered the major factor that controls ion selectivity, other microscopic factors seem to play a role. For instance, protonation of ion-coordinating acidic residues was reported to be a key regulator of K+ selectivity (5).
High cytoplasmic K+ inhibits Na+ binding to the inward facing sites, referred to as cytoplasmic K+ antagonism. Based on the assumption that cytoplasmic K+ binds with low affinity to three identical and non-interacting sites in the E1 form, a kinetic model was introduced to account for the K+ antagonism (6). Subsequently, it was shown that enzymes from different mammalian tissues have widely different sensitivities to inhibition by high K+ (7, 8), and it was proposed that cytoplasmic K+/Na+ sensitivity is a tissue-specific pump regulatory mechanism.
Based on early work on the Na+ channel (9, 10), results from voltage clamp experiments on Na+,K+-ATPase expressed in Xenopus oocytes have introduced several organic cations as congeners of Na+ and K+ (11, 12). In particular, acetamidinium (Acet+)2 and formamidinium (Form+) were shown to replace extracellular K+ (12). A congener (such as Rb+) is seemingly expected to exhibit effects on the pump identical to those of K+ (13), i.e. stimulating activity upon binding to the high affinity outward facing sites but decreasing activity by inhibiting (at high concentrations) the interaction of Na+ with the inward facing sites. Identifying ion congeners that bind to the different sites on an exclusive basis would provide mechanistic information (e.g. Ref. 14). In this study we have examined whether Acet+ and Form+ impair cytoplasmic Na+ activation of the pump as is the case with K+ (or Rb+). Other organic cations that obscure the extracellular ion binding sites without being occluded were used for comparison (Fig. 1). Surprisingly, Acet+ and Form+ were found not to interact with the inward facing sites and thereby do not antagonize cytoplasmic Na+ binding. This unprecedented one-side reactivity allows estimation of the contribution of cytoplasmic K+ to inhibition of pump function. In addition, relying on pump structures in the two major conformations together with molecular dynamics (MD) simulations, we suggest an indirect role for hydration of the ion binding cavity in modulation of the ion selectivity of the pump.
FIGURE 1.

Schematic structure of the organic cations used in this study. Organic cations that bind and occlude in the K+ sites of the Na+,K+-ATPase are acetamidinium, formamidinium, and chloroacetamidinium. Organic cations that do not occlude in the pump but inhibit K+ interaction are guanidinium (Gua), methyl guanidinium (M Gua+), dimethyl guanidinium (diM Gua), N-methyl-d-glucamine, and BTEA.
EXPERIMENTAL PROCEDURES
Enzyme Preparation, Hydrolytic Activity, and Phosphoenzyme Measurements
Plasma membranes from pig kidney red outer medulla were isolated according to a modified Jørgensen's method described earlier (15). In brief, minced tissue pieces were homogenized in 30 mm imidazole buffer, pH 7.4, containing 250 mm sucrose and 1 mm EDTA. The homogenate was subjected to several differential centrifugation steps to obtain a microsomal fraction, which were treated with a mild concentration of SDS to open sealed vesicles and dissociate several peripheral proteins from the membrane. The final preparation had a specific activity of 1.6–2.0 mmol h−1 mg−1 protein at 37 °C (standard substrate conditions: 30 mm histidine buffer, pH 7.3, 130 mm NaCl, 20 mm KCl, and 3 mm MgATP). The protein concentration was determined using a Bio-Rad detergent compatible kit (catalog number 500-0113), according to the manufacturer's instructions. ATPase activity was measured by incubating the enzyme with buffer and substrates at 37 °C followed by measuring the phosphate liberated from ATP, according to the method of Baginsky et al. (16). Control activities were measured in identical conditions in the presence of 1 mm ouabain.
Para-nitrophenyl phosphatase (pNPPase) activity was measured in a reaction mixture containing 50 mm Tris-HCl, pH 7.2, 7 mm MgCl, 7 mm pNPP (Tris salt), 5 μg of protein, and substrate ions as indicated in the figure legends. The pNPPase reaction is sensitive to ionic strength. Hence, in all pNPPase assays described in this study, the ionic strength was kept constant at 150 mm using choline chloride. Choline chloride alone had no effect on pNPP hydrolysis (data not shown). The reactions were started with the addition of enzyme, and stopped with 4.5% trichloroacetic acid. Ouabain sensitive para-nitrophenol release was determined by measuring the absorbance of the post-hydrolytic mixture at 410 nm. The calculations are based on a molar extinction coefficient for para-nitrophenol of 1.8 × 104 m−1 cm−1.
Phosphorylation from ATP (prior to pNPPase measurement) was performed in a mixture containing 200 μm ATP, 0.1 mm EGTA, 1 mm Mg2+, and 10 mm Na+. At this relatively low Na+ concentration, the conformational equilibrium is shifted toward the E2P form (7). The phosphorylation reactions were performed on ice, and aliquots of the phosphorylated enzyme were transferred to pNPPase assays (37 °C). Control experiments using [32P]ATP indicated that the steady state phosphoenzyme level is stable on ice for at least 2 h in the presence of 200 μm ATP. It was necessary to add 50 μm ATP and 5 mm NaCl to the pNPPase assays (final concentrations) to prevent complete dephosphorylation of the enzyme during the pNPPase assay. An ATP concentration of 50 μm and a Na+ concentration of 5 mm have no effect of pNPP hydrolysis, as estimated from control experiments.
Phosphorylation from inorganic phosphate was performed in a mixture containing 15 mm histidine, pH 7.3, 1 mm MgCl2, and 1 mm inorganic phosphate (phosphoric acid adjusted to pH 7.2 using Tris buffer). The reactions were incubated at room temperature for 30 min, and aliquots of the phosphorylated enzyme were transferred directly to the pNPPase assays.
Dephosphorylation was measured at 0 °C by diluting the phosphoenzyme into 1.5 mm Tris-ATP and 3 mm MgCl2 with or without 15% glucose (chasing solution), followed by acid quenching after several time intervals. The acid-stable phosphoenzyme was collected, washed twice by centrifugation, and measured by scintillation counting.
Proteolytic Cleavage, Gel Electrophoresis, and Immunoblotting
Exhaustive proteolysis of the pig kidney α-subunit was performed in a reaction mixture containing 100 μg of purified membrane protein suspended in 25 mm histidine, pH 7.2, 1 mm EDTA, and 30 mm K+, Acet+, Form+, or Cl-Acet+, in the presence of different glucose concentrations. The protein samples were preincubated with ions for 30 min at 37 °C before the addition of 20 μg of trypsin and incubation of the mixtures for a further 90 min at 37 °C. Proteolysis was terminated with an equal volume of SDS sample buffer containing 1% trichloroacetic acid to irreversibly inhibit the protease. The mixtures were analyzed by SDS-PAGE overnight at 150 V and 12 mA/gel. 5 μg of protein was loaded onto 8% SDS-PAGE, and protein fragments on the gel were transferred to polyvinylidene fluoride membranes and visualized by Western blotting using a C-terminal α-subunit antibody (raised against peptide Ile1002-Tyr1016 of the pig kidney α-subunit), as described previously (17).
MD Simulations
All-atom MD simulations were implemented with the recently published E1·AlF4−·ADP·3Na+ structure (sometimes referred to as E1 structure) of the Na+,K+-ATPase (PDB accession number 3WGU, Ref. 18) embedded in a fully hydrated 1-palmitoyl,2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane, together with sodium ions. For studies on organic cation binding, Form+ and Acet+ were placed in the binding sites to reveal the atomistic details of their interactions with the pump in the aforementioned structure.
System Construction
The AlF4− group present in the crystal structure (18) was removed, the Asp369 was manually phosphorylated, and the ADP molecule was retained. Therefore, our model represents the phosphorylated ADP-bound E1P form of the Na+,K+-ATPase. Inorganic ions in the structure (sodium in the ion binding sites and the magnesium near Asp369) as well as water molecules were retained in the simulations. The sodium coordinating Glu327 and Glu779, as well as Glu954 that contributes to the proposed transient IIIa site (19), were kept protonated, to ensure the optimal geometry of the ion binding sites. The remaining aspartates and glutamates were kept deprotonated. Subsequently, the protein was embedded in the equilibrated POPC membrane (∼460 lipid molecules), using the g_membed (20) procedure as implemented in GROMACS version 4.6.1, and hydrated with ∼60,000 water molecules. Electroneutrality was kept with sodium and magnesium ions present in the crystal structure, and with an addition of 14 randomly distributed sodium ions in the solution. The above procedure created what we refer to as sodium system. Studies on organic cation interactions were performed by insertion of two Form+ or two Acet+ ions, replacing the two sodium ions in sites I and II. The organic cations were placed in three different orientations. After a short equilibration, one promising orientation for both ions was chosen for longer simulation, referred to as FORM and ACET systems, respectively.
Force Field Parameters and Simulation Details
All simulation details were essentially the same as used in our recent work on disease-causing mutations of the Na+,K+-ATPase (21). GROMACS version 4.6.1 (22, 23) was used to propagate the MD equations of motions, using the leap frog algorithm, employing CHARMM27 force field parameters for proteins (24–26) and CHARMM36 parameters for lipids (27) and sodium ions. Parameters for phosphorylated aspartate were taken from Damjanović et al. (28). Water was modeled as TIP3P CHARMM model (24) with Lennard-Jones interactions on hydrogens. Parameters for Form+ and Acet+ were obtained in a manner similar to a previous work (12) describing interactions of Acet+ with the E2·MgF42−·2K+ structure (sometimes referred to as E2 structure) of the Na+,K+-ATPase (PDB accession number 2ZXE, Ref. 30). Periodic boundary conditions were applied in all three directions. A neighbor list with a 1.3-nm cut-off was used for treatment of non-bonded interactions and was updated every 10 ps. The van der Waals interactions were switched off from 0.8 to 1.2 nm. The Particle Mesh Ewald method (31, 32) with a 1.3-nm cut-off was employed for electrostatic interactions. Simulated systems were maintained at 310 K and 1 bar, realizing the NpT statistical ensemble. Temperature coupling was realized using the Berendsen thermostat (33) for the equilibration phases, followed by the Nose-Hoover thermostat (34, 35) for the production runs, separately for the solute (Na+,K+-ATPase, POPC, and ADP) and the solvent (water and ions). A semi-isotropic pressure coupling was applied with the Berendsen barostat for equilibration phases and Parinello-Rahman barostat (36) for the production runs. All systems were minimized with 5000 steps of steepest decent algorithm, followed by 10-ns equilibration and subsequent 50-ns production run. Trajectories were sampled every 10 ps. The analysis was carried out using GROMACS suite programs and homemade scripts. Visualizations and snapshots were rendered with visual molecular dynamics (37). The computations were done at the University of Southern Denmark node (Horseshoe) of the Danish Center for Scientific Computing and the Joint Nordic Supercomputer in Iceland, Gardar.
RESULTS
K+ and Organic Cation Dependence of ATPase Activity
According to the Post-Albers scheme, the dephosphorylated enzyme exposes inward facing sites selective for Na+, whereas the phosphorylated enzyme exposes outward facing sites selective for K+. We utilized ATPase assays to compare K+ and organic cations Acet+ and Form+. Fig. 2 depicts ATPase assays in the presence of increasing concentrations of K+/congener, measured at four different concentrations of Na+. Increasing K+ strongly inhibited enzyme activity owing to K+ competition with cytoplasmic Na+ for the inward facing sites (Fig. 2A). The inhibition is reversed by high Na+ concentrations that produce a shift to the E1 form. On the other hand, high concentrations of Acet+ did not inhibit ATPase activity even at a Na+ concentration of 5 mm (Fig. 2B). However, ATPase activity decreased with increasing Na+ concentration. The decrease in activity at higher Na+ concentrations is likely due to saturation of the outward facing sites, competing with the forward interaction of the organic cation and producing inhibition of ATP hydrolysis. Form+ produced higher ATPase activities than Acet+ but inhibited the ATPase activity at 5 mm Na+ (Fig. 2C), implying that Form+ interacts with the extracellular sites with a higher affinity than Acet+, but also inhibits Na+ stimulation of the enzyme to some extent. Chloroacetamidinium (Cl-Acet+), containing a chloride ion replacing one of the methyl group hydrogens (Fig. 1) did stimulate ATPase activity to a much lower level (∼20% of that obtained with Acet+, data not shown). Guanidinium (Gua+), methyl guanidinium (M-Gua+), dimethyl guanidinium (diM-Gua+), and N-methyl-d-glucamine (NMG+) all inhibited Na+,K+-ATPase activity by competing with K+ (data not shown). These data indicate that Cl-Acet+ is the maximum allowed size that can function as a pump substrate. Hence, although Acet+ and Form+ function as surrogates for K+ (12), they interact differently with the pump than K+.
FIGURE 2.
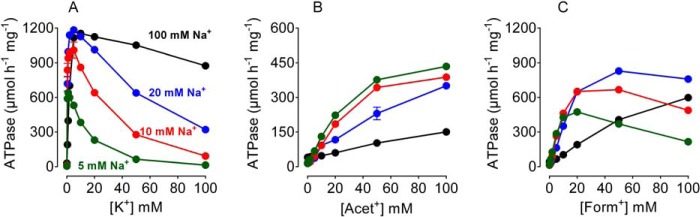
ATPase activity in the presence of K+/congeners. Ouabain-dependent ATPase activity was measured at 37 °C in the presence of 3 mm MgATP, 30 mm histidine buffer, pH 7.2, 2 μg of protein, and the indicated concentrations of K+, Acet+, or Form+. All reactions were performed in the presence of four different Na+ concentrations as indicated. Data are mean ± S.E. (smaller than symbol sizes) of four measurements. A representative of three independent measurements is shown.
Acet+ and Form+ Interact Exclusively with the Outward Facing Sites of the Na+,K+-ATPase
The K+ occluded form of the Na+,K+-ATPase (E2K+) produces ouabain-sensitive hydrolysis of pNPP, referred to as K+-phosphatase or pNPPase activity (38, 39). In the presence of Mg2+, low affinity K+ binding to the inward facing sites in the dephosphorylated E1 form (reverse or direct route, E1 → E2K+ reaction) or high affinity K+ binding to the outward facing sites in the phosphorylated E2 form (forward or physiological route, E2P → E2K+) produces pNPP hydrolysis. Increased pNPP hydrolysis reflects augmentation of K+ interaction (i.e. binding and release) with the pump (38). Thus, pNPPase provides information on a few steps among several different steps in the complete reaction cycle of the enzyme. In addition, pNPPase assays allow the estimation of the orientation of the open ion binding sites in unsided membrane preparations (39).
We have investigated the interaction of Acet+ and Form+ with Na+,K+-ATPase, employing pNPPase assays. Fig. 3 shows results of pNPPase measured in the presence of the different ions. The dephosphorylated enzyme produced K+-phosphatase activity following K+ binding to the inward facing sites (Fig. 3A). Acet+ was unable to produce pNPP hydrolysis, showing its inability to enter the inward facing sites and induce transition to the pNPP-hydrolyzing E2 form (Fig. 3B). Form+ produced negligible pNPPase at higher concentrations (Fig. 3C). The experiments were now performed using enzyme phosphorylated from ATP (E2P-ATP, see “Experimental Procedures”). This conformation exposes outward facing high affinity K+ sites. As seen in Fig. 3D, maximum K+-pNPPase was achieved at lower K+ concentrations (compare with Fig. 3A). This increased sensitivity indicates the presence of the E2P form. Interestingly, Acet+ (Fig. 3E) and Form+ (Fig. 3F) produced appreciable pNPP hydrolysis, showing that they fit into the outward sites and become occluded. Thus, the outward facing ion binding cavity should be broader than the inward facing sites enough to accommodate the large organic cations. Several control experiments were performed to ensure the contribution of E2P to pNPP hydrolysis in this set of experiments. 50 μm ATP together with 5 mm Na+ was added in the assay medium to stabilize the E2P form. Experiments in which the enzyme was preincubated in phosphorylation media lacking either Na+ or ATP showed no activation by Acet+ or Form+. Thus, Acet+ and Form+ interact almost exclusively with the outward facing sites of the pump.
FIGURE 3.

Ion-dependent pNPPase activity. pNPPase assays were performed in the presence of 50 mm Tris-HCl, 7 mm pNPP, 7 mm MgCl2, 5 μg of protein, and the indicated concentrations of ions. Panels A–C depict activity of the dephosphorylated enzyme (intracellular) in the presence of the indicated concentrations of K+ (A), Acet+ (B), or Form+ (C). Panels D–F depict activity of enzyme phosphorylated from ATP (E2P-ATP), in the presence of the indicated concentrations of K+ (D), Acet+ (E), or Form+ (F). Panels G–I depict activity of enzyme phosphorylated from inorganic phosphate (E2P-Pi), in the presence of the indicated concentrations of K+ (G), Acet+ (H), or Form+ (I). Data are presented as μmol of pNPP h−1 (mg protein)−1 and are mean ± S.E. of three measurements. Closed squares, circles, diamonds, and triangles (superimposed) indicate pNPPase activity measured in the presence of the same concentrations of NMG, Gua+, M-Gua+, and diM-Gua+, respectively. A representative of four independent measurements is shown.
Even in the presence of substrates expected to stabilize a given pump conformation, P-type pumps are known to undergo conformational fluctuations, slipping to other conformations (40). Thus, in the experiments depicted in Fig. 3, D–F, we do not mean to imply that all the pumps in the assay are stabilized in the E2P form. The incubation conditions ensure the presence of a considerable amount of the E2P form having the binding sites exposed to the extracellular medium, as indicated from the increase in the apparent K+ affinity as well as the lack of effect upon preincubation in conditions not allowing enzyme phosphorylation.
The experiments were now performed using enzyme phosphorylated from inorganic phosphate (E2P-Pi, see “Experimental Procedures”). As revealed by pNPPase assays, the E2P form produced by phosphorylation from ATP (Fig. 3D) or from inorganic phosphate (Fig. 3G) interact similarly with K+. A conspicuous observation is the inability of Acet+ and Form+ to produce pNPP hydrolysis in the case of enzyme phosphorylated from inorganic phosphate. The small fraction of pNPPase observed in the presence of Form+ (Fig. 3I) is likely due to Form+ interaction with the inward facing sites (Fig. 3C). Thus, the large difference between results in Fig. 3, E and F, and H and I, indicates that Na+ and/or the adenine nucleotide are important for configuring the outward facing sites, to be available to the organic K+ congener. A role for ADP in opening the luminal gate of sarcoplasmic reticulum Ca2+-ATPase during the E1P → E2P transition has been proposed (41). Only ions that are occluded in the K+ sites produce pNPP hydrolysis. This was confirmed by experiments showing the lack of activity in the presence of non-occluded ions (Fig. 1). Thus, Gua+, M-Gua+, diM-Gua+, and NMG+ did not produce pNPP hydrolysis under the different conditions (Fig. 3, A, D, and G).
To obtain explicit evidence for ion interaction with the inward facing sites, the quaternary organic amine benzyl triethylammonium (BTEA) was used. BTEA blocks outward facing sites thus inhibiting forward K+ interaction with the pump, but is not occluded in the binding site (42, 43). The pump does not catalyze any pNPP hydrolysis in the presence of BTEA concentrations up to 200 mm (data not shown). Hence, ion-mediated pNPPase activities obtained in the presence of high concentrations of BETA would reflect ion interaction with the inward facing sites in the pump. As seen in Fig. 4, K+-pNPPase activity was fully insensitive to BTEA, demonstrating that K+ is able to enter freely through the inward facing sites even when the external sites are blocked. In contrast, Acet+-pNPPase was almost completely inhibited by BTEA, whereas Form+-pNPPase was only ∼70% inhibited. Thus, Acet+ interacts exclusively with the outward facing sites, whereas Form+ mildly interacts with the inward facing sites. Importantly, shielding the external sites by BTEA did not seem to abrogate ion entrance to the inward facing sites, indicating that no major conformational changes take place. Recent crystallographic studies have shown that blocking the KcsA K+ channel by tetrabutylammonium occurs without inducing major structural changes (44), suggesting that quaternary organic amines may share a common mechanism of action.
FIGURE 4.
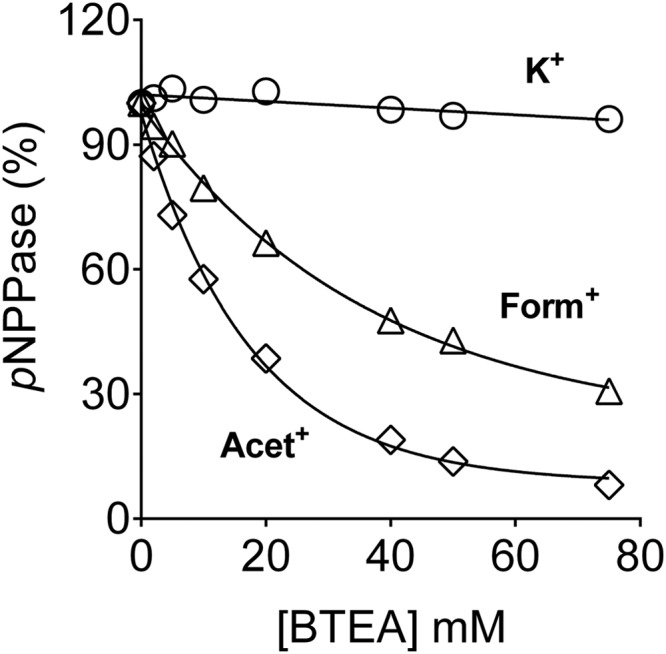
Effect of blocking the outward facing sites with BTEA on residual pNPPase activity. Assays were performed as described in the legend to Fig. 3, D–F, but in the presence of 25 mm K+, Acet+, or Form+, and the indicated concentrations of BTEA. Data are presented as percentage of control, measured in the absence of BTEA, and are mean ± S.E. of three measurements. A representative of three independent measurements is shown.
Acet+ and Form+ Poorly Antagonize Pump Stimulation by Cytoplasmic Na+
We have investigated cytoplasmic Na+/K+ selectivity by utilizing ATPase assays. Na+ activation of ATPase was measured in the presence of fixed concentrations of K+, Acet+, or Form+, and the calculated apparent affinities for Na+ (K′Na) were plotted against the K+/congener concentration (6). The slope of such a linear relationship describes how the apparent affinity of cytoplasmic Na+ changes with increasing K+/congener, providing information on cytoplasmic Na+/K+ selectivity. As shown in Fig. 5, the slope was 0.277 ± 0.01 mm for K+ and 0.017 ± 0.002 mm for Acet+. This 16-fold reduction indicates that Acet+ does not antagonize the cytoplasmic Na+ interaction as K+, consistent with its inability to interact with the inward facing sites (e.g. Fig. 3B). In the case of Form+, the curve was best fit to a sum of two components. At Form+ concentrations below 50 mm, the slope was 0.07 ± 0.01, whereas at Form+ concentrations >50 mm, the slope was 0.19 ± 0.01. It is likely that the antagonistic effect of Form+ on the cytoplasmic Na+ interaction dominates at higher Form+ concentrations, producing a biphasic increase in K′Na+. Further studies are required to understand this odd trend. The y intercept at x = 0 (KNao) denotes a hypothetical value describing Na+ affinity in the absence of the antagonistic effect of the counterion. KNao was 2.42 ± 0.4 mm for K+ and 0.036 ± 0.02 mm for Acet+. This strong difference demonstrates influence of the cytoplasmic ion antagonism on Na+ stimulation. Although we have used a model that describes inhibition of Na+ activation to ions that do not antagonize cytoplasmic Na+ binding, the outcome is informative, showing that the congeners do not compete with cytoplasmic Na+ activation, because of their restricted interaction with the inward facing sites.
FIGURE 5.
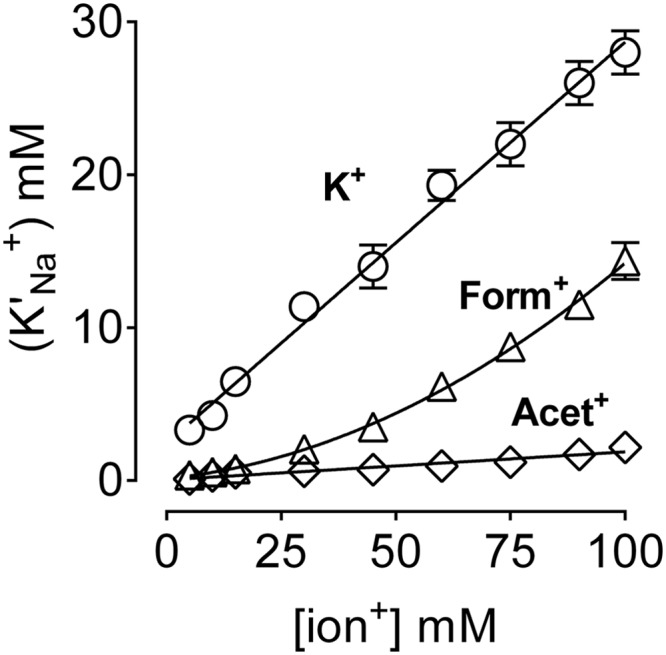
Kinetic analysis of inhibition of cytoplasmic Na+ activation. Na+ activation curves were performed in the presence of 50 mm Tris-HCl, pH 7.2, 3 mm MgATP, different K+/congener concentrations, and Na+ concentrations ranging from 0 to 100 mm. The apparent affinity of Na+ (K′Na+) was calculated as previously described (6) and plotted against the K+/congener concentration, as indicated. The curves of K+ and Acet+ were analyzed using the straight line equation: K′Na = KoNa + (KoNa/KK) (see text). Data are mean ± S.E. of four K′Na values.
Rotamer Transitions in the α-Subunit and the Role of Water
Previous MD simulations to the E2·MgF42−·2K+ structure indicated that Acet+ produces a minimum distortion of the binding site (12). CH3 in Acet+ and CH in Form+ are both unable to participate in hydrogen bonding. However, the low electronegativity of carbon would instead qualify binding via dispersion forces. This leads to the prediction that the outward facing binding sites in the E2 form contribute a van der Waals contact to the organic cation that is absent in the E1 form. Critical inspection of the two high resolution crystal structures of the sodium pump revealed that the hydroxymethyl group of Thr772 (Thr779 in shark) undergoes a backbone-independent rotamer transition (Refs. 18 and 30, see also Fig. 6). Thus, the side chain of Thr772 directs a hydroxyl group to site I in the E1 form, and a methyl group to the same site in the E2 form. In the E1 structure, a water molecule was shown to bind between the two Na+ ions in sites I and II (Ref. 18, see also Fig. 6A). Upon transition to the E2 form, water molecules seem to flood the binding site and the side chain of Thr772 is rotated, directing a methyl group to the hydrated K+ in site I (Ref. 30, see also Fig. 6B).
FIGURE 6.
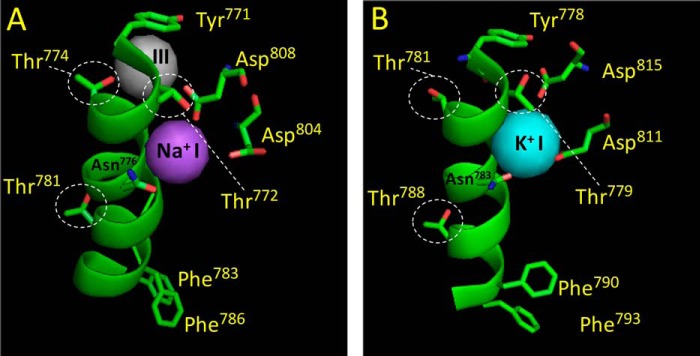
Rotamer transitions revealed by the high-resolution crystal structures of the Na+,K+-ATPase. Site I coordination in the E1·AlF4−·ADP·3Na+ structure (A, PDB code 3WGU, Ref. 18), and the E2·MgF42−·2K+ structure (B, PDB code 2ZXE, Ref. 30). The figure shows site I and the position of important M5 residues as well as the side chains of Thr781, Phe783, and Phe786 (kidney sequence), located near the extracellular surface. A, the hydroxyl group of Thr772 is coupled to a Na+ in site I. The hydroxyl group of Thr774 is contacting a Na+ in site III (shown in gray). The side chains of Phe783 and Phe786 bend toward the extracellular side. B, the methyl group of Thr772 is connected to a K+ in site I through water molecules. The side chain of Thr781 is disconnected from “empty site III.” The side chains of Phe790 and Phe793 (shark sequence) are moved toward the cytoplasm (compare with their position to that in the E1 structure). Note that the side chain of Thr781 (Thr788 in the shark sequence), located away from the cytoplasmic side, does not adopt rotamer transition, i.e. the side chain has the identical position in both structures. Note the close proximity of Asp808 (Asp815 in the shark sequence) to the Na+ in site I compared with its position in the E2 structure, this insertion (likely facilitated by the movement of M6) seems to regulate the traffic of water molecules into the binding cavity around site I. Note also that the side chain of Asn776 also seems to adopt a rotation, directing the side chain hydroxyl toward K+ in site I in the E2 structure (Asn783 in shark sequence). The figure was made using PyMOL.
To further investigate the interaction of Form+ and Acet+ with Na+,K+-ATPase, MD simulations to the E1 form were employed. The organic cations were placed in ion binding sites I and II in the crystal structure (see “Experimental Procedures”). After 50-ns simulations, we assessed the structural rearrangements in the systems containing Form+ (FORM) or Acet+ (ACET), compared with the Na+ system. Throughout the entire sodium simulations, Na+ ions occupied binding sites I and II (Fig. 7A) as seen in the E1 structure (18). However, the Na+ ion in site III was found to leave the position indicated in the structure and move slightly toward the formal charge of Asp926, an important Na+ affinity-determining residue. The stability of ion binding to selected residues that participate in ion coordination is evaluated by calculating the root mean square displacement with respect to the E1 structure (18), as summarized in Table 1.
FIGURE 7.
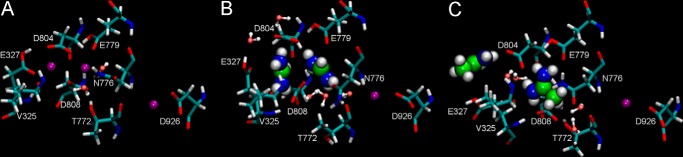
Last snapshots (t = 50 ns) from the simulated systems viewed from the cytoplasm: A, sodium; B, FORM; C, ACET. Bound sodium ions are shown as purple spheres. Note the deflection of the third Na+ ion toward Asp926, with respect to its location in the crystal structure (18). Ion binding residues are shown in licorice and organic cations as bigger spheres (carbon is shown in green, nitrogen in red, and hydrogen in white). Water molecules within the binding sites are shown as small red (oxygen)-white (hydrogen) spheres.
TABLE 1.
Root mean square displacement (RMSD) of the Na+,K+-ATPase α-subunit residues involved in ion binding in sites I and II, with respect to the crystal structure (18)
Calculated values are the average from the last 10 ns of simulations, with standard deviations given in parentheses.
| Residue | RMSD |
||
|---|---|---|---|
| Sodium | Form | Acet | |
| Å | |||
| Glu327 | 0.35 (0.09) | 0.97 (0.20) | 1.65 (0.19) |
| Glu779 | 0.36 (0.10) | 0.42 (0.10) | 0.58 (0.32) |
| Asp804 | 0.18 (0.05) | 0.79 (0.06) | 1.15 (0.03) |
| Asp808 | 0.29 (0.05) | 0.74 (0.16) | 0.66 (0.09) |
| Asn776 | 1.06 (0.07) | 1.23 (0.02) | 1.20 (0.10) |
| Thr772 | 0.31 (0.09) | 1.10 (0.07) | 0.29 (0.09) |
| Val322 | 0.36 (0.10) | 0.36 (0.11) | 0.39 (0.06) |
| Val325 | 0.47 (0.08) | 0.45 (0.10) | 1.48 (0.06) |
Ion binding residues are only marginally disturbed in the sodium system, indicating that the selected protonation state (protonated Glu327 and Glu779 and deprotonated Asp804 and Asp808) leads to the most stable system. The only exception is Asn776, which was found to have a rather high root mean square displacement value as shown by the simulations (1.06 ± 0.07, Table 1). Closer inspection revealed that the carboxamide side chain of Asn776 adopts a position (Fig. 7A) different from its position in the crystal (18). Thus, the carboxamide group is rotated in such a way that its nitrogen atom points firmly toward the sodium ion in site I, explaining the high root mean square displacement value. This unexpected rotation occurs quickly; it occurred within ∼10 ns and remained so to the end of the simulation time, suggesting that it is a naturally occurring event in the active pumping mechanism. Whether or not this unfavorable position could be part of the mechanism responsible for forward Na+ release from the high energy E1P(3Na+) state remains an open question. The Asn776 side chain rotation can clearly be captured by comparing the two high resolution crystal structures of the Na+,K+-ATPase (Refs. 18 and 30, and see also Fig. 6).
Insertion of large organic cations in sites I and II in the modified E1 structure distorts the optimal geometry of the ion binding sites. The representative snapshots from simulations are shown in Fig. 7, B and C. Besides Asn776, the most disturbed residues are Thr772, Glu327, Asp804, and Asp808 in the FORM system (Fig. 7B), and Glu327, Val325, Asp804, and Asp808 in the ACET system (Fig. 7C). In the FORM system, the hydroxymethyl side chain of Thr772 was found to undergo a 120° rotation; adapting now an orientation similar to the one observed in the E2·MgF42−·2K+ structure (30). This rotation is, at least partly, caused by water molecules. The water effect on rotation of the Thr772 side chain was revealed in the FORM system (see supplemental Movie S1). Water entered and broke the hydrogen bond between the side chains of Asp808 and Thr772. The Asp808 carboxyl group moves slightly toward site II, placing itself between the two bound Form+ molecules. The created space is now partially occupied by the Asn776 carboxamide group, whose oxygen interacts with the NH2 group of Form+. The presence of Form+ in site II destabilizes the interaction between Glu327 and Asp804 by breaking the hydrogen bond between them. However, during simulations, the Glu327 side chain rotates away from the binding site and a fair geometry is later partially rescued by a water molecule that locates itself between Glu327 and Asp804, allowing for extended hydrogen bonding (Fig. 7B).
Changes occurring in the ACET system are far more drastic. An Acet+ cation placed in site I is too big to accommodate, therefore it also partially occupies site II, effectively pushing the second Acet+ cation away. This motion opens the binding sites, compromising the hydrogen bond between Glu327 and Asp804, as well as changing the orientation of the Val325 side chain. Glu327 points now toward the cytoplasm, hence, site I is partially occupied by water molecules. The methyl group of Acet+ in site I repel the water, pushing the Asp808 carboxylic group toward the cytoplasm, and allowing it to hydrogen bond with Thr772. This architecture resembles that in the E1 structure (18). Therefore, lack of the Thr772 side chain rotation seems to be due to a preserved interaction between Asp808 and the Thr772 side chain hydroxyl. Changes in the hydration levels of Asp804 and Asp808 are measured with radial distribution functions between these residues and water molecules. The presence of the organic cations definitely increases the hydration of both of these residues (Fig. 8).
FIGURE 8.
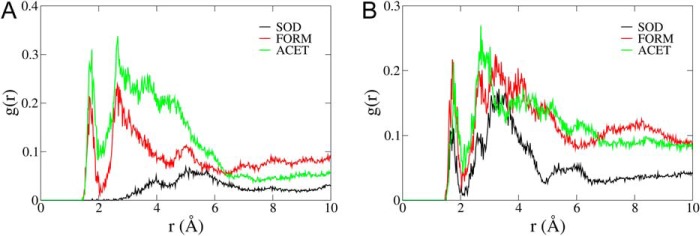
Radial distribution functions between (A) Asp804 and (B) Asp808 and water molecules. Both residues are significantly more hydrated in the presence of the organic cations.
Effect of Osmotic Stress on Ion Interaction and Membrane Domain Organization
Crystal structures showed that less water molecules are present in the ion binding cavity in the E1 structure (18) than in the E2 structure (30). MD simulations showed that water molecules are necessary to stabilize the organic cations in the binding sites (Fig. 7 and supplemental Movie S1). Initially, we anticipated that dehydration of the ion binding cavity would create space for the large organic cation to bind. To investigate the effect of dehydration on the interaction of ions with the pump, we utilized methodologies previously described by others reporting osmotic stress-mediated removal of water from membrane-embedded cavities in macromolecular systems (45, 46). Thus, we have studied the effect of osmotic stress on interaction of the K+/congener with the outward facing sites (E2P-ATP) using pNPPase assays. Interestingly, osmotic stress induced by increasing concentrations of glucose strongly increased Acet+- and Form+-pNPPase (Fig. 9A), but had no effect on K+-pNPPase (see also Ref. 47). Sucrose and polyethylene glycol 6000 (PEG 6000), at the same concentrations described in Fig. 9A, produced similar results but were less effective than glucose (data not shown). The use of such compounds that have widely different molecular mass indicated that stimulation of pNPPase is related to osmotic stress and not to solution viscosity. PEG 6000 has a large molecular mass and accordingly a lower osmotic stress compared with the same molar concentration of glucose. Hence, PEG 6000 produced the least stimulation of pNPPase, whereas glucose produced the most stimulation. Fig. 9B depicts the effect of dehydration on ion stimulation of pNPP hydrolysis, showing that dehydration increases the affinity of the organic cation as well as maximum pNPP hydrolysis. Remarkably, Cl-Acet+ now produced appreciable pNPP hydrolysis when present together with the dehydrated enzyme stabilized in the E2P form (Fig. 9B). This indicates that selectivity of the outward facing sites is compromised by dehydration. Dehydration did not produce pNPP hydrolysis in the presence of the non-occluded ions (data not shown).
FIGURE 9.

Dehydration facilitates organic cation interaction with the outward facing sites. A, assays were performed as described in the legend to Fig. 3, D–F, but in the presence of 25 mm K+/congener, 5 μg of protein (stabilized in the E2P form, see “Experimental Procedures”), and the indicated concentrations of d-glucose. B, assays were performed in the presence of 2 m glucose, and the indicated concentrations of ions. Open and closed diamonds indicate Cl-Acet+-mediated pNPPase activity in the presence or absence of glucose. A representative of four independent measurements is shown.
Evidence for dehydration-enhanced ion binding was obtained by studying the time dependence of dephosphorylation. Hence, phosphorylated enzyme (see “Experimental Procedures”) was diluted in medium containing ions with and without 15% glucose. As seen in Fig. 10, the rates of phosphoenzyme decay were 2.35 ± 0.11, 0.29 ± 0.04, and 0.35 ± 0.02 s−1 in the presence of K+, Acet+, and Form+, respectively. Dehydration was found to have no appreciable effect on the rate of dephosphorylation mediated by K+ or Form+. In contrast, dehydration induced a significant 100% increase in the rate of Acet+-mediated dephosphorylation. It should be mentioned that dehydration per se decreased the rate of spontaneous dephosphorylation measured in the absence of ions by more than 2-fold (data not shown), likely through reducing water activity with consequent retardation of phosphoenzyme hydrolysis. These results have two implications, 1) the effect of dehydration on Acet+-mediated dephosphorylation is underestimated by the decreased dephosphorylation induced by osmotic stress, and 2) dehydration also increases Form+ interaction with the outward facing sites (Fig. 9), but the effect is likely masked by a dehydration-mediated inhibition of phosphoenzyme hydrolysis (Fig. 10).
FIGURE 10.

Effect of ions and dehydration on the dephosphorylation rate. Pig kidney enzyme was phosphorylated as described under “Experimental Procedures.” Phosphorylated enzyme was diluted for the indicated time intervals in media containing K+, Acet+, or Form+, producing a final cation concentration of 1 mm (open circles), as indicated. Closed circles indicate dephosphorylation of the enzyme as above, but with diluting solution containing glucose, producing a final concentration of 15%. Data were analyzed using a monoexponential decay function, giving the indicated rate constants of phosphoenzyme hydrolysis for control (KCnt) or in the presence of glucose (KGlc).
To provide further evidence for the effect of dehydration on the interaction of ions with the binding sites, trypsin cleavage was used to probe membrane domain organization of the α-subunit. Incubation of membrane-bound enzyme at 37 °C in the presence of a high trypsin to protein ratio removes all cytoplasmic domains, leaving pairs of membrane domains connected with hairpins, as well as a C-terminal 19-kDa fragment, produced by cleavage at Asn831 (located in the intracellular loop L67), and contains intact M7M10 (48). Bound K+ tethers membrane domains together, leading to protection of the 19-kDa fragment from cleavage. On the other hand, in the absence of bound K+, the 19 kDa is further cleaved. Thus, an intact 19-kDa fragment is an indicator of ion binding to the K+ sites. In accord, membrane-bound Na+,K+-ATPase was incubated in the presence of 30 mm K+/congener with or without different glucose concentrations, followed by the addition of trypsin and incubation for a further 90 min at 37 °C. In the absence of glucose, the 19-kDa fragment was accumulated in the presence of K+, Acet+, and Form+, the ions that bind the shared sites in the pump (Fig. 11, A–C, lane 2). In the case of Cl-Acet, the amount of the 19-kDa fragment was almost absent, showing that Cl-Acet+ does not fit in the binding sites without dehydration (Fig. 11D, lane 2). However, mild dehydration (Fig. 11D, lanes 3–5) produced accumulation of the 19-kDa fragment, consistent with enhanced Cl-Acet+ binding induced by dehydration. Interestingly, increasing glucose concentrations substantially reduced the exposure of Asn831 to trypsin, decreasing accumulation of the 19-kDa fragment in the presence of the ions (Fig. 11, A–D, lanes 6 and 7). This occurs in concomitance with accumulation of a 75-kDa fragment. Thus, dehydration shields the major cleavage site at the intracellular L67 loop and exposes another site located near the N-terminal part. Dehydration does not produce significant changes in trypsin activity, as evidenced from cleavage of the soluble substrate Nα-tosyl-l-arginine methyl ester hydrochloride (data not shown). In addition, the 19-kDa fragment of the α-subunit is fully cleaved following incubation in the absence of ions and in the presence of 28.5% glucose (data not shown).
FIGURE 11.
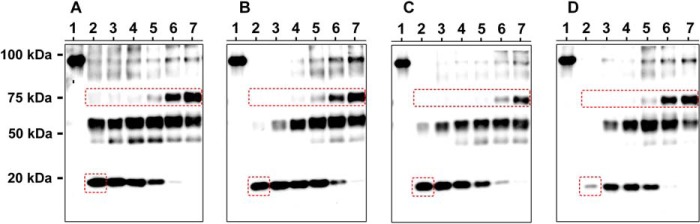
Exhaustive trypsin cleavage of the α-subunit in the presence of K+/congeners. Immunoblot showing proteolytic cleavage patterns of the α-subunit in the presence of the different ions, using an antibody against the C terminus of the α-subunit. Lanes 1 indicate controls where water replaced trypsin (intact α-subunit). Lanes 2–7 indicate proteolytic reactions performed in the presence of 0, 6.48, 12.24, 16.48, 22.24, and 28.48% glucose, respectively. The indicated approximate molecular weights of the fragments were estimated by using Bio-Rad precision plus protein standard, as indicated. Note the almost complete cleavage of the 19-kDa fragment in the case of Cl-Acet+ (D, red square) and the dehydration-mediated exposure of an N-terminal site in the α-subunit, producing a 75-kDa fragment (red rectangles). The intensity (normalized to that obtained with 30 mm K+) of the 19-kDa fragment in the absence of glucose is 100, 69 ± 5, 86 ± 6, and 3 ± 1% for panels A–D, respectively. A representative of three independent measurements is shown.
DISCUSSION
Hydration was previously shown to affect selectivity (49–51) and energetics (52) of ion channels. In addition, crystal structures of P-type ATPases indicated the participation of water molecules in ion coordination (18, 30, 53). In the E1·AlF4−·ADP·3Na+ structure of the Na+,K+-ATPase, insertion of the Asp808 (Asp815 in shark) carboxyl to site I, facilitated by Na+ binding to site III, was proposed to favor occupation of the site by a Na+ and not by a K+ (18). In the E2 structure, the interaction between Asp808 and K+ in site I is disturbed by several water molecules (30). Hence, the distance between the formal charge of Asp808 and site I seem to be the primary factor that determines which ion occupies site I (Fig. 6). Indeed, ion coordination by the side chain carboxyl is usually associated with breakage of the hydration shells around the ion, with consequent dehydration. Thus, the inability of Acet+ to enter into the binding site through the direct route is likely attributed to the insertion of Asp808, which narrows the space around site I in the E1 conformation. In the E2 structure, however, Asp808 is deflected away from the site, creating more space for binding of the organic cation, explaining why the organic cation enter the shared sites from the external side but not from the internal side. Our MD simulations to the E1 structure also provide insights on the inability of the organic cation to enter the inward facing sites (Fig. 3, B and C). Although both organic cations distorted the ion binding sites, Form+ produced much less distortion (Fig. 7B) and could fit in the binding sites. In particular, Form+ binding produces deflection of Asp808 and rotation of the Thr772 side chain, a similar arrangement to the E2 form (30). Apparently, Acet+ is unable to break the interaction between Asp808 and Thr772 and hence cannot interact with the inward facing sites. In addition, molecular size alone may qualify Form+ but not Acet+ to enter the inward facing sites (the predicted molecular volumes of Acet+ and Form+ are 60.85 and 44.15 Å3, respectively).
In this study we address the question of whether side chain rotamer transitions in the ion binding cavity in a membrane-bound pump play a role in ion selection and pumping. MD simulations revealed that water traffic in the binding site is associated with rotations of the Thr772 side chain (supplemental Movie S1). Threonine rotamer transitions are known to be regulated by solvation (54, 55). Rotation of the Thr772 side chain is likely to be a determinant of ion selectivity. The rotations revealed by the crystal structures are specific to Thr772 and Thr774. Thus, the side chain of Thr781 (Thr788 in shark), also located in M5 but closer to the extracellular side, has an identical position in both structures (Fig. 6). Interestingly, Thr772 and Thr774 are absolutely conserved in Na+,K+-ATPase and H+,K+-ATPase, the K+ transporting pumps, but are replaced by Leu and Ser in sarcoplasmic reticulum Ca2+-ATPase 1a, respectively. Side chain rotations would produce instantaneous changes in the molecular interaction type, from the relatively strong H-bond to the weaker van der Waals contact. The importance of the van der Waals contact in ion binding to the Na+,K+-ATPase was inferred from previous studies on site-directed mutants (56), showing that the methyl group of Thr772 is important in K+ binding. Although the authors suggested a role for the methyl group in direct interaction with the hydrophobic core of the membrane, the E2 structure clearly shows that the methyl group is contacting the water molecules that hold K+ in the binding sites (Fig. 6). Also, intriguing insights on the role of Thr774 was reported in the same study, showing that removal of the hydroxyl group (T774A) strongly decreased Na+ affinity (∼20-fold). The decrease in affinity was only mild (∼5-fold) in the T774S mutant (lacking a side chain methyl group but containing a hydroxyl group), showing a specific role for the hydroxyl group in determining cytoplasmic Na+ binding. On the other hand, in the T774A mutant, containing only a methyl side chain; the half-maximum concentration of K+ required to displace ATP (K+ binding by the direct route, E1 → E2K transition) was found to be 53% less in the case of the Ala mutant compared with the wild type enzyme (56). Thus, the presence of only a methyl group significantly enhanced K+ binding by the direct route. Further mutational studies on Thr772 and Thr774 are required to understand their function. The area around Thr774 has also been shown to be transiently hydrated in previous MD simulations studies (57). It is noteworthy that hydroxymethyl rotation has been reported to play important catalytic roles in pyranose 2-oxidase (58).
It is intriguing that stabilization of the organic cations in the ion binding cavity requires water molecules as revealed by the MD simulations (Fig. 7 and Supplemental Movie S1) yet dehydration by osmotic stress increased organic cation-mediated pNPPase (Fig. 9). We believe that dehydration removes the water from the external mouth of the ion binding cavity, thereby facilitating organic cation interaction. High osmotic stress would not remove water completely from the ion binding sites, as inferred from the presence of bound water molecules in protein crystals that are grown under severe osmotic stress (18, 30). A similar mechanism has been described for the K+ channel from Streptomyces lividans (46) where osmotic stress was shown to dehydrate an area located outside the selectivity filter, thereby controlling channel reactivation. Whereas osmotic stress unambiguously leads to dehydration of the Na+,K+-ATPase, our studies do not provide a quantitative measure of the amount or location of the dehydration. Dehydration inhibits steady state Na+,K+-ATPase activity as diffusion is expected to slow down in high osmotic stress. Dehydration likely affects several steps in the reaction cycle and it is necessary to measure partial reactions to determine which step in the reaction cycle is affected. Nonetheless, dehydration is expected to strongly retard the movements of the soluble cytoplasmic domains during conformational changes. Our pNPPase assays provide strong evidence that dehydration affects events occurring in the ion binding cavity. pNPP hydrolysis is not associated with pump phosphorylation and is likely to be mediated by direct interaction with the N-domain of the α-subunit. This may not require significant reorganization of cytoplasmic domain as is the case with ATP (which cross-links the N and P domains prior to phosphoryl transfer), likely explaining the insensitivity of the pNPPase reaction to the dehydration. ATPase assays have indicated that dehydration slightly increased cytoplasmic Na+ interaction with the pump (data not shown).
How to extrapolate the high osmotic stress used in this study to physiological conditions where osmotic stress is kept constant? Indeed, water interaction with the ion binding cavity does not necessarily require drastic changes in osmotic stress across the membrane. Solvation of the ion binding cavity may likely occur by transient exposure to the bulk medium during conformational transitions. In this regard, residues such as Phe783 and Phe786 may function to regulate communication of the binding cavity with bulk water (Fig. 6). Interestingly, studies on the pump mutant F786L (present in some cases of familial rapid-onset dystonia Parkinsonism) revealed the inability to bind cytoplasmic Na+ and strong inhibition by K+ without effects on the E1-E2 equilibrium (59), consistent with modified K+/Na+ selectivity. A large body of evidence indicates that the ion binding cavities in several pumps and secondary active transports accommodate a large number of water molecules than hitherto thought (60). Formation of water conducting states in membrane transporters have been recently suggested as being an inevitable imperfection associated with large-scale conformational transitions (61). Our study indicates that water flux into the binding site during conformational changes may play a catalytic role. Water molecules may function as “engine oil” to facilitate the task of formation and disruption of hydrogen bonds during the conformational changes necessary to switch ion selectivity. Indeed, water has been shown to accelerate the movements of hydrogen-bonded units in molecular nano-machines (29). Further biochemical and biophysical studies are required to understand how water interaction with the ion binding cavity of the Na+,K+-ATPase regulates enzyme function.
To conclude, we introduce Acet+ as the first K+ congener that does not compete with Na+ for the high affinity inward facing sites in the Na+,K+-ATPase. The inability of Acet+ to act as a competitive inhibitor of the cytoplasmic Na+ interaction was analyzed using the available crystal structures together with MD simulations, suggesting a pivotal role of Asp808 to solvate site I. Hydration regulates rotamer transitions of the side chain of absolutely conserved threonine residues in M5. Acet+ may be useful to investigate K+ selectivity of other transport proteins. Finally, our data highlight the relationship between the requirement of a van der Waals interaction in facilitating forward K+ binding and the susceptibility of the outward facing sites of the Na+,K+-ATPase to several unrelated organic molecules that either bind and permeate the pump (such as Acet+ and Form+), or fail to permeate and hence inhibit the pump by competing with K+ stimulation (such as BTEA). Further investigations in this direction are in progress.
Acknowledgments
We thank B. Roux and H. Yu for help in providing the parameters of the organic cations and Hanne Kidmose for excellent technical assistance.
This work was supported by a grant from the Novo Nordic Foundation (to Y. A. M.) and the Lundbeck Foundation (to H. K. and W. K.).

This article contains supplemental Movie S1.
- Acet+
- acetamidinium
- Cl-Acet+
- chloroacetamidinium
- diM-Gua+
- dimethyl guanidinium
- Form+
- formamedinium
- Gua+
- guanidinium
- M-Gua+
- methyl guanidinium
- NMG+
- N-methyl-d-glucamine
- POPC
- 1-palmitoyl,2-oleoyl-sn-glycero-3-phosphocholine
- BTEA
- benzyl triethylammonium
- MD
- molecular dynamics
- pNPP
- para-nitrophenyl phosphate.
REFERENCES
- 1. Kühlbrandt W. (2004) Biology, structure, and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 5, 282–295 [DOI] [PubMed] [Google Scholar]
- 2. Kaplan J. H. (2002) Biochemistry of the Na+,K+-ATPase. Annu. Rev. Biochem. 71, 511–535 [DOI] [PubMed] [Google Scholar]
- 3. Geering K. (2008) Functional roles of Na+,K+-ATPase subunits. Curr. Opin. Nephrol. Hypertens. 17, 526–532 [DOI] [PubMed] [Google Scholar]
- 4. Jorgensen P. L., Pedersen P. A. (2001) Structure-function relationships of Na+, K+, ATP, or Mg2+ binding and energy transduction in Na+,K+-ATPase. Biochim. Biophys. Acta 1505, 57–74 [DOI] [PubMed] [Google Scholar]
- 5. Yu H., Ratheal I. M., Artigas P., Roux B. (2011) Protonation of key acidic residues is critical for the K+ selectivity of the Na+/K+-pump. Nat. Struct. Mol. Biol. 18, 1159–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garay R. P., Garrahan P. J. (1973) The interaction of sodium and potassium with the sodium pump in red cells. J. Physiol. 231, 297–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Therien A. G., Nestor N. B., Ball W. J., Blostein R. (1996) Tissue-specific versus isoform-specific differences in cation activation kinetics of the Na+,K+-ATPase. J. Biol. Chem. 271, 7104–7112 [DOI] [PubMed] [Google Scholar]
- 8. Therien A. G., Blostein R. (1999) K+/Na+ antagonism at cytoplasmic sites of Na,K-ATPase: a tissue-specific mechanism of sodium pump regulation. Am. J. Physiol. 277, C891-C898 [DOI] [PubMed] [Google Scholar]
- 9. Hille B. (1971) The hydration of sodium ions crossing the nerve membranes. Proc. Natl. Acad. Sci. U.S.A. 68, 280–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hille B. (1971) The permeability of the sodium channel to organic cations in myelinated nerve. J. Gen. Physiol. 58, 599–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yaragatupalli S., Olivera J. F., Gatto C., Artigas P. (2009) Altered Na+ transport after an intracellular α-subunit deletion reveal strict external sequential release of Na+ from the Na+/K+ pump. Proc. Natl. Acad. Sci. U.S.A. 106, 15507–15512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ratheal I. M., Virgin G. K., Yu H., Roux B., Gatto C., Artigas P. (2010) Selectivity of externally facing ion-binding sites in the Na/K pump to alkali metals and organic cations. Proc. Natl. Acad. Sci. U.S.A. 107, 18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. González-Lebrero R. M., Kaufman S. B., Montes M. R., Nørby J. G., Garrahan P. J., Rossi R. C. (2002) The occlusion of Rb+ in the Na+, K+-ATPase: I. the identity of occluded states formed by the physiological or the direct routes: occlusion/deocclusion kinetics through the direct route. J. Biol. Chem. 277, 5910–5921 [DOI] [PubMed] [Google Scholar]
- 14. Mahmmoud Y. A., Shattock M., Cornelius F., Pavlovic D. (2014) Inhibition of K+ transport through Na+,K+-ATPase by capsazepine: role of membrane span 10 of the α-subunit in the modulation of ion gating. PLoS One 9, e96909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klodos I., Esmann M., Post R. L. (2002) Large-scale preparation of sodium-potassium ATPase from kidney outer medulla. Kidney Int. 62, 2097–2100 [DOI] [PubMed] [Google Scholar]
- 16. Baginsky E. S., Foà P. P., Zak B. (1967) Microdetermination of inorganic phosphate, phospholipids, and total phosphate in biological materials. Clin. Chem. 13, 326–332 [PubMed] [Google Scholar]
- 17. Mahmmoud Y. A., Cramb G., Maunsbach A. B., Cutler C. P., Meischke L., Cornelius F. (2003) Regulation of Na+,K+-ATPase by PLMS, the phospholemman-like protein from shark: molecular cloning, sequence, expression, cellular distribution, and functional effects of PLMS. J. Biol. Chem. 278, 37427–37438 [DOI] [PubMed] [Google Scholar]
- 18. Kanai R., Ogawa H., Vilsen B., Cornelius F., Toyoshima C. (2013) Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature 502, 201–206 [DOI] [PubMed] [Google Scholar]
- 19. Nyblom M., Poulsen H., Gourdon P., Reinhard L., Andersson M., Lindahl E., Fedosova N., Nissen P. (2013) Crystal structure of Na+, K+-ATPase in the Na+-bound state. Science 342, 123–127 [DOI] [PubMed] [Google Scholar]
- 20. Wolf M. G., Hoefling M., Aponte-Santamaría C., Grubmüller H., Groenhof G. (2010) g_membed: efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 31, 2169–2174 [DOI] [PubMed] [Google Scholar]
- 21. Kopec W., Loubet B., Poulsen H., Khandelia H. (2014) Molecular mechanism of Na+,K+-ATPase malfunction in mutations characteristic of adrenal hypertension. Biochemistry 53, 746–754 [DOI] [PubMed] [Google Scholar]
- 22. Berendsen H. J. C., van der Spoel D., van Drunen R. (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comp. Phys. Commun. 91, 43–56 [Google Scholar]
- 23. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. J. C. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 24. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 25. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd. (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 26. Bjelkmar P. R., Larsson P., Cuendet M. A., Hess B., Lindahl E. (2010) Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theo. Comput. 6, 459–466 [DOI] [PubMed] [Google Scholar]
- 27. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D., Jr., Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Damjanović A., García-Moreno E. B., Brooks B. R. (2009) Self-guided Langevin dynamics study of regulatory interactions in NtrC. Proteins 76, 1007–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Panman M. R., Bakker B. H., den Uyl D., Kay E. R., Leigh D. A., Buma W. J., Brouwer A. M., Geenevasen J. A., Woutersen S. (2013) Water lubricates hydrogen-bonded molecular machines. Nat. Chem. 5, 929–934 [DOI] [PubMed] [Google Scholar]
- 30. Shinoda T., Ogawa H., Cornelius F., Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4-Å resolution. Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 31. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 32. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 33. Berendsen H. J. C., Postma J. P. M., Van Gunsteren W. F., Dinola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 34. Hoover W. G. (1985) Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 35. Nose S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 [Google Scholar]
- 36. Parrinello M., Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Physiol. 52, 7182–7190 [Google Scholar]
- 37. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 38. Gatto C., Arnett K. L., Milanick M. A. (2007) Divalent cation interactions with Na,K-ATPase cytoplasmic cation sites: implications for the para-nitrophenyl phosphatase reaction mechanism. J. Membr. Biol. 216, 49–59 [DOI] [PubMed] [Google Scholar]
- 39. Drapeau P., Blostein R. (1980) Interaction of K+ with Na+,K+-ATPase: orientation of K+-phosphatase sites studied with inside-out red cell membrane vesicles. J. Biol. Chem. 255, 7827–7834 [PubMed] [Google Scholar]
- 40. Inesi G., Lewis D., Toyoshima C., Hirata A., de Meis L. (2008) Conformational fluctuations of the Ca2+-ATPase in the native membrane environment: effects of pH, temperature, catalytic substrates, and thapsigargin. J. Biol. Chem. 283, 1189–1196 [DOI] [PubMed] [Google Scholar]
- 41. Toyoshima C., Nomura H., Tsuda T. (2004) Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 432, 361–368 [DOI] [PubMed] [Google Scholar]
- 42. Forbush B. (1988) The interaction of amines with the occluded state of the Na,K-pump. J. Biol. Chem. 263, 7979–7988 [PubMed] [Google Scholar]
- 43. Peluffo R. D., González-Lebrero R. M., Kaufman S. B., Kortagere S., Orban B., Rossi R. C., Berlin J. R. (2009) Quaternary benzyltriethylammonium ion binding to the Na+,K+-ATPase: a tool to investigate extracellular K+ binding reactions. Biochemistry 48, 8105–8119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yohannan S., Hu Y., Zhou Y. (2007) Crystallographic study of the tetrabutylammonium block to the KcsA K+ channel. J. Mol. Biol. 366, 806–814 [DOI] [PubMed] [Google Scholar]
- 45. Parsegian V. A., Rand R. P., Fuller N. L., Rau D. C. (1986) Osmotic stress for the direct measurement of intermolecular forces. Methods Enzymol. 127, 400–416 [DOI] [PubMed] [Google Scholar]
- 46. Ostmeyer J., Chakrapani S., Pan A. C., Perozo E., Roux B. (2013) Recovery from slow inactivation in K+ channels is controlled by water molecules. Nature 501, 121–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Esmann M., Fedosova N. U., Marsh D. (2008) Osmotic stress and viscous retardation of the Na,K-ATPase ion pump. Biophys. J. 94, 2767–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karlish S. J., Goldshleger R., Stein W. D. (1990) A 19 kDa C-terminal tryptic fragment of the α chain of Na+/K+-ATPase is essential for occlusion and transport of cations. Proc. Natl. Acad. Sci. U.S.A. 87, 4566–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Noskov S. Y., Roux B. (2007) Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J. Gen. Physiol. 129, 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dudev T., Lim C. (2009) Determinants of K+ vs Na+ selectivity in potassium channels. J. Am. Chem. Soc. 131, 8092–8101 [DOI] [PubMed] [Google Scholar]
- 51. Dudev T., Lim C. (2010) Factors governing the Na+ vs K+ selectivity in sodium ion channels. J. Am. Chem. Soc. 132, 2321–2332 [DOI] [PubMed] [Google Scholar]
- 52. Morais-Cabral J. H., Zhou Y., MacKinnon R. (2001) Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature 414, 37–42 [DOI] [PubMed] [Google Scholar]
- 53. Sugita Y., Ikeguchi M., Toyoshima C. (2010) Relationship between Ca2+ affinity and shielding of bulk water in the Ca2+-pump from molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 107, 21465–21469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kroon-Batenburg L. M. J., Kroon J. (1990) Solvent effect on the conformation of the hydroxymethyl group established by molecular dynamics simulations of methyl-β-d-glucoside in water. Biopolymers 29, 1243–1248 [Google Scholar]
- 55. Kirschner K. N., Woods R. J. (2001) Solvent interactions determine carbohydrate conformation. Proc. Natl. Acad. Sci. U.S.A. 98, 10541–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pedersen P. A., Nielsen J. M., Rasmussen J. H., Jorgensen P. L. (1998) Contribution to Tl+, K+, and Na+ binding of Asn776, Ser775, Thr774, Thr772, and Tyr771 in cytoplasmic part of fifth transmembrane segment in α-subunit of renal Na,K-ATPase. Biochemistry 37, 17818–17827 [DOI] [PubMed] [Google Scholar]
- 57. Poulsen H., Khandelia H., Morth J. P., Bublitz M., Mouritsen O. G., Egebjerg J., Nissen P. (2010) Neurological disease mutations compromise a C-terminal ion pathway in the Na+,K+-ATPase, Nature 467, 99–102 [DOI] [PubMed] [Google Scholar]
- 58. Pitsawong W., Sucharitakul J., Prongjit M., Tan T.-C., Spadiut O., Haltrich D., Divne C., Chaiyen P. (2010) A conserved active site threonine is important for both sugar and flavin oxidations of pyranose 2-oxidase. J. Biol. Chem. 285, 9697–9705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rodacker V., Toustrup-Jensen M., Vilsen B. (2006) Mutations F785L and T618M in Na+,K+-ATPase, associated with familial rapid-onset dystonia Parkinsonism, interfere with Na+ interaction by distinct mechanisms. J. Biol. Chem. 281, 18539–18548 [DOI] [PubMed] [Google Scholar]
- 60. Zeuthen T. (2010) Water-transporting proteins. J. Membr. Biol. 234, 57–73 [DOI] [PubMed] [Google Scholar]
- 61. Li J., Shaikh S. A., Enkavi G., Wen P. C., Huang Z., Tajkhorshid E. (2013) Transient formation of water-conducting states in membrane transporters. Proc. Natl. Acad. Sci. U.S.A. 110, 7696–7701 [DOI] [PMC free article] [PubMed] [Google Scholar]