

Background: The catalytic rates for yeast Pol ϵ are unknown.
Results: The catalytic core and holoenzyme have comparable catalytic rates, but the loading onto primer termini differs.
Conclusion: The accessory subunits and C terminus of the catalytic subunit do not influence the catalytic rates.
Significance: The catalytic rates of Pol ϵ provide a benchmark for future mechanistic studies of leading strand synthesis.
Keywords: DNA Polymerase, DNA Repair, DNA Replication, Enzyme Catalysis, Enzyme Kinetics
Abstract
The holoenzyme of yeast DNA polymerase ϵ (Pol ϵ) consists of four subunits: Pol2, Dpb2, Dpb3, and Dpb4. A protease-sensitive site results in an N-terminal proteolytic fragment of Pol2, called Pol2core, that consists of the catalytic core of Pol ϵ and retains both polymerase and exonuclease activities. Pre-steady-state kinetics showed that the exonuclease rates on single-stranded, double-stranded, and mismatched DNA were comparable between Pol ϵ and Pol2core. Single-turnover pre-steady-state kinetics also showed that the kpol of Pol ϵ and Pol2core were comparable when preloading the polymerase onto the primer-template before adding Mg2+ and dTTP. However, a global fit of the data over six sequential nucleotide incorporations revealed that the overall polymerization rate and processivity were higher for Pol ϵ than for Pol2core. The largest difference between Pol ϵ and Pol2core was observed when challenged for the formation of a ternary complex and incorporation of the first nucleotide. Pol ϵ needed less than 1 s to incorporate a nucleotide, but several seconds passed before Pol2core incorporated detectable levels of the first nucleotide. We conclude that the accessory subunits and the C terminus of Pol2 do not influence the catalytic rate of Pol ϵ but facilitate the loading and incorporation of the first nucleotide by Pol ϵ.
Introduction
All eukaryotes have three replicative DNA polymerases (Pol2 ϵ, Pol δ, and Pol α) that are responsible for the synthesis of the leading and lagging strands during DNA replication (1–3). Pol α and Pol δ are the major DNA polymerases that replicate the lagging strand. Pol α synthesizes short primers that are extended by Pol δ in a cyclical manner to produce stretches of DNA known as Okazaki fragments (4). Under normal conditions, Pol ϵ is primarily responsible for the synthesis of the leading strand (5). Both Pol δ and Pol ϵ have an exonuclease domain that provides a proofreading function and allows the polymerases to replicate DNA with high fidelity (6).
Kinetic studies of prokaryotic, archaeal, and eukaryotic DNA polymerases have shown that they all follow the same basic mechanism when incorporating nucleotides into the nascent DNA strand (7, 8). The first step involves binding of the enzyme to the DNA, and the second step is the binding of a dNTP and two metal ions into the active site of the enzyme. The third step involves a conformational change from an open to a closed state that aligns the incoming dNTP, the 3′-OH of the nascent DNA strand, and the metal ions in a precise arrangement to allow phosphodiester bond formation. Following the transfer of the phosphoryl group to the growing DNA chain, a second conformational change occurs that allows for the release of pyrophosphate. The length of the DNA strand has now increased by one nucleotide. The DNA polymerase can either remain bound to the DNA and continue synthesis (called “processive synthesis”) or dissociate from the DNA and then bind again for the next nucleotide incorporation (called “distributive synthesis”) (9).
The eukaryotic B-family polymerases are composed of several subunits. Yeast Pol ϵ consists of four subunits, Pol2, Dpb2, Dpb3, and Dpb4 (10, 11). Pol2 can be further divided into two domains, the N-terminal catalytic domain and a C-terminal domain that interacts with Dpb2, Dpb3, and Dpb4. The catalytic domain of Pol2 contains the polymerase site for synthesizing DNA and a 3′–5′ exonuclease site that is responsible for proofreading the newly synthesized DNA (12, 13). We will refer to the catalytic domain (amino acids 1–1228) as Pol2core and to the holoenzyme with all four subunits as Pol ϵ for the remainder of this work. The recently solved high resolution structure of Pol2core (amino acids 1–1228) revealed a domain that is not found among other DNA polymerases (13). This so-called P domain allows Pol ϵ to encircle double-stranded DNA as the DNA leaves the polymerase active site, and this domain was shown to be important for the processivity and polymerase activity of Pol2core.
Pol ϵ replicates DNA with high fidelity. In general, it has been shown that the fidelity of replicative DNA polymerases is determined by three factors: the selection of correct nucleotides at the polymerase active site, the low likelihood to extend a mismatch, and the ability to excise the mismatch in the exonuclease site if incorporated. In vitro studies of Pol ϵ have suggested that the exonuclease activity increases the fidelity by 100-fold or more (14, 15). Mice deficient in the exonuclease activity of Pol ϵ were first shown to have increased incidence of tumors (16). It has now been established that mutations in POLE, which inactivates the exonuclease activity, are associated with cancer (17–20).
The exonuclease and polymerase rates for yeast Pol ϵ have not yet been reported. In order to better understand the mechanism of DNA synthesis by Pol ϵ, it is important to determine its various kinetic rate constants. This will expand our knowledge of how Pol ϵ maintains high fidelity and processivity inside the cell. In addition, the C-terminal domain of Pol2 and the three accessory subunits have previously been implicated in checkpoint activation, initiation of DNA replication, positioning of Pol ϵ on the leading strand, processive DNA synthesis, and replication fidelity (21–26). The processivity of Pol ϵ is likely to be influenced by amino acids located in the thumb domain and palm domains, the P domain, the Dpb3 and Dpb4 subunits, interaction with proliferating cell nuclear antigen, interactions with the CMG complex (the replicative helicase), and dNTP concentrations. However, the relative contributions of the different determinants are still unclear. In this work, we have explored the kinetic mechanisms of DNA synthesis by yeast Pol ϵ and yeast Pol2core to ask if the C terminus and the accessory subunits influence the exonuclease and polymerase rates and/or the loading of yeast Pol ϵ. We found that Pol ϵ has high polymerization and exonuclease rates as would be expected from a replicative polymerase. We also found that the C terminus and the accessory subunits do not influence the catalytic rates but do influence the processivity of polymerization and facilitate the loading of yeast Pol ϵ onto the DNA.
EXPERIMENTAL PROCEDURES
Purification of Proteins
Saccharomyces cerevisiae Pol ϵ exo−, Pol2core exo− (amino acids 1–1228), and Pol2core (amino acids 1–1228) were tagged with glutathione S-transferase (GST tag) at the N terminus and purified as described (13, 27) (Fig. 1). The GST tag was removed during the purification. The exonuclease-deficient variants, Pol ϵ exo− and Pol2core exo−, carry two amino acid substitutions, D290A and E292A, that are present in the previously studied pol2-4 allele (28). S. cerevisiae Pol ϵ was purified by conventional chromatography as described previously (29) (Fig. 1). All variants were expressed under the control of an inducible Gal1-10 galactose promoter in S. cerevisiae strain Py116. Proteins were typically purified from 90–120 liters of yeast grown in glycerol-lactate medium. After a final gel filtration step over a Sephadex 200 column, the purified proteins were concentrated to 5–17 mg/ml in a buffer containing 25 mm Hepes-NaOH (pH 7.6), 10% glycerol, 1 mm tris(2-carboxyethyl) phosphine hydrochloride, and 800 mm NaAC (pH 7.8).
FIGURE 1.
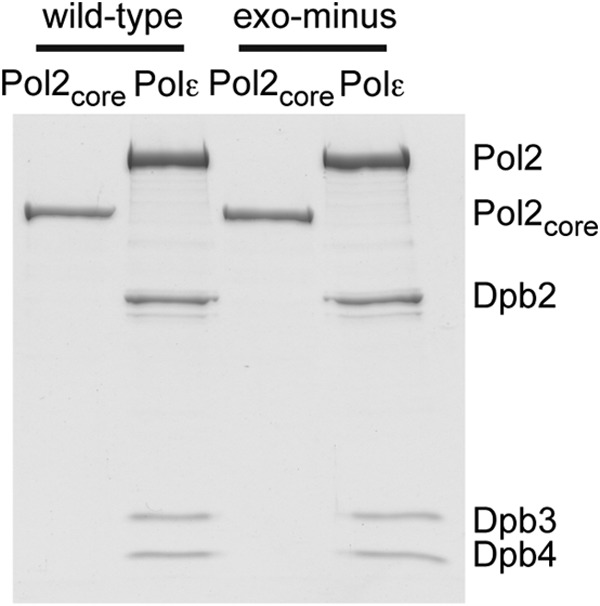
Protein preparations. 8 pmol of Pol ϵ and Pol2core (amino acids 1–1228), respectively, were resolved on 8% SDS-PAGE and stained with Coomassie Brilliant Blue. Exonuclease-deficient variants, Pol ϵ exo− and Pol2core exo−, carry two amino acid substitutions, D290A and E292A, that are present in the previously studied pol2-4 allele (28).
DNA Substrates
Oligonucleotides were purchased from MWG Operon (Ebersberg, Germany) and gel-purified before use. Primer-template duplexes were prepared by mixing 6 μm primer strand with 7.2 μm template strand in a buffer containing 100 mm Tris-HCl (pH 7.5) and 100 mm NaCl, heating to 85 °C for 5 min in a heating block, and slow cooling to room temperature.
Pre-steady-state Kinetics
All experiments were performed on a Rapid Quench QFM-400 (Bio-Logic, Claix, France) in RQ buffer containing 20 mm Tris-HCl (pH 7.8), 100 μg/ml bovine serum albumin (BSA), and 1 mm dithiothreitol (DTT) at 25 °C unless otherwise specified. A reaction mixture containing DNA, enzyme, and RQ buffer was loaded into syringe A, and a reaction mixture containing magnesium acetate, deoxyribonucleotides (dNTPs), and RQ buffer was loaded in syringe B of the rapid quench machine (specific reaction ingredients and concentrations are described below). Syringe C was loaded with water for washing, and syringe D was loaded with 3 m HCl to quench the reactions. Each time point consisted of mixing 20 μl from syringe A and 20 μl from syringe B in the reaction chamber, allowing the reaction to occur for the preset time, quenching with 20 μl of 3 m HCl, and collecting the reaction products from the exit line. The reaction products were neutralized with 100 μl of 1 m Tris-HCl (pH 8) and mixed with 150 μl of formamide containing 20 mm EDTA and 0.1% bromphenol blue. A total of 8 μl of this reaction mixture was loaded onto a 10% polyacrylamide gel containing 8 m urea and 25% formamide in 1× TBE (Tris/borate/EDTA) buffer. The gel was scanned with a Typhoon Scanner 9400 (GE Healthcare) at the Alexa 532-nm setting to excite the fluorophore (tetrachlorofluorescein) that was covalently bound to the 5′-end of the primer DNA. The band intensities were quantified with ImageQuant version 5.2 software (GE Healthcare), and relative intensities were calculated by dividing the intensity of a specific band by the total intensity of all observed bands. The data were analyzed and plotted with GraphPad Prism version 5.01.
Exonuclease Rates
Exonuclease rates were determined by loading syringe A with 400 nm enzyme, 38 nm DNA, and 1 mm EDTA in RQ buffer and syringe B with 16 mm magnesium acetate in RQ buffer. The final concentrations of reactants were 200 nm enzyme, 19 nm DNA substrate, 0.5 mm EDTA, and 8 mm magnesium acetate. The data were fit to the following equation.
Polymerase Rates
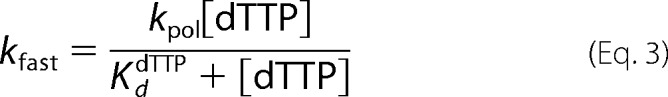
Polymerase rates were determined by loading syringe A with 400 nm enzyme, 38 nm primer-template (50/80-mer), and 1 mm EDTA in RQ buffer and loading syringe B with 16 mm magnesium acetate and dTTP in RQ buffer. The final concentration of the reactants was 200 nm enzyme, 19 nm primer-template, 0.5 mm EDTA, and 8 mm magnesium acetate. The final concentration of dTTP ranged from 0.5 to 500 μm to measure the dependence of nucleotide concentration on the rate of incorporation. Data were fit to the following equations.
 |
Processivity Assay and Polymerase Rates over Five Nucleotides
Syringe A was loaded with 60 nm enzyme, 100 nm DNA (50/80-mer), and 1 mm EDTA in RQ buffer, and syringe B was loaded with 16 mm magnesium acetate and 2× physiological dNTP concentrations (dCTP = 78 μm, dTTP = 132 μm, dATP = 44 μm, and dGTP = 22 μm) in RQ buffer. The final concentrations of reactants were 30 nm enzyme, 50 nm DNA, 0.5 mm EDTA, and estimated in vivo dNTP concentrations (dCTP = 39 μm, dTTP = 66 μm, dATP = 22 μm, and dGTP = 11 μm) (30). Data were analyzed using the global fit functions in the KinTek software (Kintek Corp.).
Loading of the 3′ Terminus of the Primer into the Active Site
Syringe A was loaded with 80 nm enzyme in RQ buffer, and syringe B was loaded with 38 nm DNA, 16 mm magnesium acetate, and 100 μm dTTP in RQ buffer. The final concentrations of the reactants were 40 nm enzyme, 19 nm DNA, 8 mm magnesium acetate, and 50 μm dTTP. The band intensities of the reaction products were quantified with ImageQuant version 5.2 software (GE Healthcare), and relative intensities were calculated by dividing the intensity of a specific band by the total intensity of all observed bands. The data were analyzed and plotted with GraphPad Prism version 5.01.
Active Site Titration
A preincubated solution of Pol ϵ exo− (65 nm) or Pol2core exo− (65 nm) and the indicated concentrations of 50/80-mer (5–150 nm) was rapidly mixed with a solution containing 100 μm dTTP and magnesium acetate. The reactions were quenched after 50 ms with 3 m HCl, neutralized with Tris-HCl (pH 8), and mixed with formamide dye before loading onto the sequencing gel. The products formed were plotted in the quadratic equation given below (31).
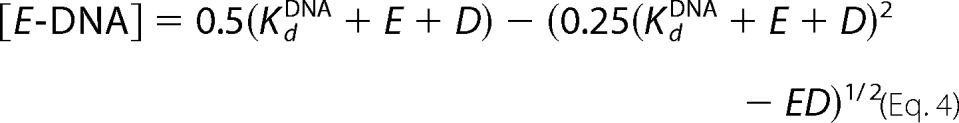 |
KdDNA represents the equilibrium dissociation constant for the binary complex (E-DNA), D is the DNA concentration, and E is the active enzyme concentration. The concentrations given are final.
RESULTS
The Exonuclease Rate of Pol ϵ
The high fidelity of Pol ϵ is in part due to its 3′–5′ exonuclease activity, which allows for efficient removal of misincorporated nucleotides (6, 14). To examine the kinetics of the exonuclease reaction and to clarify whether the accessory subunits and/or C terminus of Pol ϵ influence the exonucleolytic removal of the 3′-terminal nucleotide, we used four different substrates: single-stranded DNA, a perfectly primed template, and primer-templates ending with either one or two mispaired nucleotides at the 3′ terminus of the primer (Table 1). Exonuclease rates were determined by preincubating the respective DNA substrate with either saturating concentrations of wild-type Pol2core or Pol ϵ and then initiating the reaction by mixing the enzyme-DNA complexes with Mg2+. Loss of the 3′-nucleotide from single-stranded DNA, primed template, single-mismatched DNA, and double-mismatched DNA was quantified and plotted against time (Fig. 2). The resulting curves were fitted to a biphasic decay equation (Equation 1) with a fast phase and a slow phase. It has previously been suggested that the slow phase is observed due to the primer terminus switching between the exonuclease and polymerase active sites of replicative DNA polymerases or the binding of free polymerase to DNA (32–35).
TABLE 1.
Oligonucleotides used in the primer extension assays
| Oligonucleotide | Sequence |
|---|---|
| 51T-mer | 5′-GATCAGACTGTCCTTAGAGGATACTCGCTCGCAGCCGTCCACTCAACTCATT-3′ |
| 50/80-mer | 5′-GATCAGACTGTCCTTAGAGGATACTCGCTCGCAGCCGTCCACTCAACTCA-3′ |
| 3′-CTAGTCTGACAGGAATCTCCTATGAGCGAGCGTCGGCAGGTGAGTTGAGTAGGTCTTGTTGCAGTGACTGATAGTTCGAC-5′ | |
| 51T/80-mer | 5′-GATCAGACTGTCCTTAGAGGATACTCGCTCGCAGCCGTCCACTCAACTCATT-3′ |
| 3′-CTAGTCTGACAGGAATCTCCTATGAGCGAGCGTCGGCAGGTGAGTTGAGTAGGTCTTGTTGCAGTGACTGATAGTTCGAC-5′ | |
| 51TT/80-mer | 5′-GATCAGACTGTCCTTAGAGGATACTCGCTCGCAGCCGTCCACTCAACTCATTT-3′ |
| 3′-CTAGTCTGACAGGAATCTCCTATGAGCGAGCGTCGGCAGGTGAGTTGAGTAGGTCTTGTTGCAGTGACTGATAGTTCGAC-5′ | |
| 24/54-mer | 5′-GCTCGCAGCCGTCCACTCAACTCA-3′ |
| 3′-CGAGCGTCGGCAGGTGAGTTGAGTAGGTCTTGTTGCAGTGACTGATAGTTCGAC-5′ | |
| 12/18-mer | 5′-CCACTCAACTCA-3′ |
| 3′-GGTGAGTTGAGTAGGTCT-5′ |
FIGURE 2.
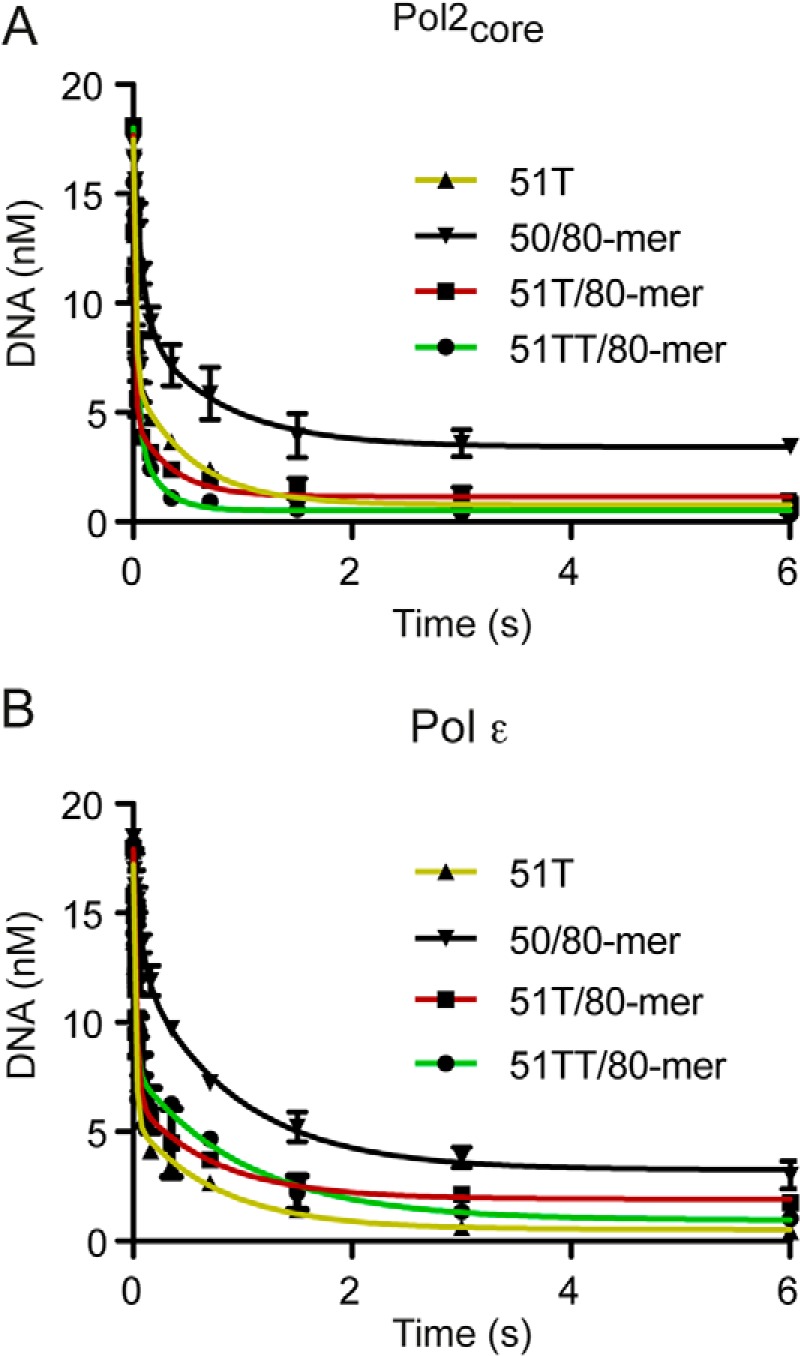
Exonuclease kinetics. Preformed enzyme-DNA complexes were rapidly mixed with Mg2+. The loss of primer was plotted as a function of time and fit to a biphasic decay equation to obtain rate constants for the fast phase. The rate constants obtained are given in Table 2. A and B, exonucleolytic degradation of DNA substrates by Pol2core and Pol ϵ, respectively. Shown are single-stranded DNA (51T) (yellow, ▴), correctly primed template (50/80) (black, ▾), and single mismatch at the primer end (51T/80) (red, ■). Green lines (●), exonucleolytic degradation of double mismatch at the primer end (51TT/80). Error bars, S.E.
We found that Pol ϵ has a Kfast of 49 s−1 and a Kslow of 1.2 s−1 when excising one nucleotide from 61 and 26%, respectively, of the single-stranded DNA (Table 2). Excising one nucleotide from a single-mismatched and double-mismatched DNA gave a Kfast of 33 and 46 s−1 when excising 61 and 49%, respectively. A correctly paired primer-end was removed at a slower rate, 11 s−1, and only about 33% of the substrate was removed during the fast phase. In contrast to the variation seen for the Kfast, the Kslow was measured to be about 1 s−1 for all four substrates. The fast phase was the major contributor to the exonuclease reaction, so only the rate obtained for the fast phase was considered below.
TABLE 2.
Kinetics of exonucleolytic degradation
| 51Ta | 50/80b | 51T/80c | 51TT/80d | |
|---|---|---|---|---|
| Pol2core | ||||
| kfast (s−1) | 47 ± 5 | 12 ± 3 | 65 ± 8 | 35 ± 3 |
| Afast (nm) | 11 | 8.8 | 12.9 | 13.5 |
| Pol ϵ | ||||
| kfast (s−1) | 49 ± 5 | 11 ± 2 | 33 ± 4 | 46 ± 8 |
| Afast (nm) | 11.6 | 6.2 | 11.6 | 9.3 |
a Single-stranded DNA.
b Correctly paired primer-template.
c Single mismatch at 3′-end of primer-template.
d Double mismatch at 3′-end of primer-template kfast represents the exonuclease rates of the fast phase, whereas Afast denotes the amplitude of the fast phase.
The observed rate constants for Pol2core and Pol ϵ were comparable except for the removal of a single mismatch in which the rate constant of Pol2core is greater than that of Pol ϵ (Table 2). This difference was not seen when the two last nucleotides in the primer were mismatched.
The Polymerase Rate and KddTTP of Pol ϵ
To measure the maximum rate of incorporation by Pol ϵ and the ground state affinity of dTTP for the binary complex of Pol ϵ and DNA (Table 1), we saturated the DNA substrate with enzyme (200 nm Pol ϵ and 19 nm DNA) and started the reaction by the addition of Mg2+ and dTTP ranging in concentration from 0.5 to 500 μm (final concentrations). The amount of product formed was plotted against time using a double-exponential equation with a fast phase and a slow phase for each dTTP concentration (Equation 2). As for the exonuclase reactions, the slower phase is probably observed due to switching of the primer terminus between the polymerase and exonuclease active sites or association of free Pol ϵ with DNA substrate. The single-turnover rates were determined for both the Pol2core exo− and Pol ϵ exo− enzymes (Tables 3 and 4). The fast phase was the major contributor to the polymerization reaction, and only the rate constants for the fast phase were plotted against dTTP concentration according to Equation 3 to calculate kpol and KddTTP (Fig. 3). The maximum polymerization rate constant (kpol) was ∼352 s−1, and the KddTTP was ∼25 μm for Pol2core exo−, and similar values of kpol ∼319 s−1 and KddTTP ∼21 μm were found for Pol ϵ exo− (Table 5). The similarities in kinetic constants between the two enzymes indicate that the accessory subunits of Pol ϵ do not influence the ground state affinity for dTTP or the maximal rate of polymerization when adding a single nucleotide.
TABLE 3.
kobs obtained at different dTTP concentrations for Pol ϵ exo−
| dNTP |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 μm | 1 μm | 2.5 μm | 5 μm | 7.5 μm | 10 μm | 15 μm | 20 μm | 30 μm | 50 μm | 75 μm | 100 μm | 200 μm | 500 μm | |
| kfast (s−1) | 4.1 ± 1.0 | 9.9 ± 1.6 | 27.6 ± 4.3 | 62.3 ± 6.1 | 91 ± 11 | 114 ± 20 | 131 ± 27 | 150 ± 26 | 194 ± 26 | 205 ± 21 | 246 ± 22 | 276 ± 35 | 291 ± 49 | 304 ± 42 |
| Afast (nm) | 3.9 | 4.8 | 5.6 | 6.4 | 6.8 | 6.8 | 8.2 | 7.8 | 9.0 | 9.4 | 9.8 | 9.5 | 10 | 11 |
| kslow (s−1) | 0.3 ± 0.1 | 0.5 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 1.0 | 0.6 ± 0.2 | 2.9 ± 1.0 | 1.6 ± 0.5 | 1.1 ± 0.3 | 1.3 ± 0.3 | 0.8 ± 0.2 | 2.9 ± 0.7 | 1.4 ± 0.6 | 1.0 ± 0.4 |
| Aslow (nm) | 5.5 | 5.7 | 5.7 | 5.5 | 5.1 | 5.0 | 4.6 | 4.4 | 4.3 | 3.9 | 3.7 | 3.8 | 3.3 | 3.0 |
TABLE 4.
kobs obtained at different dTTP concentrations for Pol2core exo−
| dNTP |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 μm | 1 μm | 2.5 μm | 5 μm | 7.5 μm | 10 μm | 15 μm | 20 μm | 30 μm | 50 μm | 75 μm | 100 μm | 200 μm | 500 μm | |
| kfast (s−1) | 5.6 ± 1.4 | 20.6 ± 7.9 | 44.6 ± 6.4 | 57. 3 ± 7.0 | 89 ± 11 | 126 ± 14 | 132 ± 21 | 153 ± 15 | 164 ± 18 | 232 ± 25 | 244 ± 31 | 312 ± 28 | 323 ± 38 | 325 ± 22 |
| Afast (nm) | 5.2 | 6.2 | 8.0 | 9.3 | 10.4 | 10.9 | 10.7 | 11.2 | 11.4 | 12.4 | 12.6 | 13.6 | 12.9 | 13.8 |
| kslow (s−1) | 0.5 ± 0.1 | 0.9 ± 0.3 | 1.6 ± 0.3 | 2.4 ± 0.5 | 1.2 ± 0.5 | 1.4 ± 0.5 | 6.7 ± 2.2 | 7.2 ± 0.7 | 9.5 ± 2.6 | 10.8 ± 3.0 | 6.2 ± 2.4 | 1.7 ± 0.7 | 10.3 ± 3.0 | 3.5 ± 1.0 |
| Aslow (nm) | 8.8 | 7.9 | 6.2 | 6.1 | 3.6 | 3.3 | 4.8 | 4.1 | 4.1 | 3.0 | 3.4 | 2.1 | 3.0 | 2.3 |
FIGURE 3.

Maximum rate of dTTP incorporation. Top, the amount of product formed was plotted against time using a double-exponential equation with a fast phase and a slow phase for each dTTP concentration. In the resulting plot, seven selected dTTP concentrations are shown as examples. Bottom, the observed rate constants for the fast phase were plotted as a function of dTTP concentration and fit to a hyperbola. Error bars, S.E. obtained from three independent experiments. The maximum rate constants obtained were kpol ∼352 s−1 and KddTTP ∼25 μm for Pol2 exo− and kpol ∼319 s−1 and KddTTP ∼21 μm for Pol ϵ exo−.
TABLE 5.
Comparison of polymerization rates with other polymerases
Elemental Effect of Nucleotide Incorporation
The rate-limiting step during DNA polymerization has been a matter of debate for some time (7, 36–39). One approach to identifying this step has been to compare the polymerization rate of an incoming normal nucleotide (dNTP) with that of a nucleotide that has replaced a non-bridging oxygen with sulfur on the α-phosphate of the incoming nucleotide ((Sp)-dNTPαS). The observed rate of incorporation has been shown to decrease by about 4–11-fold for the (Sp)-dNTPαS if the chemistry step is the rate-limiting step during polymerization (40, 41). It was previously shown that the elemental effect of (Sp)-dTTPαS incorporation by human Pol2core exo− (amino acids 1–1189) was 0.9, suggesting that the chemistry step was not rate-limiting (31). We carried out similar experiments with yeast Pol2core exo− in the presence of 15 μm dTTP or 15 μm (Sp)-dTTPαS (Fig. 4). We observed rate constants of 115 ± 22 s−1 and 68.4 ± 11 s−1 in the presence of dTTP and (Sp)-dTTPαS, respectively, to give an elemental effect of 1.6. The experiment was repeated with Pol ϵ exo− to determine whether the accessory subunits and the C terminus of Pol2 affect the chemistry step. The rate constants were 131 ± 20 s−1 for dTTP and 151 ± 21 s−1 for (Sp)-dTTPαS to give an elemental effect of 0.86 (Fig. 4), which may suggest that the chemistry step is not rate-limiting in the presence of all three accessory subunits and an intact Pol2 subunit.
FIGURE 4.
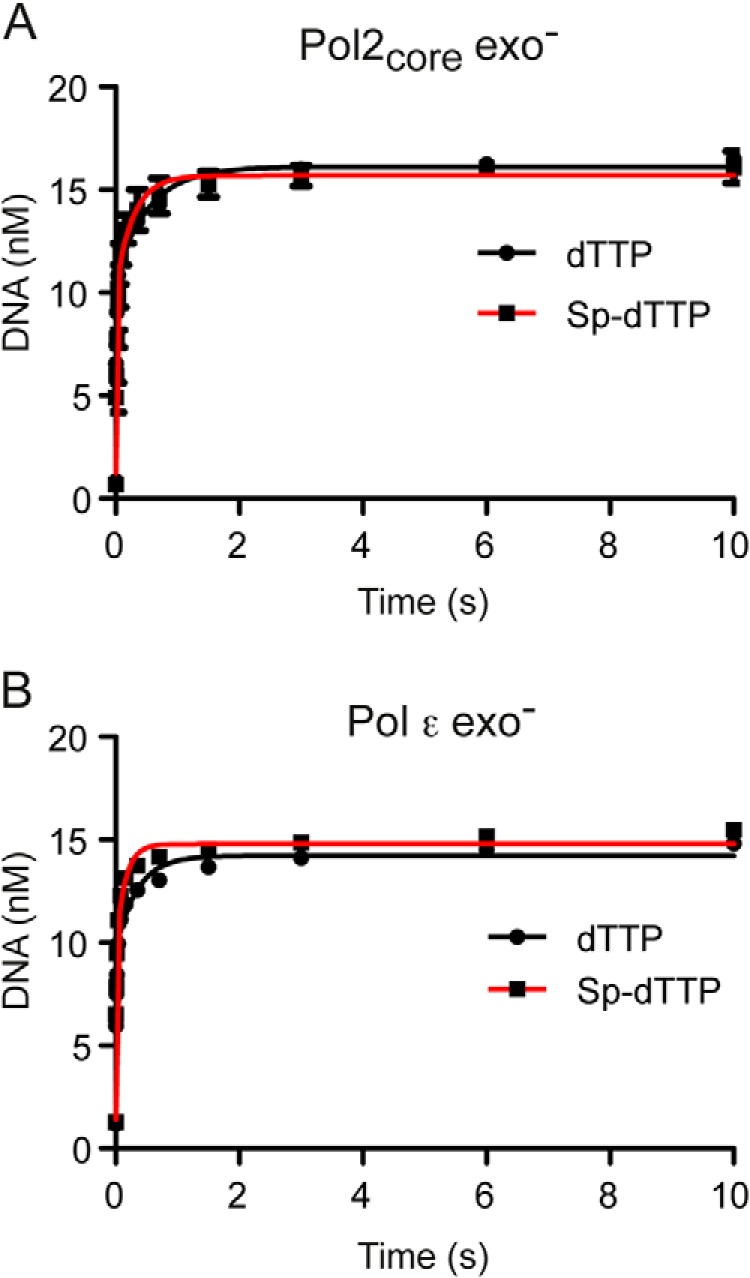
Elemental effect. Single-turnover nucleotide incorporation reactions were performed with Pol2core exo− (A) and Pol ϵ exo− (B) either in the presence of 15 μm dTTP (black line, ●) or in the presence of 15 μm (Sp)-dTTPαS (red line, ■). The amount of product formed is plotted against time and fit in a biphasic association equation. Mean values of the two independent experiments are plotted.
The Processive Polymerization Rate of Pol ϵ
The rate at which DNA polymerases incorporate multiple nucleotides in the nascent strand varies with the sequence context and is determined by multiple steps, including translocation along the DNA. To assess the rate of processive polymerization by Pol2core exo− and Pol ϵ exo−, we quantified the replication products from a primer extension reaction under single-turnover pre-steady-state conditions. We preloaded the DNA polymerase onto the DNA in the presence of excess DNA and started the reaction by the addition of what are considered to be physiological concentrations of dNTPs in yeast (dCTP = 39 μm, dTTP = 66 μm, dATP = 22 μm, and dGTP = 11 μm) (30). The reaction was quenched after 3–800 ms, and the replication products were separated on a denaturing polyacrylamide gel (Fig. 5A). The data were modeled using the global fit algorithm in the KinTek Explorer software (42) to determine the rates of polymerization and dissociation at the first six positions on the template (Fig. 5B and Table 6). The model fits the data based on the assumption that during sequential incorporation events, there is a kinetic partitioning between incorporation of the next nucleotide and dissociation from the DNA. The processivity of a polymerase is influenced by this partitioning. DNA polymerases become processive when elongation is favored over dissociation of the E-DNA complex. DNA polymerases may not be in a fully processive mode when adding the first nucleotide, which is why we only consider polymerization rates after the addition of the first nucleotide. We found that Pol2core exo− had an average polymerization rate of 176 s−1 over the second to fifth incorporated nucleotide and that Pol ϵ exo− had an average polymerization rate of 242 s−1. Both Pol2core exo− and Pol ϵ exo− were sufficiently processive to synthesize 30 nucleotides without dissociating from the template. It took Pol2core exo− about 0.1 s and Pol ϵ exo− about 0.04 s to synthesize equal amounts of 30-nucleotide product (Fig. 5), suggesting that the difference in polymerization rates between the two enzymes is maintained over longer distances than the first five nucleotides. The dissociation rates at each position on the template influence the processivity. We found that Pol2core exo− had an average dissociation rate of 4.6 s−1 over the first five nucleotides, whereas Pol ϵ exo− had an average dissociation rate of 5.1 s−1 (Table 6). In the case of Pol ϵ exo−, the average dissociation rate might be overestimated due to the much higher than average dissociation rate at the fifth position.
FIGURE 5.

Processive polymerization rates. A, multiple nucleotide incorporations were carried out to measure the time taken by Pol2core exo− and Pol ϵ exo− to incorporate 30 nucleotides under single-turnover conditions. Excess 50/80-mer (100 nm) was preincubated with enzyme (60 nm) and rapidly mixed with Mg2+ and physiological dNTP concentrations. B, the amount of remaining substrate (50-mer in red) and each intermediate product (51-mer in red, 52-mer in yellow, 53-mer in maroon, 54-mer in green, 55-mer in light blue, and 56-mer in black) were plotted with the KinTek global simulation software using a model defining the incorporation of the first six nucleotides and six DNA dissociation events. The polymerization rate constants obtained for Pol2core exo− and Pol ϵ exo− are listed in Table 6.
TABLE 6.
Kinetic parameters obtained by global fitting of processive polymerization
| Nucleotide | Rates of polymerization |
Dissociation rates |
||
|---|---|---|---|---|
| Pol2core exo− | Pol ϵ exo− | Pol2core exo− | Pol ϵ exo− | |
| s−1 | s−1 | |||
| 1st | 54 ± 1 | 92 ± 1 | 0.8 ± 0.48 | 1.00 ± 0.12 |
| 2nd | 151 ± 3 | 174 ± 4 | 4.4 ± 1.3 | 1.58 ± 0.20 |
| 3rd | 142 ± 5 | 175 ± 7 | 4.4 ± 0.39 | 2.04 ± 0.04 |
| 4th | 251 ± 35 | 345 ± 92 | 3.3 ± 0.29 | 3.79 ± 3.00 |
| 5th | 162 ± 4 | 275 ± 11 | 6.3 ± 0.50 | 13 ± 7.5 |
The Loading of Pol ϵ onto the 3′ Terminus of the Primer
The mechanism by which Pol ϵ loads onto the 3′ terminus of the primer-template is poorly understood. The high resolution structure of Pol2core revealed a domain that allows Pol ϵ to encircle the double-stranded DNA (13), but this P domain might also restrict the loading of Pol ϵ onto the 3′ terminus of the nascent strand because the active site of Pol ϵ is less accessible. Our kinetics experiments show that Pol2core is clearly able to bind to DNA with the 3′ terminus in the polymerase active site, but it is unclear whether the accessory subunits facilitate the loading of Pol ϵ onto the DNA. To test this, we synthesized oligonucleotide primer-templates of different lengths, including a 12/18-mer, a 24/54-mer, and a 50/80-mer (Table 1). Primer extension assays were performed in which the DNA polymerase was not preincubated with DNA. Instead, we added the polymerase to a complete mix with dNTPs and primer-template that allowed us to measure the time it took to load either Pol2core exo− or Pol ϵ exo− onto the 3′ terminus and extend the primer by at least one nucleotide (Fig. 6). We found that the time for loading onto the primer terminus and incorporation of the first nucleotide varied for the three different substrates. Both Pol2core exo− and Pol ϵ exo− were efficiently loaded onto the primer termini of the 12/18-mer, and extension products had already started accumulating by 100–200 ms. Pol2core exo− was slower to bind and extend the 24/54- and 50/80-mer compared with the 12/18-mer in the given time range. A barely detectable product appeared after 1.5 s on the 50/80-mer. Pol ϵ exo− also bound and extended the 24/54- and 50/80-mer with slower kinetics than the 12/18-mer, but in both cases, there was detectable product at 400 ms. As described above, when the polymerase is preloaded onto the primer-template DNA, the catalytic rates for the first incorporated nucleotide are similar for Pol2core exo− and Pol ϵ exo− (Table 5). Thus, the loading and extension of the first nucleotide appears to be facilitated by the C terminus of Pol2 and/or the accessory subunits when longer DNA substrates are used. The comparable rates on the short 12/18-mer are probably due to diffusion onto the ends of the short oligonucleotides, something that is suppressed as the oligonucleotides become longer.
FIGURE 6.
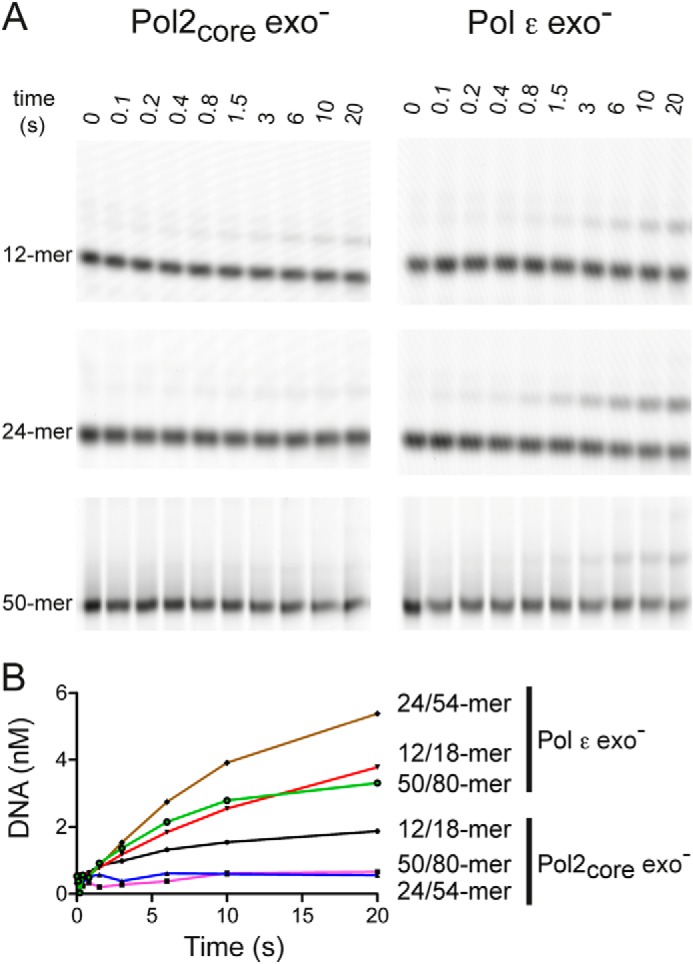
Loading of Pol ϵ onto the 3′-primer terminus. Primer extension reactions were carried out by rapidly mixing enzyme with DNA, Mg2+, and dTTP and then quenching the reactions at the time points indicated in A. dTTP incorporation was indicative of ternary complex formation and subsequent phosphodiester bond formation. Different lengths of DNA substrates were used to monitor the length dependence on the accessibility for loading the 3′-primer terminus into the polymerase active site and to extend the primer by one nucleotide. The relative band intensities of the incorporation of dTTP (B) were plotted against time for Pol2 core exo− and Pol ϵ exo−.
Active Site Titration
To ask whether the accessory subunits and C terminus of Pol2 affect the equilibrium dissociation constant of the DNA-Pol ϵ exo− binary complex, we carried out an active site titration. This experiment also determines whether both enzyme preparations (Pol2core exo− and Pol ϵ exo−) are equally active and if the differences observed in the loading experiments are not an artifact due to less active Pol2core exo−. DNA-polymerase complex dissociation is a slow process, and it is for that reason possible to estimate the active polymerase concentration and dissociation constant of a binary complex by examining the dependence of product formation on DNA concentration. To determine the KdDNA and active concentration of the respective enzymes, a 65 nm concentration of either Pol ϵ exo− or Pol2core exo− was preincubated with DNA (50/80-mer) ranging from 5 to 150 nm and rapidly mixed with 100 μm dTTP and magnesium acetate. The reactions were quenched after 50 ms, and the amount of product formed correlates with the active enzyme concentration. The products formed at each DNA concentration were plotted against the DNA concentration in a quadratic equation to calculate the dissociation constants of the binary complex and active enzyme concentration (Equation 4 and Fig. 7). For Pol2core exo−, the KdDNA was 15.6 ± 3 nm, and the active enzyme concentration was 45 ± 2 nm, which means that 70% of our Pol2core exo− preparation was active. The KdDNA for Pol ϵ exo− was 11.6 ± 2 nm, and the concentration of active enzyme was 52 ± 2 nm, which means that 80% of our Pol ϵ exo− preparation was active. These results show that both Pol2core exo− and Pol ϵ exo− are equally active. We conclude that the differences observed in the loading of Pol ϵ exo− and Pol2core exo− onto the 50/80-mer are inherent properties of the two variants and not an artifact observed because of inactive Pol2core exo− (Fig. 6).
FIGURE 7.
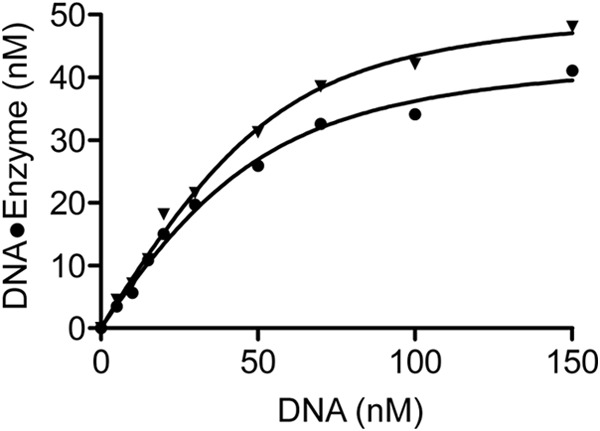
Active site titration. 65 nm Pol2core exo− (●) or Pol ϵ exo− (▴) was preincubated with different concentrations of DNA and rapidly mixed with dTTP and Mg2+ for 50 ms. The mean product formed was plotted against DNA concentration in a quadratic equation to determine the KdDNA of 15.65 ± 3 nm and active enzyme concentration of 45 ± 2 nm for Pol2core exo− and KdDNA of 11.6 ± 2 nm and active enzyme concentration of 52 ± 2 nm for Pol ϵ exo−. Values are S.E. obtained from three independent experiments.
DISCUSSION
The rate of DNA replication is determined by several factors, including the rate at which the helicase unwinds the DNA, the rates at which different protein complexes exchange positions on the leading and lagging strands, the concentrations of dNTPs, and the rate of the chemical reaction in which the DNA polymerase adds a nucleotide to the nascent strand. Here we have established kinetic properties of yeast Pol ϵ in the absence of other proteins that function as part of the replisome.
Fidelity measurements have shown that yeast Pol ϵ is highly accurate when incorporating deoxyribonucleotides during DNA replication (14, 44). Interestingly, Pol ϵ relies to a larger extent than Pol δ on its proofreading activity to achieve such high fidelity (14, 44). Here we determined the exonuclease rates of Pol2core and Pol ϵ on four different DNA substrates to explore whether the accessory subunits and/or C terminus of Pol2 may influence the exonuclease rate of Pol ϵ. Single-stranded DNA, duplex DNA with a single mismatch at the 3′ terminus, and duplex DNA with a double mismatch at the 3′ terminus were all degraded at a higher rate than a correctly paired primer-template. This was expected from measurements of other DNA polymerases with proofreading capacity and is dependent on the partitioning between the exonuclease site and polymerase site, which are located about 40 Å apart (3, 35). A comparison between the rates of Pol2core and Pol ϵ shows that the rates for removal of nucleotides from single-stranded DNA, a double-mismatched duplex, and a correctly paired primer-template were comparable between the two enzymes (Table 2). In contrast, Pol2core had an increased exonuclease rate when removing a single mismatch at the 3′ terminus compared with Pol ϵ. The small differences in exonuclease rates are in agreement with in vitro fidelity measurements on gapped substrates that showed that the two forms of Pol ϵ have comparable levels of fidelity (45). The exonucleolytic rate obtained for yeast Pol2core and Pol ϵ (∼11 s−1 at 25 °C) on a correctly base-paired primer-template are more than 10-fold higher than the reported value for the human Pol2core (0.86 s−1 at 37 °C) (46). The difference between the studies might in part be due to differences in the design of the experiments as well as to differences in sequence context. Both polymerase and exonuclease activities are influenced by the local DNA sequences contexts, which affect the base stacking interactions between adjacent bases and can alter the geometry of the duplex DNA. The DNA sequences present at the primer terminus junction can also influence the rate at which the two strands are separated and the rate at which the mismatched primer terminus can be positioned within the exonuclease active site (15, 47, 48). Our results suggest that yeast Pol ϵ has a very potent exonuclease activity even on a matched template, and there is only about a 3-fold difference between the removal of a mismatched and matched 3′ terminus. The determined rates of 33 and 11 s−1 may include the melting of DNA and transition of the 3′-end from the polymerase to the exonuclease site via an intramolecular mechanism because the dissociation rates during processive polymerization were determined to be 1–4 s−1. The observed dissociation rates also suggest that the observed Kslow for both the polymerase rate (0.1–3 s−1) and exonuclease rate (0.2–5 s−1) under pre-steady-state kinetics represents the association of free enzyme with DNA that is present at equilibrium during the reaction. In comparison, there is a 10- and 5-fold difference between the removal of a mismatched and matched 3′ terminus in exonuclease rates for T7 DNA polymerase and T4 DNA polymerase, respectively (33, 35). The measured polymerization rate of about 250 s−1 for yeast Pol ϵ is only about 25-fold higher than the determined exonuclease rate of a matched 3′ end. This should be compared with a >400- and >115-fold ratio for T4 and T7 DNA polymerases, respectively (33, 35, 40). Thus, it appears that the balance between the polymerase and exonuclease activity is shifted more toward the exonuclease site for yeast Pol ϵ, when compared with T4 and T7 DNA polymerase. These results also agree well with previous observations that Pol ϵ, even in the presence of 100 μm dNTPs, will degrade a portion of the matched primer during primer extension assays (29). Fidelity measurements have shown that 92% of base-base mismatches and 99% of single-nucleotide deletions made by yeast Pol ϵ are corrected by the associated exonuclease activity (14). Germ line mutations in the exonuclease domain of Pol ϵ are associated with colon cancer (17). In sporadic cancer, mutations in POLE are predominantly localized in the exonuclease domain and this reinforces the importance of proofreading by Pol ϵ (18–20, 49). It was recently shown that these exonuclease mutant polymerases have decreased exonuclease activity, which contributes to the mutator phenotype of the polymerase (20). The high exonuclease activity on double-stranded DNA could also allow Pol ϵ to participate in nascent strand excision during repair of mismatched DNA as previously suggested in genetic studies (50).
The polymerization rate of the replicative DNA polymerases is critical for the rate at which the replication fork can advance. It was previously reported that yeast Pol δ has an apparent kpol of about 1 s−1 for the chemical step, which is too slow, considering that the fork movement has been estimated to be ∼100 nucleotides s−1 (51, 52). The apparent kpol and KddTTP for yeast Pol2core exo− and Pol ϵ exo− were comparable with each other, with a kpol above 300 s−1. Similar values of kpol and KddTTP were recently determined for human Pol2core (15, 31). The kpol and KddTTP of Pol ϵ are also similar to kpol values reported for other polymerases, such as T4, T7, and RB69 gp43 (33, 40, 53) (Table 5). The KddTTP value of 21 μm obtained for the incorporation of dTTP indicates that the maximum rate of incorporation is achieved at half of the physiological dTTP concentration.
The measured catalytic rates suggest that the chemistry performed by Pol ϵ is not rate-limiting for replication fork progression. To make an overall assessment of the polymerization rate of Pol ϵ exo−, including presumed conformational changes and translocation steps, we measured the polymerization rates and dissociation rates over a distance of five nucleotides. We found that the processive polymerization rate varied from nucleotide to nucleotide, ranging from 142 to 345 s−1 (Table 6). This reflects the influence of the sequence context on the polymerization rate, and the measured values across a short distance of five nucleotides might not represent the average rate across an entire genome. The average rate was 176 s−1 for Pol2core exo− and 242 s−1 for Pol ϵ exo− from the second to fifth nucleotide. These rates are higher than the estimated fork rate of 100 nucleotides s−1 (51, 54). The difference in overall rate persists over 30 nucleotides because it takes Pol2core exo− longer to reach full-length products than it does for Pol ϵ exo−. Altogether, these results suggests that the fork rate is not limited by the synthesis of new DNA by Pol ϵ. The fork rate will instead be limited by the turnover of enzymes at the fork, the rate of unwinding by the helicase, or synthesis of DNA by another polymerase.
It was recently shown that in vivo nucleotide concentrations might influence the polymerization rate (55). For this reason, we chose to use physiological concentrations of dNTPs when measuring the polymerization rate and processivity. An arbitrary value to estimate the processivity of a DNA polymerase can be calculated by dividing the polymerization rate by the dissociation rate (56). Comparing Pol2core exo− and Pol ϵ exo− over five nucleotides in the presence of physiological concentrations of nucleotides showed that Pol ϵ exo− has a 47 times higher likelihood to incorporate another nucleotide versus to dissociate from the template. Pol2core exo− is limited to a 38 times higher likelihood to extend the 3′-end versus dissociate from the template. This is in agreement with previous reports suggesting that Dpb3 and Dpb4 contribute to the processivity of Pol ϵ (24, 29). It also supports previous reports showing that Pol2core has an intrinsically high processivity compared with Pol δ, presumably in part supported by the P domain (13).
It has been shown that Dpb3 and Dpb4 have affinity for double-stranded DNA (43), and this could contribute not only to the processivity but also to the loading of Pol ϵ. For this reason, we measured the time it took for Pol2core exo− and Pol ϵ exo− to form a ternary complex with the DNA and the incoming nucleotide and then to incorporate the nucleotide. The polymerization rates for Pol2core exo− and Pol ϵ exo− were comparable when a binary complex with DNA was established before the addition of dTTP (Table 5). In addition, the active site titration experiments showed that the amounts of active enzyme in the Pol2core exo− and Pol ϵ exo− preparations were comparable. Thus, any differences in the rate by which Pol2core exo− and Pol ϵ exo− load onto the DNA and incorporate one nucleotide should be due to differences in the rate at which a binary complex with DNA is formed, here defined as the loading of Pol2core exo− and Pol ϵ exo− onto the primer termini. We compared the ability to load and extend the primer on three oligonucleotide substrates of increasing length, and it was clear that the length of the primer-template influenced the loading and extension.
Both Pol2core exo− and Pol ϵ exo− were loaded with equal efficiency onto the shortest oligonucleotide substrates. However, as the oligonucleotide substrates became longer, the loading of Pol ϵ became much more efficient than on the smaller oligonucleotide substrates (Fig. 6B). In contrast, the loading of Pol2core exo− was almost negligible on longer oligonucleotide substrates. We conclude, therefore, that the C terminus of Pol2 and the Dpb2, Dpb3, and Dpb4 subunits facilitate the loading and extension of the primer. We hypothesize that the difference in loading onto different lengths of oligonucleotide substrates might be due to a steric clash between the DNA and the P domain and thumb domain and that conformational changes may be needed to allow Pol2core to load onto the 3′-primer terminus on longer oligonucleotide substrates. The loading of Pol ϵ on the longer substrates might be more efficient due to the presence of the accessory subunits and/or C terminus of Pol2 that may overcome this steric constraint. However, it takes Pol ϵ about 1.5 s to load and extend one nucleotide on the 50/80-mer substrate (Fig. 6). This is very slow, considering that estimates suggest that entire Okazaki fragments are synthesized and processed in under 1 s (57, 58). The loading of Pol ϵ might be faster in vivo due to the activities of other proteins, such as GINS in the preloading complex/CMG helicase in yeast and TIM-Tipin in human cells (25, 59, 60). Recent in vitro reconstitution experiments showed that CMG helicase selects Pol ϵ over Pol δ on the leading strand in yeast (61). Having established the kinetic properties of Pol ϵ, we are now prepared to explore how other proteins might influence the properties of Pol ϵ.
Acknowledgment
We thank Matthew Hogg, Ph.D., for technical assistance with the rapid quench experiments and for critically reading the manuscript.
This work was supported in part by grants from the Swedish Research Council, the Swedish Cancer Society, the Kempe Foundations, the Knut and Alice Wallenberg Foundation, and Insamlingstiftelsen at the medical faculty of Umeå Universitet.
- Pol
- polymerase
- exo−
- exonuclease-deficient
- (Sp)-dNTPαS
- 2′-deoxynucleoside 5′-O-(1-thiotriphosphate)
- (Sp)-dTTPαS
- 2′-deoxythymidine 5′-O-(1-thiotriphosphate)
- CMG
- Cdc45-Mcm2–7-GINS.
REFERENCES
- 1. Kunkel T. A., Burgers P. M. (2008) Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 18, 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burgers P. M. (2009) Polymerase dynamics at the eukaryotic DNA replication fork. J. Biol. Chem. 284, 4041–4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johansson E., Macneill S. A. (2010) The eukaryotic replicative DNA polymerases take shape. Trends Biochem. Sci. 35, 339–347 [DOI] [PubMed] [Google Scholar]
- 4. Nick McElhinny S. A., Gordenin D. A., Stith C. M., Burgers P. M., Kunkel T. A. (2008) Division of labor at the eukaryotic replication fork. Mol. Cell 30, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pursell Z. F., Isoz I., Lundström E. B., Johansson E., Kunkel T. A. (2007) Yeast DNA polymerase ϵ participates in leading-strand DNA replication. Science 317, 127–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morrison A., Sugino A. (1994) The 3′ → 5′ exonucleases of both DNA polymerases δ and ϵ participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol. Gen. Genet. 242, 289–296 [DOI] [PubMed] [Google Scholar]
- 7. Johnson K. A. (2010) The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rothwell P. J., Waksman G. (2005) Structure and mechanism of DNA polymerases. Adv. Protein Chem. 71, 401–440 [DOI] [PubMed] [Google Scholar]
- 9. Goodman M. F., Creighton S., Bloom L. B., Petruska J. (1993) Biochemical basis of DNA replication fidelity. Crit. Rev. Biochem. Mol. Biol. 28, 83–126 [DOI] [PubMed] [Google Scholar]
- 10. Hamatake R. K., Hasegawa H., Clark A. B., Bebenek K., Kunkel T. A., Sugino A. (1990) Purification and characterization of DNA polymerase II from the yeast Saccharomyces cerevisiae: identification of the catalytic core and a possible holoenzyme form of the enzyme. J. Biol. Chem. 265, 4072–4083 [PubMed] [Google Scholar]
- 11. Chilkova O., Stenlund P., Isoz I., Stith C. M., Grabowski P., Lundström E. B., Burgers P. M., Johansson E. (2007) The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic Acids Res. 35, 6588–6597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Budd M. E., Sitney K. C., Campbell J. L. (1989) Purification of DNA polymerase II, a distinct DNA polymerase, from Saccharomyces cerevisiae. J. Biol. Chem. 264, 6557–6565 [PubMed] [Google Scholar]
- 13. Hogg M., Osterman P., Bylund G. O., Ganai R. A., Lundström E. B., Sauer-Eriksson A. E., Johansson E. (2014) Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat. Struct. Mol. Biol. 21, 49–55 [DOI] [PubMed] [Google Scholar]
- 14. Shcherbakova P. V., Pavlov Y. I., Chilkova O., Rogozin I. B., Johansson E., Kunkel T. A. (2003) Unique error signature of the four-subunit yeast DNA polymerase ϵ. J. Biol. Chem. 278, 43770–43780 [DOI] [PubMed] [Google Scholar]
- 15. Zahurancik W. J., Klein S. J., Suo Z. (2014) Significant contribution of the 3′ → 5′ exonuclease activity to the high fidelity of nucleotide incorporation catalyzed by human DNA polymerase. Nucleic Acids Res. 42, 13853–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Albertson T. M., Ogawa M., Bugni J. M., Hays L. E., Chen Y., Wang Y., Treuting P. M., Heddle J. A., Goldsby R. E., Preston B. D. (2009) DNA polymerase ϵ and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 17101–17104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palles C., Cazier J. B., Howarth K. M., Domingo E., Jones A. M., Broderick P., Kemp Z., Spain S. L., Guarino Almeida E., Salguero I., Sherborne A., Chubb D., Carvajal-Carmona L. G., Ma Y., Kaur K., Dobbins S., Barclay E., Gorman M., Martin L., Kovac M. B., Humphray S., CORGI Consortium, WGS 500 Consortium, Lucassen A., Holmes C. C., Bentley D., Donnelly P., Taylor J., Petridis C., Roylance R., Sawyer E. J., Kerr D. J., Clark S., Grimes J., Kearsey S. E., Thomas H. J., McVean G., Houlston R. S., Tomlinson I. (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 45, 136–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Church D. N., Briggs S. E., Palles C., Domingo E., Kearsey S. J., Grimes J. M., Gorman M., Martin L., Howarth K. M., Hodgson S. V., NSECG Collaborators, Kaur K., Taylor J., Tomlinson I. P. (2013) DNA polymerase ϵ and δ exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 22, 2820–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cancer Genome Atlas Research Network, Kandoth C., Schultz N., Cherniack A. D., Akbani R., Liu Y., Shen H., Robertson A. G., Pashtan I., Shen R., Benz C. C., Yau C., Laird P. W., Ding L., Zhang W., Mills G. B., Kucherlapati R., Mardis E. R., Levine D. A. (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kane D. P., Shcherbakova P. V. (2014) A common cancer-associated DNA polymerase ϵ mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 74, 1895–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Navas T. A., Zhou Z., Elledge S. J. (1995) DNA polymerase ϵ links the DNA replication machinery to the S phase checkpoint. Cell 80, 29–39 [DOI] [PubMed] [Google Scholar]
- 22. Jaszczur M., Flis K., Rudzka J., Kraszewska J., Budd M. E., Polaczek P., Campbell J. L., Jonczyk P., Fijalkowska I. J. (2008) Dpb2p, a noncatalytic subunit of DNA polymerase ϵ, contributes to the fidelity of DNA replication in Saccharomyces cerevisiae. Genetics 178, 633–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng W., Rodriguez-Menocal L., Tolun G., D'Urso G. (2003) Schizosacchromyces pombe Dpb2 binds to origin DNA early in S phase and is required for chromosomal DNA replication. Mol. Biol. Cell 14, 3427–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aksenova A., Volkov K., Maceluch J., Pursell Z. F., Rogozin I. B., Kunkel T. A., Pavlov Y. I., Johansson E. (2010) Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymerase ϵ. PLoS Genet. 6, e1001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muramatsu S., Hirai K., Tak Y. S., Kamimura Y., Araki H. (2010) CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ϵ, and GINS in budding yeast. Genes Dev. 24, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sengupta S., van Deursen F., de Piccoli G., Labib K. (2013) Dpb2 integrates the leading-strand DNA polymerase into the eukaryotic replisome. Curr. Biol. 23, 543–552 [DOI] [PubMed] [Google Scholar]
- 27. Sabouri N., Johansson E. (2009) Translesion synthesis of abasic sites by yeast DNA polymerase ϵ. J. Biol. Chem. 284, 31555–31563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morrison A., Bell J. B., Kunkel T. A., Sugino A. (1991) Eukaryotic DNA polymerase amino acid sequence required for 3′–5′ exonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 88, 9473–9477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Asturias F. J., Cheung I. K., Sabouri N., Chilkova O., Wepplo D., Johansson E. (2006) Structure of Saccharomyces cerevisiae DNA polymerase ϵ by cryo-electron microscopy. Nat. Struct. Mol. Biol. 13, 35–43 [DOI] [PubMed] [Google Scholar]
- 30. Sabouri N., Viberg J., Goyal D. K., Johansson E., Chabes A. (2008) Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 36, 5660–5667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zahurancik W. J., Klein S. J., Suo Z. (2013) Kinetic mechanism of DNA polymerization catalyzed by human DNA polymerase ϵ. Biochemistry 52, 7041–7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bloom L. B., Otto M. R., Eritja R., Reha-Krantz L. J., Goodman M. F., Beechem J. M. (1994) Pre-steady-state kinetic analysis of sequence-dependent nucleotide excision by the 3′-exonuclease activity of bacteriophage T4 DNA polymerase. Biochemistry 33, 7576–7586 [DOI] [PubMed] [Google Scholar]
- 33. Capson T. L., Peliska J. A., Kaboord B. F., Frey M. W., Lively C., Dahlberg M., Benkovic S. J. (1992) Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry 31, 10984–10994 [DOI] [PubMed] [Google Scholar]
- 34. Hogg M., Rudnicki J., Midkiff J., Reha-Krantz L., Doublié S., Wallace S. S. (2010) Kinetics of mismatch formation opposite lesions by the replicative DNA polymerase from bacteriophage RB69. Biochemistry 49, 2317–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Donlin M. J., Patel S. S., Johnson K. A. (1991) Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 30, 538–546 [DOI] [PubMed] [Google Scholar]
- 36. Joyce C. M., Benkovic S. J. (2004) DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry 43, 14317–14324 [DOI] [PubMed] [Google Scholar]
- 37. Rothwell P. J., Mitaksov V., Waksman G. (2005) Motions of the fingers subdomain of klentaq1 are fast and not rate limiting: implications for the molecular basis of fidelity in DNA polymerases. Mol. Cell 19, 345–355 [DOI] [PubMed] [Google Scholar]
- 38. Dunlap C. A., Tsai M. D. (2002) Use of 2-aminopurine and tryptophan fluorescence as probes in kinetic analyses of DNA polymerase β. Biochemistry 41, 11226–11235 [DOI] [PubMed] [Google Scholar]
- 39. Johnson K. A. (1993) Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 62, 685–713 [DOI] [PubMed] [Google Scholar]
- 40. Patel S. S., Wong I., Johnson K. A. (1991) Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 [DOI] [PubMed] [Google Scholar]
- 41. Herschlag D., Piccirilli J. A., Cech T. R. (1991) Ribozyme-catalyzed and nonenzymatic reactions of phosphate diesters: rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry 30, 4844–4854 [DOI] [PubMed] [Google Scholar]
- 42. Johnson K. A. (2009) Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods Enzymol. 467, 601–626 [DOI] [PubMed] [Google Scholar]
- 43. Tsubota T., Tajima R., Ode K., Kubota H., Fukuhara N., Kawabata T., Maki S., Maki H. (2006) Double-stranded DNA binding, an unusual property of DNA polymerase epsilon, promotes epigenetic silencing in Saccharomyces cerevisiae. J. Biol. Chem. 281, 32898–32908 [DOI] [PubMed] [Google Scholar]
- 44. Fortune J. M., Pavlov Y. I., Welch C. M., Johansson E., Burgers P. M., Kunkel T. A. (2005) Saccharomyces cerevisiae DNA polymerase δ: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J. Biol. Chem. 280, 29980–29987 [DOI] [PubMed] [Google Scholar]
- 45. Pursell Z. F., Isoz I., Lundström E. B., Johansson E., Kunkel T. A. (2007) Regulation of B family DNA polymerase fidelity by a conserved active site residue: characterization of M644W, M644L and M644F mutants of yeast DNA polymerase ϵ. Nucleic Acids Res. 35, 3076–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Göksenin A. Y., Zahurancik W., LeCompte K. G., Taggart D. J., Suo Z., Pursell Z. F. (2012) Human DNA polymerase ϵ is able to efficiently extend from multiple consecutive ribonucleotides. J. Biol. Chem. 287, 42675–42684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mendelman L. V., Boosalis M. S., Petruska J., Goodman M. F. (1989) Nearest neighbor influences on DNA polymerase insertion fidelity. J. Biol. Chem. 264, 14415–14423 [PubMed] [Google Scholar]
- 48. Joyce C. M., Sun X. C., Grindley N. D. (1992) Reactions at the polymerase active site that contribute to the fidelity of Escherichia coli DNA polymerase I (Klenow fragment). J. Biol. Chem. 267, 24485–24500 [PubMed] [Google Scholar]
- 49. Shinbrot E., Henninger E. E., Weinhold N., Covington K. R., Göksenin A. Y., Schultz N., Chao H., Doddapaneni H., Muzny D. M., Gibbs R. A., Sander C., Pursell Z. F., Wheeler D. A. (2014) Exonuclease mutations in DNA polymerase ϵ reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 24, 1740–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tran H. T., Gordenin D. A., Resnick M. A. (1999) The 3′ → 5′ exonucleases of DNA polymerases δ and ϵ and the 5′ → 3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol. Cell Biol. 19, 2000–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raghuraman M. K., Winzeler E. A., Collingwood D., Hunt S., Wodicka L., Conway A., Lockhart D. J., Davis R. W., Brewer B. J., Fangman W. L. (2001) Replication dynamics of the yeast genome. Science 294, 115–121 [DOI] [PubMed] [Google Scholar]
- 52. Dieckman L. M., Johnson R. E., Prakash S., Washington M. T. (2010) Pre-steady state kinetic studies of the fidelity of nucleotide incorporation by yeast DNA polymerase δ. Biochemistry 49, 7344–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hogg M., Cooper W., Reha-Krantz L., Wallace S. S. (2006) Kinetics of error generation in homologous B-family DNA polymerases. Nucleic Acids Res. 34, 2528–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rivin C. J., Fangman W. L. (1980) Replication fork rate and origin activation during the S phase of Saccharomyces cerevisiae. J. Cell Biol. 85, 108–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yao N. Y., Schroeder J. W., Yurieva O., Simmons L. A., O'Donnell M. E. (2013) Cost of rNTP/dNTP pool imbalance at the replication fork. Proc. Natl. Acad. Sci. U.S.A. 110, 12942–12947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brown J. A., Suo Z. (2009) Elucidating the kinetic mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase B1. Biochemistry 48, 7502–7511 [DOI] [PubMed] [Google Scholar]
- 57. Ayyagari R., Gomes X. V., Gordenin D. A., Burgers P. M. (2003) Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J. Biol. Chem. 278, 1618–1625 [DOI] [PubMed] [Google Scholar]
- 58. Jin Y. H., Ayyagari R., Resnick M. A., Gordenin D. A., Burgers P. M. (2003) Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′-5′-exonuclease activities of Pol δ in the creation of a ligatable nick. J. Biol. Chem. 278, 1626–1633 [DOI] [PubMed] [Google Scholar]
- 59. Aria V., De Felice M., Di Perna R., Uno S., Masai H., Syväoja J. E., van Loon B., Hübscher U., Pisani F. M. (2013) The human Tim-Tipin complex interacts directly with DNA polymerase ϵ and stimulates its synthetic activity. J. Biol. Chem. 288, 12742–12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cho W. H., Kang Y. H., An Y. Y., Tappin I., Hurwitz J., Lee J. K. (2013) Human Tim-Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 110, 2523–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Georgescu R. E., Langston L., Yao N. Y., Yurieva O., Zhang D., Finkelstein J., Agarwal T., O'Donnell M. E. (2014) Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat. Struct. Mol. Biol. 21, 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]